
Abstract
Aims
While nintedanib treatment has been shown to slow the progression of idiopathic pulmonary fibrosis (IPF) in patients across varying levels of lung function, the effect of treatment timing on outcomes has not been examined. We assessed hospitalization risk and medical costs among patients with IPF based on the timing of nintedanib initiation after IPF diagnosis.
Materials and methods
This retrospective administrative claims study included data from 04/01/2014–09/30/2019 for patients aged ≥40 years who initiated nintedanib within 1 year of IPF diagnosis. Patients were assigned to study cohorts based on the time from IPF diagnosis to nintedanib initiation. All-cause hospitalization and all-cause medical costs were modeled using marginal structural models including inverse probability weights to adjust for both baseline and time-varying characteristics.
Results
Of 11,195 patients diagnosed with IPF during the identification period, 449 met the study selection criteria (mean age 72.3 years, 68% male, mean follow-up time 13.3 months). Adjusted hospitalization risk and medical costs both varied significantly by the timing of nintedanib initiation (p < .001 and p = .020, respectively). Adjusted weighted hospitalization risk was higher among untreated vs. treated patients in months 2–3, months 4–6, and months 7–12 after diagnosis (hazard ratio [95% CI] 1.97 [1.09–3.56], p = .026; 2.62 [1.22–5.63], p = .014; and 5.57 [2.31–13.45], p < .001, respectively). Medical costs were 69% higher for patients initiating treatment in months 2–3 vs. month 1 (cost ratio [95% CI] 1.69 [1.20–2.38], p = .003).
Limitations
Disease severity could not be assessed because clinical data were unavailable; however, proxies such as oxygen use were included to adjust for between-cohort differences in disease severity.
Conclusions
Patients who initiate nintedanib promptly after IPF diagnosis may have reduced hospitalization risk and medical costs compared with those who start treatment later. Additional studies are warranted to improve understanding of the impact of prompt antifibrotic therapy on patient outcomes.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, incurable lung disease characterized by progressive lung function declineCitation1. IPF is more common among men than women and occurs primarily among older adultsCitation2–4. Patients with IPF experience diminished quality of life related to dyspnea, cough, and fatigue, and have increased incidence of comorbidities such as pulmonary hypertension, sleep apnea, emphysema, pulmonary embolism, cardiovascular disease, and heart failureCitation5,Citation6. Taken together, these factors contribute to a poor prognosis for patients with IPF, who have a median survival time of only 3–5 years after diagnosis if not treatedCitation7–9.
Until relatively recently, there were no effective pharmacological treatments available for IPF. However, the treatment landscape changed significantly with the emergence of the novel antifibrotics nintedanib and pirfenidone, which were shown in multiple placebo-controlled clinical trials to reduce lung function decline among patients with IPFCitation10–12. On the strength of these findings, nintedanibCitation13 and pirfenidone were approved for the treatment of IPF by the US Food and Drug Administration in 2014 and are now the primary therapeutic options recommended in contemporary IPF treatment guidelinesCitation14.
Results from clinical trials demonstrating the efficacy of nintedanib have since been corroborated by real-world data showing reduced disease progression among patients with IPF treated with nintedanib in clinical practiceCitation15–19. Moreover, results from both real-world studies and post-hoc analyses of pivotal trial data have indicated that nintedanib is beneficial regardless of lung function (measured by forced vital capacity, FVC) at the time of treatment initiation, effectively reducing IPF progression among patients with preserved lung function (FVC percent predicted above 70 or 90%) as well as those with more severe disease than was typical of clinical trial participantsCitation16,Citation19–22. This reduction in FVC decline has also been consistent across patient subgroups defined by sex, age, race, smoking status, health-related quality of life, medication use, and baseline gas-exchange impairment (assessed via diffusing capacity of the lungs for carbon monoxide, DLCO)Citation21–24.
Based on the evidence of consistent benefit associated with nintedanib, current recommendations suggest that the majority of individuals diagnosed with IPF receive antifibrotic treatmentCitation14,Citation25. Nevertheless, some clinicians remain hesitant to prescribe antifibrotics for patients who are perceived as having “stable” disease and/or do not have severe symptoms, despite IPF being the prototypical progressive fibrosing ILDCitation26,Citation27. Given the observed variability in the management of patients with this deadly condition, it is important to better understand the effects of timing of treatment initiation for IPF; for example, while it is plausible that taking earlier action to slow lung function decline may translate into better patient outcomes, this question has not yet been examined. This study was performed to help address this knowledge gap by assessing hospitalization risk and medical costs among patients with IPF based on the timing of nintedanib initiation after IPF diagnosis.
Methods
Study design and data source
This was a retrospective observational study conducted using administrative claims data from the Optum Research Database (ORD) from 01 April 2014 through 30 September 2019 (study period). The ORD is a deidentified patient-level database containing medical and pharmacy claims data and linked enrollment information for individuals enrolled in health plans across the US. Medical claims in the ORD include diagnosis codes from the International Classification of Diseases, 9th and 10th Revisions, Clinical Modification (ICD-9-CM and ICD-10-CM); procedure codes from the ICD-10 Procedure Coding System; Current Procedural Terminology or Healthcare Common Procedure Coding System codes; and site of service codes. Pharmacy claims include drug name, National Drug Code, dosage form, drug strength, and fill date for a health plan–paid outpatient pharmacy services.
Institutional review board approval or waiver of approval was not required for this study, as no identifiable protected health information was accessed.
Patient selection
This study included commercial and Medicare Advantage with Part D (MAPD) health plan enrollees who were diagnosed with IPF (≥2 non-diagnostic medical claims on separate dates with ICD-9-CM code 516.31 and/or ICD-10-CM code J84.112) from 01 October 2014 through 30 June 2019 (identification period) and initiated nintedanib within 1 year of the first IPF diagnosis date (defined as the index date). Included patients were also required to have ≥1 medical claim with a procedure code for high-resolution computed tomography (HRCT) or lung biopsy during the identification period or the 6 months before the index date (baseline period), to be aged ≥40 years as of the index year, and to have continuous health plan enrollment with medical and pharmacy benefits during the 6-month baseline period and for at least 3 months after and including the index date, or less if due to death (follow-up period).
Patients were excluded if they had prior IPF diagnosis and/or treatment (≥1 medical claim with an IPF diagnosis code and/or ≥1 pharmacy claim for nintedanib or pirfenidone during the baseline period); evidence of connective tissue disease (≥2 medical claims on different dates of service during the baseline or follow-up periods with a diagnosis code for rheumatoid arthritis, sarcoidosis, systemic lupus erythematosus, dermato/polymyositis, systemic sclerosis, Sjogren’s disease, or mixed connective tissue disease; codes available upon request); or unknown or missing demographic information (age, sex, geographic region, insurance type).
Study variables
The time from IPF diagnosis to nintedanib initiation was captured as a categorical variable with initiation in month 1, months 2–3, months 4–6, or months 7–12 after the index date. Included patients were assigned to study cohorts based on these categories ().
Figure 1. Patient identification and attrition. Abbreviations. HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; MAPD, Medicare Advantage with Part D.
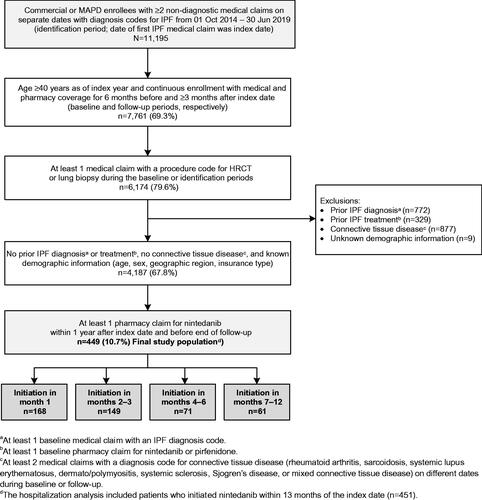
Variables assessed during the baseline period included patient demographic and clinical characteristics, baseline all-cause healthcare resource utilization (HCRU), and baseline all-cause healthcare costs. Demographic characteristics (age, sex, geographic region, health plan type) were identified from patient enrollment information. Clinical characteristics included Charlson comorbidity scoreCitation28, comorbidity categories identified using Clinical Classifications Software from the Agency for Healthcare Research and QualityCitation29, and IPF-related assessments and treatments. These clinical characteristics were identified from medical and pharmacy claims in the baseline period, as were baseline all-cause HCRU and costs. Time-dependent covariates were also captured during each month of follow-up and included all-cause ED visits; all-cause office visits; and receipt of spirometry testing, HRCT, oxygen therapy, or pulmonary rehabilitation.
Study outcomes assessed during the follow-up period included all-cause hospitalization and all-cause medical costs. Patients were considered to have an all-cause hospitalization if there was an occurrence of an inpatient stay of any type during the follow-up period. All-cause medical costs comprised the combined health plan–paid and patient-paid costs related to any ambulatory visits, emergency department (ED) visits, inpatient stays, and other medical costs (e.g. laboratory, durable medical equipment, long-term care). All costs were adjusted to 2019 US$ using the annual medical care component of the Consumer Price IndexCitation30.
Statistical analysis
Descriptive analysis included numbers and percentages for categorical variables and means, medians, and standard deviations for continuous variables. Results were presented overall and stratified by treatment initiation cohort. Between-cohort differences were evaluated using Fisher exact tests for categorical variables and two-sample t-tests or analysis of variance for continuous variables. Statistical significance was defined as p < .05.
Marginal structural modeling
Marginal structural modeling between treatment timing and outcomes of hospitalization and cost was used to adjust for confounding from both baseline characteristics and time-varying treatments and assessments. Unlike traditional multivariable modeling, this approach accounts for confounding due to changes in disease severity over time, which can affect both treatment decisions and study outcomes. Each outcome model included weights to adjust for censoring of patients during the follow-up period (censoring weight) and to balance groups of patients who initiated treatment in different months (treatment weight). Monthly weights were calculated using logistic regression with independent variables including month, age, sex, geographic region, index year, Charlson comorbidity score, HCRU, and IPF-related treatments and assessments (see the footnotes to Supplementary Table 1 for a detailed variable list). The dependent variable was censoring in the next month for censoring weights, and initiation of nintedanib in the current month for treatment weights. It was assumed that patients who initiated treatment remained on treatment.
For all-cause hospitalization, the unadjusted weighted outcome model was estimated with a repeated-measures logistic generalized estimating equation including weights (combined censoring and treatment) and a robust variance estimator. Initiation of nintedanib was included as an explanatory variable with outcomes compared between patients who were not yet treated vs. patients treated before the end of month 1, months 2–3, months 4–6, and months 7–12. The hospitalization model included patients who initiated nintedanib within 13 months of the index date (n = 451) to compare outcomes for patients who were untreated vs. treated through the end of month 12. For the adjusted weighted model, the following baseline covariates were included: patient demographics, Charlson comorbidity score category, HCRU, and IPF-related treatments and assessments, including spirometry testing, HRCT, and oxygen therapy.
For all-cause medical costs, the unadjusted weighted outcome was estimated with a generalized estimating equation with log link, gamma distribution, and a robust variance estimator. Initiation of nintedanib was included as an explanatory variable with outcomes compared for treatment initiation in month 1 vs. initiation in months 2–3, months 4–6, or months 7–12. The cost model included patients who initiated nintedanib within 12 months of the index date. The model was run among the subset of patients who had 12 months of follow-up continuous enrollment and included weights to represent the entire sample of patients who met the initial study selection criteria (n = 449). For the adjusted weighted model, the following additional baseline covariates were included: patient demographics, Charlson comorbidity score category, healthcare resource utilization, and IPF-related treatments and assessments, including spirometry testing, HRCT, oxygen therapy, and pulmonary rehabilitation.
For both outcomes of interest, sensitivity models were also run for the exposure of interest (e.g. comparing initiation in month 1 vs. initiation in months 2–12) and to examine the impact of the censoring and treatment weighting model specifications on the results.
Results
Study sample
Of 11,195 commercial and MAPD enrollees with IPF diagnoses during the identification period, a total of 449 met the study selection criteria (). Mean (SD) age was 72.3 (7.6) years, 68% were male, and 78% had MAPD insurance (). Comorbidity burden was high in the overall patient population, with 43% of patients having Charlson comorbidity scores of 1–2, 24% having scores of 3–4, and 5% having scores of 5 or above. Prevalent comorbidities included hypertension, lipid metabolism disorders, and heart disease (more than 60% of patients); chronic obstructive pulmonary disease and upper gastrointestinal disorders (more than one-third of patients); obstructive sleep apnea and heart failure (18% each); and pulmonary hypertension (10%). Baseline patient demographic and clinical characteristics, baseline IPF-related assessments and treatments, and baseline all-cause HCRU/costs were not significantly different for patients across the treatment cohorts, with a few exceptions (index year, presence of baseline spirometry testing, and presence of a baseline HCRT). Mean observation time in the overall population was 13.3 months (range: 1.0–58.8 months).
Table 1. Patient characteristics.
Study outcomes
All-cause hospitalization
The unadjusted weighted risk of all-cause hospitalization during follow-up differed by the timing of nintedanib initiation (p < .001) (; see Supplemental Table 1 for full model). Among patients with IPF who were untreated in the first month after diagnosis, unadjusted weighted all-cause hospitalization risk relative to those who were treated was higher but not statistically different (hazard ratio [HR] [95% CI] 2.12 [0.98–4.59], p = .058). In months 2–3, months 4–6, and months 7–12, patients who were not yet treated vs. patients treated before the end of each time period had significantly higher unadjusted weighted all-cause hospitalization risk (HR [95% CI] 2.04 [1.15–3.64], p = .016; 2.44 [1.21–4.96], p = .013; and 5.82 [2.59–13.08], p < .001; respectively).
Figure 2. Weighted marginal structural models of first all-cause hospitalization.
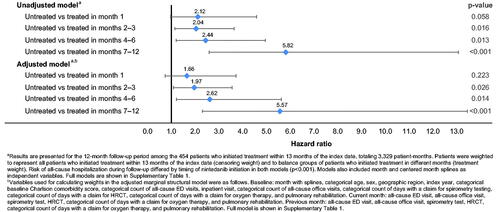
After adjustment for baseline and time-varying characteristics using MSM, all-cause hospitalization risk still varied significantly by the timing of nintedanib initiation (p < .001) (; see Supplemental Table 1 for full model). The adjusted weighted risk (95% CI) of an all-cause hospitalization among patients with IPF who were untreated vs. treated in month 1 was higher but not statistically different (HR [95% CI] 1.66 [0.74–3.73], p = .223). However, in months 2–3, months 4–6, and months 7–12, adjusted weighted all-cause hospitalization risk was significantly higher among patients who were not yet treated vs. patients treated before the end of the time period, similar to the unadjusted weighted results (HR [95% CI] 1.97 [1.09–3.56], p = .026; 2.62 [1.22–5.63], p = .014; and 5.57 [2.31–13.45], p < .001, respectively).
The results of sensitivity analyses in which the exposure of interest was changed (comparing patients who were treated in month 1 vs. months 2–12) (Supplementary Tables 2, 3) or the specifications for censoring and treatment weighting were varied (data not shown) were similar to those of the primary analysis.
All-cause medical costs
Unadjusted weighted 12-month all-cause medical costs were not statistically different by nintedanib initiation month (p = .289) (). After adjustment for baseline characteristics, 12-month medical costs differed significantly by the timing of nintedanib initiation (p = .020) and were 69% higher for patients who initiated nintedanib in months 2–3 vs. month 1 with estimated mean costs of $29,423 for patients initiating in months 2–3 vs. $17,428 for month 1 (cost ratio [95% CI] 1.69 [1.20–2.38], p = .003) (). Costs for patients initiating in months 4–6 ($21,045) and months 7–12 ($23,459) vs. month 1 were higher by 21% and 35%, respectively; however, these differences were not statistically significant (cost ratio [95% CI] 1.21 [0.81–1.81], p = .359; and cost ratio [95% CI] 1.35 [0.91–2.00], p = .140, respectively).
Table 2. Unadjusted weighted marginal structural model of 12-month all-cause medical costsa.
Table 3. Adjusted weighted marginal structural model of 12-month all-cause medical costsa,b.
The results of sensitivity analyses in which the exposure of interest was changed (comparing patients who initiated treatment in month 1 vs. months 2–12) (Supplementary Tables 4, 5) or the specifications for censoring and treatment weighting were varied (data not shown) were similar to those of the primary analysis.
Discussion
This retrospective claims analysis is the first study to our knowledge to examine the effect of timing of treatment initiation with nintedanib on outcomes among patients with IPF. Among 449 patients with IPF who initiated nintedanib, both all-cause hospitalization risk and all-cause medical costs varied significantly by the length of time from IPF diagnosis to treatment. Adjusted weighted all-cause hospitalization risk was significantly higher among untreated vs. treated patients in months 2–3, months 4–6, and months 7–12 after diagnosis. Medical costs were 69% higher for patients initiating treatment in months 2–3 vs. month 1. Compared with initiation in month 1, costs were also numerically higher for initiation in months 4–6 and months 7–12; however, these differences were not statistically significant. Our findings suggest that treatment delays may facilitate unabated disease progression, leading to increases in hospitalization risk and associated healthcare costs.
These results are congruent with an existing body of evidence indicating that patients with IPF may benefit from prompt antifibrotic treatmentCitation16,Citation19–22. Post-hoc analyses of data from pivotal trials have shown that nintedanib reduces disease progression and may improve survival among patients across the spectrum of baseline lung function, including those with FVC ≤70 vs. >70% or ≤90 vs. >90% and those with DLCO ≤40 vs. >40%Citation21–24,Citation31. A subsequent retrospective claims analysis showed that patients treated with nintedanib or pirfenidone as part of normal clinical practice had a lower risk of all-cause mortality and hospitalization than untreated patients, confirming the real-world effectiveness of antifibroticsCitation32. Despite these findings, a variety of perspectives regarding IPF management persists in current practice, with some clinicians hesitating to offer antifibrotic therapy to patients soon after diagnosisCitation26,Citation27. Common reasons cited for this approach include perceptions that the patient has “stable” disease, does not have severe symptoms, and/or has good lung functionCitation26. The concept of stable IPF is problematic, however, as IPF is progressive by nature; data from both clinical trials and real-world studies indicate that patients with IPF, regardless of their FVC at diagnosis, inevitably experience lung function decline over timeCitation16,Citation21,Citation33. For example, in phase III trials of nintedanib, more than 75% of placebo-treated patients with FVC >90% at baseline had substantial lung function decline over the subsequent yearCitation21. In addition, antifibrotic therapy has been shown to decrease the incidence of acute exacerbationsCitation10,Citation23,Citation34, which are potentially deadly and can occur suddenly even among individuals not presently considered to have severe diseaseCitation21,Citation27,Citation35. Improved understanding of the clinical signs of IPF has helped individuals with this condition to be identified at earlier stages in the disease process, when lung function may still be largely preservedCitation14,Citation36. As fibrotic lung damage in IPF is irreversible, delayed treatment of patients diagnosed with this condition may miss a window of opportunity to start slowing lung function decline earlier in the disease course, reduce the incidence of acute exacerbations, and potentially even extend survivalCitation37. Our results lend support to this concept by demonstrating that prompt antifibrotic treatment after IPF diagnosis may translate into a measurable benefit for real-world patients.
Although all adjusted outcomes examined in this study were directionally consistent with lower all-cause hospitalization risk and all-cause medical costs among patients initiating nintedanib treatment in the first month after diagnosis vs. later, some comparisons did not reach statistical significance. Nevertheless, our findings suggest that further investigation of treatment timing in IPF is warranted, particularly in light of burgeoning advocacy for treatment approaches that offer antifibrotic therapy to patients at the time of diagnosisCitation16,Citation21,Citation27,Citation38–41.
Study limitations
The results of this study should be considered in light of certain limitations. First, the study sample comprised only US commercial and MAPD health plan enrollees; therefore, the findings are most applicable to insured US patients and the generalizability to other populations is unknown. Given the selection criteria, generalizability is further restricted to patients who initiate nintedanib treatment within a year of IPF diagnosis. Second, results may have been affected by the specifications for censoring, treatment weighting, and outcome models used in the marginal structural models of all-cause hospitalization and all-cause medical costs; however, sensitivity analyses in which these specifications were varied had results similar to those of the main analysis (data not shown). Third, although the use of MSM to adjust for time-varying confounders is a strength of this study, there is some potential for residual confounding or unobserved confounding by factors not measured in the study. Fourth, small sample sizes for patients who initiated treatment later after diagnosis may have contributed to the wide confidence intervals observed for some study outcomes. Future studies conducted among larger patient populations may provide more precise estimates of the effect of nintedanib treatment timing on relative hospitalization risk and healthcare costs.
This study was also subject to several limitations inherent to claims analyses. The presence of ICD-9-CM or ICD-10-CM diagnosis codes on medical claims does not confirm the diagnosis, as codes may have been entered incorrectly or included as rule-out conditions. To minimize this risk, patients were selected using non-diagnostic medical claims only, and required to have ≥2 non-diagnostic medical claims on separate dates with diagnosis codes for IPF as well as IPF-related treatment and testing. Similarly, the presence of a claim for a filled prescription does not indicate that the medication was consumed or taken as prescribed. Finally, because laboratory values and results from assessments, such as FVC, DLCO, and HCRT are not readily available in claims data, proxies such as oxygen use were utilized as surrogate markers for disease severity. While these proxies were intended to help account for any differences in disease severity across cohorts, it is uncertain how well they were able to account for differences that could not be observed with imaging results or lung function measures.
Conclusions
Our findings suggest that patients who initiate nintedanib promptly after IPF diagnosis may have reduced hospitalization risk and medical costs compared with those who start treatment later. Additional investigation of the effect of antifibrotic treatment timing on outcomes for this fatal condition is warranted.
Transparency
Declaration of funding
This work was funded by Boehringer Ingelheim Pharmaceuticals, Inc (BIPI).
Declaration of financial/other relationships
CC and SS are employees of Boehringer Ingelheim (BI). DS was an employee of BI at the time the study was conducted. JDA was a paid consultant for BI for this study. LGSB, AA, and LB are employees of Optum, which was contracted by BI to conduct the study.
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
DS, LGSB, CC, and SS conceptualized and designed the study. LGSB, AA, and LB collected and analyzed the data. All authors interpreted the data, critically revised the manuscript, and approved the final version for publication.
Supplemental Material
Download MS Word (31.9 KB)Acknowledgements
Medical writing assistance was provided by Yvette Edmonds, Ph.D. (Optum), and was contracted and funded by BIPI.
References
- Hiwatari N, Shimura S, Takishima T. Pulmonary emphysema followed by pulmonary fibrosis of undetermined cause. Respiration. 1993;60(6):354–358.
- Raghu G, Chen SY, Hou Q, et al. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J. 2016;48(1):179–186.
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566–572.
- Raimundo K, Chang E, Broder MS, et al. Clinical and economic burden of idiopathic pulmonary fibrosis: a retrospective cohort study. BMC Pulm Med. 2016;16(2):2.
- Collard HR, Ward AJ, Lanes S, et al. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ. 2012;15(5):829–835.
- Caminati A, Lonati C, Cassandro R, et al. Comorbidities in idiopathic pulmonary fibrosis: an underestimated issue. Eur Respir Rev. 2019;28(153):190044.
- Flaherty KR, Toews GB, Travis WD, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur Respir J. 2002;19(2):275–283.
- King TE Jr., Tooze JA, Schwarz MI, et al. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001;164(7):1171–1181.
- Rudd RM, Prescott RJ, Chalmers JC, et al. Fibrosing alveolitis subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax. 2007;62(1):62–66.
- Richeldi L, Cottin V, Du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS((R)) trials. Respir Med. 2016;113:74–79.
- Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087.
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769.
- OFEV (nintedanib) capsules, for oral use [Package insert]. 2014 [cited 2022 Mar 7]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205832s000lbl.pdf.
- Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–e19.
- Kang J, Han M, Song JW. Antifibrotic treatment improves clinical outcomes in patients with idiopathic pulmonary fibrosis: a propensity score matching analysis. Sci Rep. 2020;10(1):15620.
- Noor S, Nawaz S, Chaudhuri N. Real-World study analysing progression and survival of patients with idiopathic pulmonary fibrosis with preserved lung function on antifibrotic treatment. Adv Ther. 2021;38(1):268–277.
- Brunnemer E, Walscher J, Tenenbaum S, et al. Real-World experience with nintedanib in patients with idiopathic pulmonary Fibrosis. Respiration. 2018;95(5):301–309.
- Rivera-Ortega P, Hayton C, Blaikley J, et al. Nintedanib in the management of idiopathic pulmonary fibrosis: clinical trial evidence and real-world experience. Ther Adv Respir Dis. 2018;12:1753466618800618.
- Hughes G, Toellner H, Morris H, et al. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med. 2016;5(9):78.
- Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082.
- Kolb M, Richeldi L, Behr J, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72(4):340–346.
- Costabel U, Inoue Y, Richeldi L, et al. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. Am J Respir Crit Care Med. 2016;193(2):178–185.
- Richeldi L, Kolb M, Jouneau S, et al. Efficacy and safety of nintedanib in patients with advanced idiopathic pulmonary fibrosis. BMC Pulm Med. 2020;20(1):3.
- Brown KK, Flaherty KR, Cottin V, et al. Lung function outcomes in the INPULSIS® trials of nintedanib in idiopathic pulmonary fibrosis. Respir Med. 2019;146:42–48.
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68.
- Maher TM, Swigris JJ, Kreuter M, et al. Identifying barriers to idiopathic pulmonary fibrosis treatment: a survey of patient and physician views. Respiration. 2018;96(6):514–524.
- Maher TM, Molina-Molina M, Russell AM, et al. Unmet needs in the treatment of idiopathic pulmonary fibrosis-insights from patient chart review in five European countries. BMC Pulm Med. 2017;17(1):124.
- Quan H, Li B, Couris CM, et al. Updating and validating the Charlson Comorbidity Index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am J Epidemiol. 2011;173(6):676–682.
- Clinical Classifications Software (CCS) for ICD-9-CM/ICD-10/CM. Rockville (MD): Agency for Healthcare Research and Quality. http://www.hcup-us.ahrq.gov/toolssoftware/ccs/ccs.jsp.
- US Department of Labor, Bureau of Labor Statistics. Consumer Price Index. Medical Care. Series ID: CUUR0000SAM; 2019 [cited 2020 Nov 29]. http://data.bls.gov/cgi-bin/surveymost?cu
- Kolb M, Raghu G, Wells AU, et al. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2018;379(18):1722–1731.
- Dempsey TM, Sangaralingham LR, Yao X, et al. Clinical effectiveness of antifibrotic medications for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200(2):168–174.
- Russell AM, Adamali H, Molyneaux PL, et al. Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194(8):989–997.
- Collard HR, Richeldi L, Kim DS, et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur Respir J. 2017;49(5):1601339.
- Kolb M, Bondue B, Pesci A, et al. Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180071.
- Cottin V, Richeldi L. Neglected evidence in idiopathic pulmonary fibrosis and the importance of early diagnosis and treatment. Eur Respir Rev. 2014;23(131):106–110.
- Reichmann WM, Yu YF, Macaulay D, et al. Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm Med. 2015;15(1):167.
- Torrisi SE, Pavone M, Vancheri A, et al. When to start and when to stop antifibrotic therapies. Eur Respir Rev. 2017;26(145):170053.
- Wuyts WA, Wijsenbeek M, Bondue B, et al. Idiopathic pulmonary fibrosis: best practice in monitoring and managing a relentless fibrotic disease. Respiration. 2020;99(1):73–82.
- Molina-Molina M, Aburto M, Acosta O, et al. Importance of early diagnosis and treatment in idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2018;12(7):537–539.
- Maher TM, Strek ME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir Res. 2019;20(1):205.