
ABSTRACT
Background
MERTK (MER proto-oncogene, tyrosine kinase) is a transmembrane protein essential in regulating photoreceptor outer segment phagocytosis. Biallelic mutations in MERTK cause retinal degeneration. Here we present the retinal phenotype of three patients with missense variants in MERTK.
Materials and methods
All patients underwent a full clinical examination, fundus photography, short-wavelength fundus autofluorescence and optical coherence tomography imaging. Two patients also underwent Goldmann visual field testing and electroretinography was undertaken for the third patient. Molecular genetic testing was undertaken using next generation or whole-exome sequencing with all variants confirmed by Sanger sequencing.
Results
The first patient was a 29-year-old female heterozygous for a missense variant (c.1133C>T, p.Thr378 Met) and a nonsense variant (c.1744_1751delinsT, p.Ile582Ter) in MERTK. The second patient was a 26-year-old male homozygous for a c.2163T>A, p.His721Gln variant in MERTK. The third patient was an 11-year-old female heterozygous for a deletion of exons 5–19 and a missense variant (c.1866 G>C, p.Lys622Asn) in MERTK. Reduced night vision was the initial symptom in all patients. Fundoscopy revealed typical signs of retinitis pigmentosa (RP) with early-onset macular atrophy. All three MERTK missense variants affect highly conserved residues within functional domains, have low population frequencies and are predicted to be pathogenic in silico.
Conclusions
We report three missense variants in MERTK and present the associated phenotypic data, which are supportive of non-syndromic RP. MERTK is a promising candidate for viral-mediated gene replacement therapy. Moreover, one variant represents a single nucleotide transition, which is theoretically targetable with CRISPR-Cas9 base-editing.
Introduction
Retinitis pigmentosa (RP) is a set of clinically and genetically heterogenous inherited retinal dystrophies characterised by progressive primary rod photoreceptor degeneration with concurrent, or later degeneration of cones (Citation1). It typically manifests with difficulties in dark adaptation and night vision, followed by progressive peripheral visual field loss, with subsequent loss of central vision. A vast number of heterogenous genetic defects have been implicated in the pathogenesis of non-syndromic RP (Citation2,Citation3).
The MER proto-oncogene, tyrosine kinase (MERTK) gene encodes a transmembrane protein expressed in the retinal pigment epithelium (RPE), which plays a critical role in photoreceptor homeostasis by regulation of phagocytosis of shed photoreceptor outer segment discs (, panel A) (Citation4,Citation5). Numerous mutations in MERTK have been identified as pathogenic for retinal dystrophies (Citation6,Citation7). MERTK mutations cause a rod-cone dystrophy with early macular atrophy, with RP being the most common retinal phenotype, although cases of Leber congenital amaurosis have been reported (Citation7).
Figure 1. Panel A: structure of the MERTK transmembrane protein. The extracellular portion includes two immunoglobulin-like (Ig-like) domains (green) and two fibronectin type III (FN-III) domains (blue). The intracellular region contains a highly conserved kinase domain (yellow). The location of the Three mutations discussed are indicated by red arrows. The respective amino acid residues corresponding to the domains affected are indicated below the protein schematic. Panel B: Conservation across species of the amino acid residue subject to mutation in the c.1133c>t, p.Thr378 met variant (B1), the c.2163t>a, p.His721gln variant (B2) and the c.1866G>C, p.Lys622Asn variant (B3). Panel C: Pedigrees for Case 1 (C1) and Case 2 (C2). Pedigree not available for Case 3. Note the pedigrees are based on clinical presentation and segregation has not been possible.
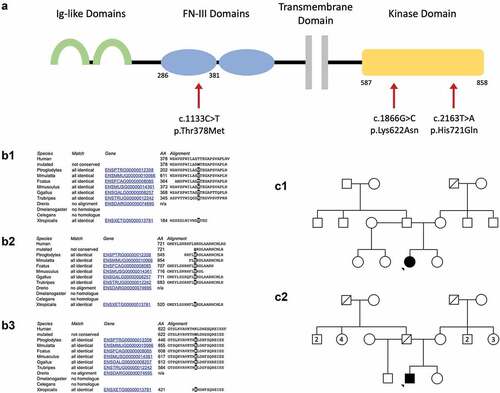
Herein, we describe three patients who presented with an autosomal recessive RP phenotype and were identified to have biallelic MERTK mutations. These missense variants in MERTK have been previously reported in isolation without detailed phenotypic data (Citation7–10). Reporting rare missense mutations and describing the associated phenotype builds the evidence base for the pathogenicity of a given variant, identifies exons that are essential for retinal function and identifies variants that are targetable by currently available gene-editing tools.
Materials and methods
The three patients were referred to the Oxford Eye Hospital, a tertiary referral centre for inherited retinal degenerations. Data were collected retrospectively using clinical information and investigations undertaken as part of their routine clinical care. These included best-corrected visual acuity, Goldmann kinetic visual field testing, slit lamp and fundoscopic examination. Retinal images included digital fundus photography (Optos, Dunfermline, Scotland); short-wavelength fundus autofluorescence (FAF) with optical coherence tomography (OCT) imaging (Heidelberg Spectralis, Heidelberg Engineering GmbH, Heidelberg, Germany). Full-field electroretinography was undertaken according to ISCEV standards for Case 3.
Molecular genetic testing was undertaken as previously described (Citation11). For Cases 1 and 2, a panel of 111 genes associated with RP or RP-like phenotypes was utilised for genetic characterisation. A customised HaloPlex Target Enrichment system (Agilent Technologies, Santa Clara, United States) was used, and sequencing was done with the Illumina MiSeq technology. For Case 3, TWIST Singleton whole-exome sequencing with targeted analysis of a panel of 199 genes associated with R32 Retinal disorders was used. The Twist Human Core Exome Multiplex Hybridization Kit (Twist Bioscience, San Francisco, United States) was used as the method of enrichment and the Illumina NovaSeq6000 technology was used for sequencing. Both sequencing technologies involve data generated by the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics, Oxford. Data were analysed using a validated in-house analysis pipeline. All variants identified were confirmed by Sanger sequencing. For Case 3, SNP microarray was used to confirm the large deletion. SNP microarray processing was carried out using the Illumina GSAv3 microarray (>665,000 probes, mean OMIM gene resolution 15kb minimum). Analysis and interpretation were carried out using NxClinical 6.1 (BioDiscovery) and the FASST2 CNV calling algorithm on human genome build GRCh37(hg19).
Results
Case 1
A 29-year-old female was referred to our clinic for assessment of RP which was diagnosed 6 years earlier. She reported progressive nyctalopia from childhood with worsening daytime vision and glare. No other family member had a history of RP (, panel C1) and there was no known consanguinity in her family who was originally from Cyprus. Ophthalmic assessment performed abroad as a young child did not detect any abnormalities.
On examination, best corrected visual acuity was 6/24 in the right eye and 6/36 in the left eye. She had normal anterior segments with no cataract. There were a few vitreous cells bilaterally. Retinal examination demonstrated bilateral attenuated vessels and sparse bone spicule pigmentation, consistent with RP. FAF imaging showed patchy hypoautofluorescent spots most marked in the nasal midperiphery and along the supero-temporal arcade. OCT demonstrated extensive photoreceptor loss with a partially preserved foveal ellipsoid zone (). Goldmann visual fields were constricted bilaterally ().
Figure 2. Retinal imaging studies for Case 1: (a, b) colour photographs of right and left fundi demonstrating attenuated vessels and waxy disc pallor but not capturing sparse bone spicule pigmentations; (c, d) widefield FAF imaging (55 degrees) demonstrating patchy autofluorescence in the nasal midperiphery and along the supero-temporal arcades; (e, f) OCT imaging showing extensive photoreceptor loss and outer retinal degeneration, with a partially preserved foveal ellipsoid zone.
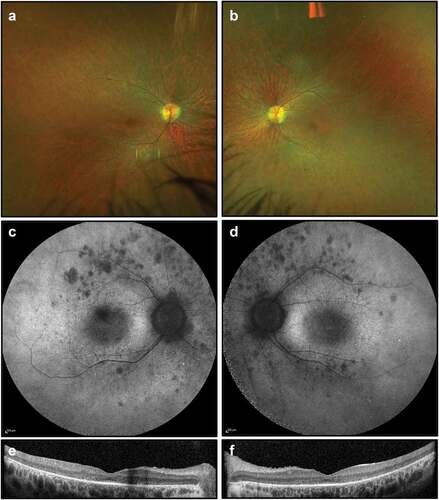
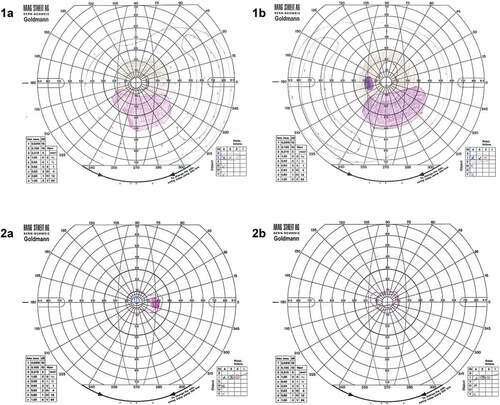
Figure 3. Goldmann visual fields for Case 1 (1a) right and (1b) left eyes showing a bilateral residual temporal island as is often seen in advanced RP with a bilateral inferior Scotoma that matches the pattern of degeneration on FAF imaging in . Case 2 (2a) right and (2b) left demonstrates generalised, severe visual field constriction with the V4 isopter constricted to approximately 10 degrees in both eyes.
Molecular genetic testing identified a missense variant in MERTK (c.1133C>T, p.Thr378 Met, genomic coordinates Chr2.hg19:g. 112,733,038) and a heterozygous nonsense variant in MERTK (c.1744_1751delinsT, p.Ile582Ter, genomic coordinates Chr2.hg19:g. 112760722_112760728). The c.1133C>T variant affects the highly conserved amino acid residue threonine in position 378, found within the extracellular fibronectin type III functional domain (, panel B1). It has a low population frequency in genetic databases and is predicted to be pathogenic in silico (, for additional details refer to Supplements). It was not possible to obtain segregation studies for this patient.
Table 1. Summary of the characteristics of the MERTK missense variants.
Case 2
A 26-year-old male was seen in our clinic with gradual visual loss over the preceding 2 years. He first noticed night and colour vision problems as a teenager. His vision subsequently declined over recent years with marked glare and reading difficulties. There was no known family history of vision problems (, panel C2). His parents were from the same region in Albania, but there was no known consanguinity. The younger sister was also examined and had a normal fundoscopic examination.
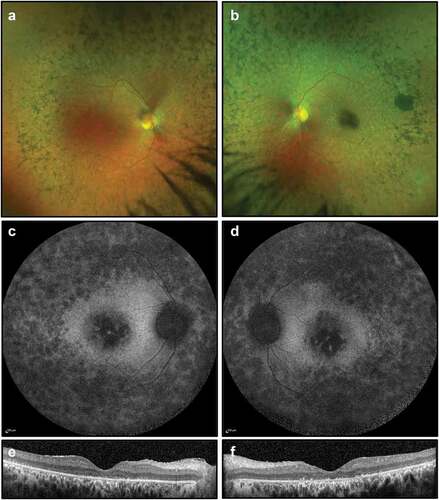
At the time of presentation (26 years of age), best-corrected visual acuity was 6/12–1 in the right eye and 6/60 in the left eye. This deteriorated over 3 years to 6/48 in the right eye and counting fingers in the left eye. On examination, he had bilateral mild posterior subcapsular cataract, vitreous cells, retinal arteriolar constriction, atrophic retinal changes with bone spicule pigmentation and marked macular atrophy (). FAF imaging demonstrated widespread peripheral and macular atrophy, with no ring of increased autofluorescence. OCT showed extensive photoreceptor loss, outer retinal degeneration, presence of subretinal material and absence of foveal ellipsoid zone (). 18 months later, OCT imaging showed mild macular cystoid changes, more pronounced in the right eye. Dorzolamide drops, used three times daily, were trialled for degenerative cystoid macular oedema with minimal clinical improvement. His visual acuity, dark adaptation and visual fields deteriorated significantly to the point where he was unable to work.
Figure 4. Retinal imaging for Case 2: (a, b) colour photographs of fundi demonstrating widespread peripheral bone spicule pigmentations and attenuated vessels; (c, d) widefield FAF imaging (55 degrees) demonstrating widespread autofluorescence atrophy and macular atrophy with no ring of increased autofluorescence; (e, f) OCT imaging showing extensive photoreceptor loss, outer retinal degeneration, absence of foveal ellipsoid zone and presence of subretinal material, thought to be secondary to the defective phagocytic process by the RPE.
Molecular genetic testing identified a homozygous variant in MERTK (c.2163T>A, p.His721Gln, genomic coordinates Chr2.hg19:g.112,777,073). This causes a substitution of the amino acid residue in position 721 from histidine to glutamine within the highly conserved intracellular tyrosine kinase domain (, panel B2). It has only been identified in 1 of 56,868 Non-Finnish European individuals in genetic databases and is predicted to be pathogenic in silico (, for additional details refer to Supplements).
Case 3
An 11-year-old female was referred to our clinic for specialist assessment due to parental concerns regarding poor night vision. She reported problems with low light and night vision for as long as she could remember. There was no known family history of any retinal dystrophies or eye conditions and no known consanguinity.
Best corrected visual acuity was 6/9 in both eyes, with no improvement on pinhole testing. On examination, she had healthy anterior segments, thin retina with pigment migration in both eyes. FAF imaging showed bilateral foveal hyper-autofluorescence and OCT demonstrated macular thinning with outer retinal degeneration (). Full-field electroretinography demonstrated absent rod responses and severely attenuated cone responses.
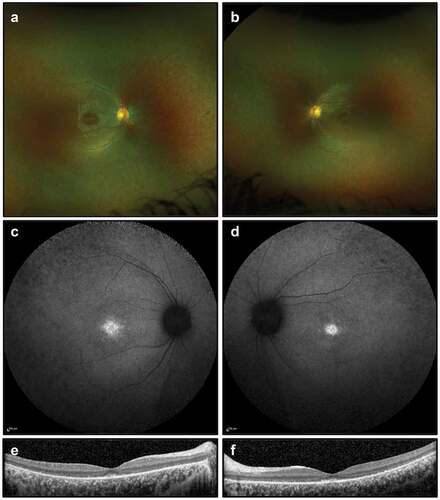
Figure 5. Retinal imaging for Case 3: (a, b) colour photographs of fundi demonstrating sparse peripheral pigment migration; (c, d) widefield FAF imaging (55 degrees) showing foveal hyper-autofluorescence; (e, f) OCT imaging showing outer retinal degeneration with some preservation of the foveal ellipsoid zone.
Molecular genetic testing identified a pathogenic heterozygous deletion of exons 5–19 in MERTK (genomic coordinates Chr2.hg19:g 112,722,768– 112,786,441), confirmed using a SNP microarray. NGS dosage data suggested that the deletion event may have included exons 3 and 4; however due to probe coverage on the SNP microarray it was only possible to confirm a deletion of MERTK exons 5–19. It also identified a heterozygous missense variant in MERTK (c.1866 G>C, p.Lys622Asn, genomic coordinates Chr2.hg19:g. 112,761,560) of uncertain significance, previously reported by Ellingford et al (Citation8). The c.1866C>G missense variant affects the highly conserved lysine amino acid residue in position 622 (, panel A and B3). It has a low population frequency in genetic databases and is predicted to have a deleterious effect upon protein function in silico (, for additional details refer to Supplements). It was not possible to obtain segregation studies for the patient.
Discussion
The MERTK gene is located on the long arm of chromosome 2 (2q14.1). It encodes the MER receptor tyrosine kinase, a 999 amino acid transmembrane protein that functions as a signalling molecule in the phagocytic pathway in the RPE (Citation4). As part of the process of photoreceptor outer segment disc renewal, shed outer segments discs are phagocytosed by the RPE cells (Citation5–7). This is a continual process essential in preventing accumulation of toxic debris in the subretinal space. The association between retinal degeneration and the MERTK gene was first identified in the Royal College of Surgeons rat (Citation12,Citation13). In this naturally occurring model of retinal dystrophy, the failure the RPE to phagocytose shed discs (Citation14) leads to accumulation of debris and photoreceptor cell degeneration (Citation15,Citation16). Autosomal recessive variants in MERTK were subsequently identified in humans with an RP phenotype, confirming the mechanism of defective RPE-mediated phagocytosis in human retinal disease for the first time (Citation6). To date, numerous MERTK variants have been reported in patients with inherited retinal dystrophies, with 79 variants identified up to 2018 (Citation7) and further variants reported in more recent years (Citation17–20). Inherited retinal dystrophies associated with MERTK mutations are characterised by early macular involvement in addition to more peripheral retinal abnormalities (Citation7,Citation21), and the reduced outer segment phagocytosis seems to result in reduced lipofuscin accumulation (Citation22).
The c.1133C>T, p.Thr378 Met missense variant identified in Case 1 has only been previously reported in a homozygous 21-year-old male, without presentation of detailed clinical data (Citation9). The variant involves a change in polarity from a polar uncharged to a non-polar hydrophobic amino acid. The rarity of the variant, its position within the fibronectin type III (FN III) functional domain (Citation23) and the high degree of sequence conservation of the amino acid residue across species, increase the likelihood of its pathogenicity ().
The c.1744_1751delinsT non-sense truncating variant also present in the first patient has been previously described in a case of autosomal recessive Leber Congenital Amaurosis in Turkey (Citation24). It is predicted to cause nonsense-mediated mRNA decay or premature termination of translation producing a shorter protein missing the kinase domain. Consequently, this mutation is predicted to be an autosomal recessive pathogenic variant with deleterious effect on protein structure and function (Citation7), which suggests this is a functional null allele. Therefore, in the presence of a severe truncating mutation and a moderately severe RP phenotype, we hypothesise that c.1133C>T is possibly a hypomorphic variant that might account for reduced protein function.
The second patient was homozygous for the c.2163T>A, p.His721Gln variant. This has been described in a female of Albanian descent by Audo et al. (Citation7) and in a homozygous 28-year-old male by Colombo et al (Citation10). The phenotype presented by Audo et al is similar to that of the case we report here, mindful of differences in severity due to age at evaluation (26 years versus 43 years). The variant results in the substitution of a histidine residue at position 721 (polar, positively charged) to glutamine (polar, uncharged) within the highly conserved intracellular tyrosine kinase domain (Citation23). The rarity of the variant, conservation of the residue across species, presence in a functional domain and in silico prediction tools support the likelihood that this variant is indeed pathogenic for RP and may represent a founder mutation from Albania or the surrounding region ().
The third case was heterozygous for the c.1866G>C, p.Lys622Asn missense variant. This has been described once in the literature by Ellingford et al (Citation8) in a homozygous patient with a reported RP/rod-cone dystrophy phenotype from a British cohort. The variant is predicted to be deleterious by way of substitution of a basic lysine residue by a polar uncharged asparagine residue within the highly conserved intracellular tyrosine kinase domain (Citation23). Our report adds valuable phenotypic and clinical data to characterise this variant and further the evidence regarding its possible pathogenicity.
In recent years, remarkable advances in the field of ocular gene therapy have generated promising therapeutic opportunities for inherited retinal dystrophies (Citation25–28). MERTK has been the object of investigation for treatment strategies including adeno-associated viral (AAV)-mediated retinal gene therapy. This was first investigated in animal models, which yielded successful results (Citation29–31). Vollrath et al (Citation25) found that subretinal administration of recombinant adenovirus encoding MERTK reversed the RPE phagocytosis defect and rescued photoreceptors from degeneration in juvenile rats, so that treated areas appeared near normal in animal models that already demonstrated significant pathology (Citation31). In 2016, Ghazi et al. conducted a phase I clinical trial in a cohort of six patients with RP who exhibited biallelic mutations in MERTK. They established that AAV-mediated subretinal gene therapy had acceptable ocular and systemic safety profile, although the clinical benefit was variable and limited (Citation32). MERTK continues to be a promising target for gene therapy for several reasons: 1) there are validated pre-clinical models available for vector validation; 2) it is expressed in the RPE, which can be targeted efficiently for gene transfer (Citation33,Citation34); 3) the MERTK cDNA sequence (3kb) is encodable within AAV (4.7kb) with sufficient space for the required transcriptional and regulatory elements (Citation35).
A more recent alternative to gene therapy is the utilisation of RNA-programmable CRISPR-associated (CRISPR-Cas) nuclease technology, a powerful tool which allows precise and site-specific modifications of genomic DNA. Base-editing tools consist of catalytically inactivated Cas-nucleases programmed to localize target DNA loci, paired with a base-modification enzyme (Citation36). They permit the direct and irreversible conversion of a specific target DNA base, theoretically enabling the correction of point missense transition mutations in a programmable manner (Citation37–39). The cytosine to thymine single base mutation in the c.1133C>T variant here described might therefore be suitable for targeting with adenine base editors to revert the mutated A•T base pair back to G•C on the reverse strand (Citation39). Prime-editing—a complementary DNA-editing tool that utilises the CRISPR-Cas9 targeting specificity with an RNA template that is inserted following reverse transcription—may edit transversion mutations (Citation40). Therefore, the c.2163T>A and c.1866 G>C variants are theoretically correctable using this technology, or alternatively, by gene replacement therapy.
The identification of missense mutations in MERTK is important to better describe the spectrum of clinical phenotypes, to build the evidence base for pathogenicity of the variant and to identify potential candidates who may benefit from inclusion in clinical trials for retinal gene therapy. Base-editing and prime-editing tools are powerful technologies in which editing of pathogenic missense variants is possible.
Supplemental Material
Download PDF (106 KB)Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/13816810.2022.2113541
Additional information
Funding
References
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–809. doi:10.1016/S0140-6736(06)69740-7.
- RetNet: summaries of genes and loci causing retinal diseases. 2021. https://sph.uth.edu/retnet/sum-dis.htm#A-genes.
- Verbakel SK, van Huet RA, Boon CJ, den Hollander AI, Collin RW, Klaver CC, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86.
- Feng W, Yasumura D, Matthes MT, LaVail MM, Vollrath D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J Biol Chem. 2002;277(19):17016–22. doi:10.1074/jbc.M107876200.
- Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5(11):a009076. doi:10.1101/cshperspect.a009076.
- Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, Apfelstedt-Sylla E, Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26(3):270–71. doi:10.1038/81555.
- Audo I, Mohand‐said S, Boulanger‐scemama E, Zanlonghi X, Condroyer C, Démontant V, Boyard F, Antonio A, Méjécase C, El Shamieh S, Sahel JA. MERTK mutation update in inherited retinal diseases. Hum Mutat. 2018;39(7):887–913. doi:10.1002/humu.23431.
- Ellingford JM, Barton S, Bhaskar S, O’Sullivan J, Williams SG, Lamb JA, Panda B, Sergouniotis PI, Gillespie RL, Daiger SP, et al. Molecular findings from 537 individuals with inherited retinal disease. J Med Genet. 2016;53(11):761–67. doi:10.1136/jmedgenet-2016-103837.
- Takahashi VK, Xu CL, Takiuti JT, Apatoff MBL, Duong JK, Mahajan VB, Tsang SH. Comparison of structural progression between ciliopathy and non-ciliopathy associated with autosomal recessive retinitis pigmentosa. Orphanet J Rare Dis. 2019;14(1):1–9. doi:10.1186/s13023-019-1163-9.
- Colombo L, Maltese PE, Castori M, El Shamieh S, Zeitz C, Audo I, Zulian A, Marinelli C, Benedetti S, Costantini A, et al. Molecular epidemiology in 591 Italian probands with nonsyndromic retinitis pigmentosa and usher syndrome. Invest Ophthalmol Vis Sci. 2021;62(2):13. doi:10.1167/iovs.62.2.13.
- Shanks ME, Downes SM, Copley RR, Lise S, Broxholme J, Hudspith KA, Kwasniewska A, Davies WI, Hankins MW, Packham ER, et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet. 2013;21(3):274–80. doi:10.1038/ejhg.2012.172.
- D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9(4):645–51. doi:10.1093/hmg/9.4.645.
- Nandrot E, Dufour EM, Provost AC, Péquignot MO, Bonnel S, Gogat K, Marchant D, Rouillac C, Sépulchre de Condé B, Bihoreau M-T, et al. Homozygous deletion in the coding sequence of the c-mer gene in RCS rats unravels general mechanisms of physiological cell adhesion and apoptosis. Neurobiol Dis. 2000;7(6):586–99. doi:10.1006/nbdi.2000.0328.
- Mullen RJ, LaVail MM. Inherited retinal dystrophy: primary defect in pigment epithelium determined with experimental rat chimeras. Science. 1976;192(4241):799–801. doi:10.1126/science.1265483.
- Dowling JE, Sidman RL. Inherited retinal dystrophy in the rat. J Cell Biol. 1962;14(1):73–109. doi:10.1083/jcb.14.1.73.
- Bok D, Hall MO. The role of the pigment epithelium in the etiology of inherited retinal dystrophy in the rat. J Cell Biol. 1971;49(3):664–82. doi:10.1083/jcb.49.3.664.
- Jonsson F, Burstedt M, Kellgren TG, Golovleva I Non-Homologous recombination between Alu and LINE-1 repeats results in a 91 kb deletion in MERTK causing severe retinitis pigmentosa. Mol Vis. 2018;24:667.
- Liu S, Bi JG, Hu Y, Tang D, Li B, Zhu P, Peng W, Du D, He H, Zeng J, et al. Targeted next generation sequencing identified novel loss-of-function mutations in MERTK gene in Chinese patients with retinitis pigmentosa. Mol Genet Genomic Med. 2019;7(4):e00577. doi:10.1002/mgg3.577.
- Jespersgaard C, Bertelsen M, Arif F, Gellert-Kristensen HG, Fang M, Jensen H, Rosenberg T, Tümer Z, Møller LB, Brøndum-Nielsen K, et al. Bi-allelic pathogenic variations in MERTK including deletions are associated with an early onset progressive form of retinitis pigmentosa. Genes. 2020;11(12):1517. doi:10.3390/genes11121517.
- Birtel J, Eisenberger T, Gliem M, Müller PL, Herrmann P, Betz C, Zahnleiter D, Neuhaus C, Lenzner S, Holz FG, et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci Rep. 2018;8(1):1–11. doi:10.1038/s41598-018-22096-0.
- Issa PC, Bolz HJ, Ebermann I, Domeier E, Holz FG, Scholl HP. Characterisation of severe rod–cone dystrophy in a consanguineous family with a splice site mutation in the MERTK gene. Br J Ophthalmol. 2009;93(7):920–25. doi:10.1136/bjo.2008.147397.
- Gliem M, Müller PL, Birtel J, Herrmann P, McGuinness MB, Holz FG, Charbel Issa P. Quantitative fundus autofluorescence and genetic associations in macular, cone, and cone–rod dystrophies. Ophthalmol Retina. 2020;4(7):737–49. doi:10.1016/j.oret.2020.02.009.
- Strick DJ, Vollrath D. Focus on molecules: MERTK. Exp Eye Res. 2010;91(6):786. doi:10.1016/j.exer.2010.05.006.
- Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C, Bieg A, Gliem M, Issa PC, Holz FG, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PloS One. 2013;8(11):e78496. doi:10.1371/journal.pone.0078496.
- Cehajic-Kapetanovic J, Xue K, de la Camara, A Nanda, Martinez-Fernandez C, Nanda LJ, Davies A, Wood, LJ, Fischer, MD, Aylward, JW, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med. 2020;26(3):354–59. doi:10.1038/s41591-020-0763-1.
- Xue K, Jolly JK, Barnard AR, Rudenko A, Salvetti AP, Patrício MI, Edwards, TL, Groppe, M, Orlans, HO, Tolmachova, T, et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat Med. 2018;24(10):1507–12. doi:10.1038/s41591-018-0185-5.
- Russell S, Bennett J, Wellman JA, Chung DC, Yu Z, Tillman A, Wittes, J, Pappas, J, Elci, O, McCague, S, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRpe65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–60. doi:10.1016/S0140-6736(17)31868-8.
- Askou AL, Jakobsen TS, Corydon TJ Retinal gene therapy: an eye-opener of the 21st century. Gene Ther. 2021;28(5):209–16. doi:10.1038/s41434-020-0168-2.
- Tschernutter M, Schlichtenbrede FC, Howe S, Balaggan KS, Munro PM, Bainbridge J, Thrasher, AJ, Smith, AJ, Ali, RR Long-Term preservation of retinal function in the RCS rat model of retinitis pigmentosa following lentivirus-mediated gene therapy. Gene Ther. 2005;12(8):694–701. doi:10.1038/sj.gt.3302460.
- Smith AJ, Schlichtenbrede FC, Tschernutter M, Bainbridge JW, Thrasher AJ, Ali RR AAV-Mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8(2):188–95. doi:10.1016/S1525-0016(03)00144-8.
- Vollrath D, Feng W, Duncan JL, Yasumura D, D’Cruz PM, Chappelow A, Matthes, MT, Kay, MA, LaVail, MM Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Nat Acad Sci. 2001;98(22):12584–89. doi:10.1073/pnas.221364198.
- Ghazi NG, Abboud EB, Nowilaty SR, Alkuraya H, Alhommadi A, Cai H, Hou, R, Deng, W-T, Boye, SL, Almaghamsi, A, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. 2016;135(3):327–43. doi:10.1007/s00439-016-1637-y.
- Li T, Adamian M, Roof DJ, Berson EL, Dryja TP, Roessler BJ, Davidson, BL In vivo transfer of a reporter gene to the retina mediated by an adenoviral vector. Invest Ophthalmol Vis Sci. 1994;35(5):2543–49.
- Bennett J, Wilson J, Sun D, Forbes B, Maguire A Adenovirus vector-mediated in vivo gene transfer into adult murine retina. Invest Ophthalmol Vis Sci. 1994;35(5):2535–42.
- Dong J, Fan P, Frizzell RA Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum Gene Ther. 1996;7(17):2101–12. doi:10.1089/hum.1996.7.17-2101.
- Rees HA, Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–88. doi:10.1038/s41576-018-0059-1.
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–24. doi:10.1038/nature17946.
- Cox DB, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang, F. RNA editing with CRISPR-Cas13. Science. 2017;358(6366):1019–27. doi:10.1126/science.aaq0180.
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu, DR Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–71. doi:10.1038/nature24644.
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen, PJ, Wilson, C, Newby, G A, Raguram, A., et al. Search-And-Replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–57. doi:10.1038/s41586-019-1711-4.