
ABSTRACT
Background
Distal Arthrogryposis type 5D (DA5D) is a rare genetic disease, expressed phenotypically by skeletal and ocular abnormalities.
Materials and methods
Two sisters, ages 42 and 46 years old, were ascertained, both diagnosed with arthrogryposis and unusual ophthalmic late expressions of the disease. They were examined and followed up by both ophthalmologists and medical geneticists. Molecular analysis was performed and population screening followed among healthy individuals of the same ethnic background who reside in the same village.
Results
The two sisters expressed myogenic ptosis with poor levator palpebrae function, limitation in up gaze, lagophthalmos, refractive errors, corneal scarring and vascularization along with severe distal arthrogryposis. The newly reported features were: significant lower lid retraction, causing inferior scleral show. Sanger sequencing of the coding regions of ECEL1 gene revealed a homozygous deletion of 46 bps. The carrier frequency is 1:24 (4.2% carriers) in the probands’ village.
Conclusions
We diagnosed two patients with DA5D carrying a homozygous pathogenic genetic variant previously reported only once. We report the late ophthalmologic manifestations of this rare disorder and emphasize the importance to recognize possible long-term ophthalmic complications. Measures are needed to diagnose this rare disorder at a younger age and to address ophthalmic and orthopedic complications that might be prevented. We revealed the causative genetic variant and a carrier frequency of 1:24 for DA5D, in the probands’ village, thus enabling accurate genetic counselling and justifying genetic testing to the residents of this village as a diagnostic and preventive measure.
Introduction
Arthrogryposis Multiplex Congenita (AMC; OMIM 108,110) is a heterogeneous group of disorders characterized by congenital contractures of more than two joints in multiple parts of the body (Citation1). Ocular manifestations are also commonly observed in AMC. Previous studies suggest that 66% of patients with arthrogryposis had significant ocular disease including most commonly astigmatism, strabismus and ptosis to less commonly Brown syndrome, and cortical blindness (Citation2). Eyelids abnormalities that have been described include down‐slanting and/or narrow palpebral fissures (Citation3) and ptosis (Citation2,Citation4). To date the pathophysiology of the ocular phenotype is poorly understood (Citation5).
AMC affects both sexes equally with an incidence of 1 in 3000 live births. Both genetic and environmental factors are implicated in its etiology (Citation6–8). When contractures are non-progressive and affect mainly the distal joints (i.e. hands and feet) with limited involvement of proximal joints, the disorder is known as distal arthrogryposis, DA (Citation9,Citation10).
DAs, a subgroup of AMC, are genetically and phenotypically heterogeneous, and to date 13 distinct OMIM-listed DA subtypes have been defined on the basis of phenotypic features. Known DA genes typically encode for sarcomeric proteins of the contractile apparatus: MYH3 (DA2A, 2B and DA8), MYH8 (DA7), MYBPC1 (DA1B), TPM2 (DA1A), TNNI2 (DA2B), TNNT3 (DA2B), PIEZO2 (DA3 and DA5), all transmitted with autosomal dominant inheritance with the exception of rare families segregating recessive pathogenic PIEZO2 variants (Citation11). There is a considerable genetic and clinical overlap with the congenital myopathy spectrum, in particular nemaline myopathies and myosinopathies (Citation12).
In 2013, recessive mutations in the Endothelin-converting Enzyme-Like 1 (ECEL1) gene, a membrane-bound zinc metalloprotease (Citation13,Citation14), were identified in patients presenting with features suggestive of DA5D.
Distal Arthrogryposis type 5D is a rare genetic disease, an autosomal recessive (AR) variant of DA, characterized by congenital limited knee flexion, club feet, hip dislocation, short stature, scoliosis and ocular manifistations (Citation13,Citation14). DA might also include camptodactyly, overriding fingers, clenched fists, ulnar deviation of the wrist and/or clubfeet (Citation8,Citation15).
We report here the ocular, skeletal and genetic abnormalities in two DA5D patients presented to our clinic at their fifth decade of life, thus expanding the clinical phenotype and describing the natural history of the rare and possible underdiagnosed DA5D.
Materials and methods
We have ascertained two patients who presented with skeletal and ophthalmic manifestations probably since birth: Two sisters (: III2 & III4) of ages 42 and 46 years old that were referred to ophthalmologic clinic for evaluation.
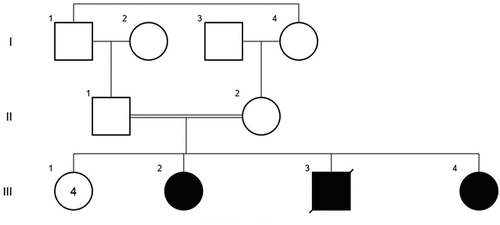
Figure 1. Pedigree of the two patients described with Distal Arthrogryposis type 5D (DA5D), III2 and III4.
This is a case series study that was held since 1/2017 when the younger sister was referred to the ophthalmologists. Her older sister presented in 7/2019. Medical and family history were taken. The two patients were examined and followed up in both the ophthalmologic and genetic clinics within the Galilee Medical Center, Nahariya, Israel.
This study has been performed in accordance with the principles stated in the Declaration of Helsinki. A written informed consent was obtained from both patients to participate in this observational study and for photographs to be taken and published.
No further ethical approvement is needed as this is a non-interventional study.
Genetic analyses
Molecular analysis was performed by Sanger sequencing:
ECEL1 (NM_004826.3) coding exons were PCR amplified using gDNA purified from peripheral blood of the patient. The PCR products were Sanger sequenced with the BigDye Terminator cycle sequencing kit version 1.1 (Applied Biosystems), and analyzed using the ABI PRISM 3500 XL Genetic Analyzer (Applied Biosystems), according to the manufacturer’s instructions. Sequence data (coding region +30 flanking bps) was analyzed using NCBI Nucleotide Blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi) by alignment to the human reference genome (GRCh38 assembly).
Population Screening was performed on a cohort of anonymous healthy individuals who reside in the same village as the sisters.
The presence of the ECEL1 causative variant was assessed in 48 healthy ethnically matched control individuals by PCR reaction and direct size inspection on 2% agarose gel. PCR product from normal allele is 656 bp long whereas the mutant allele generates 610 bp long product.
Results
Clinical findings
Two sisters (: III2 & III4) of ages 42 and 46 years old, born to consanguineous (1st degree cousins) parents of Arab Christian origin were referred to ophthalmologic clinic for evaluation.
Both of them were diagnosed at birth with joint contractures involving the upper and lower limbs in various degrees. The older sister had an orthopedic reconstructive surgery for her hand and the younger one was operated too, after being treated with casts on her feet since birth.
Both sisters have scoliosis and were consequently operated in their teenage years. Today both of them suffer from restrictive lung disease due to their severe back deformation and cannot tolerate general anesthesia. The younger sister suffers from Obstructive Sleep Apnea Syndrome (OSAS) as well and is currently being treated with Continuous Positive Airway Pressure (CPAP) during her night sleep.
Their cognitive function is normal—both possess higher education degrees, but there is a history of motor developmental delay. They never married.
They have four healthy sisters and had a male sibling () who died 40 days after birth because of fever, and was reported to have congenital joint contractures as well.
On general physical examination, the two sisters share similar facial features () that include down-slanting palpebral fissures, ptosis and lagophthalmos, arched eyebrows, high palate, small mouth, and furrowed tongue ().
Figure 2. Clinical photographs of patient III2 with Distal Arthrogryposis type 5D (DA5D). Clinical images of III2 showing childhood phenotype (a) and current phenotype including down-slanting palpebral fissures, ptosis and arched eyebrows (b), camptodactyly and adducted thumbs (c), inferior scleral show and corneal scarring (d).
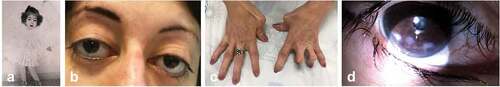
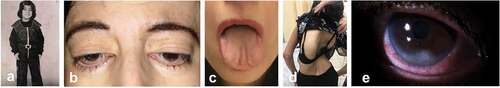
Figure 3. Clinical photographs of patient III4 with Distal Arthrogryposis type 5D (DA5D). Clinical images of III4 showing childhood phenotype (a) and current phenotype including down-slanting palpebral fissures, ptosis and arched eyebrows (b), small mouth and furrowed tongue (c), scoliosis (d), inferior scleral show and corneal scarring (e).
Their hand fingers show camptodactyly and adducted thumbs (). Severe scoliosis is prominent and cause deviation of the entire body axis ().
Ophthalmologic examination
Index patients III2 and III4 (), were referred to our ophthalmology clinic because of dry eyes and foreign body sensation in both eyes. Patient III2 had no past medical history while patient III4 sustained right eye penetrating trauma at a young age, and extra-capsular cataract extraction. Refraction measurements showed against-the rule astigmatism for both patients with hyperopic refractive error for III2 with and myopic refractive error for III4. Both patients demonstrated notable up gaze limitation in movements of both eyes, while other eye movements were intact. Forced duction test demonstrated a certain amount of restriction while trying to move the eyeball in upward direction. Significant myogenic ptosis was encountered with levator palpebrae function of 2 mm in both eyes, which is considered poor levator function. Despite this poor function, the visual axis was free in both patients allowing vision. Severe inferior scleral show was seen due to significant inferior eyelid retraction. Presence of lagophthalmos, lower eyelid retraction and inferior scleral show contribute to long-lasting exposure of both corneas that were scarred, in the lower 3 mm () Corneal sensation was reduced. Down slanting of palpebral fissure and pseudo exophthalmos were also notable. Otherwise, the anterior and posterior segments were normal in both eyes, but for III4‘s right eye that had an intraocular lens (IOL) and posterior segment could not be evaluated due to the post traumatic eye. Macular OCT showed normal foveal contour with unremarkable inner and outer retinal layers. To repair the lower lid retraction, we performed permanent tarsorrhaphy in both eyes for patient III2, and for patient III4 -lower eyelid retractors recession and elongation of posterior lamella using ear cartilage. Later on, we performed permanent tarsorrhaphy in both eyes for this patient.
Sanger sequencing
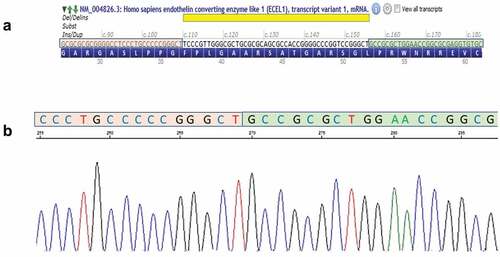
Sanger sequencing of the coding regions of ECEL1 gene (17 exons ±30 bps, NM_004826.3) revealed a homozygous pathogenic deletion of 46 bps at the first coding exon: c.110_155del (). This pathogenic variant causes a frameshift starting at codon Phenylalanine 37, which is changed to a Cysteine residue, and creates a premature stop codon at position 151 of the new reading frame, p. (Phe37fs*). This change in the protein reading frame is predicted to cause loss of the protein function either through the truncation of the protein or by nonsense-mediated mRNA decay. This variant is reported as pathogenic in ClinVar by four different clinical laboratories (accession: RCV000503413).
Figure 4. Graphical illustration of the variant c.110_155del p.(phe37cysfs *151) in ECEL1. (a) the genomic region of the 46 bp deletion (yellow) in Exon 2 showing the upstream (orange) and downstream (green) flanking sequences—from alamut visual (interactive bio software, SOPHIATM) (b) Electropherograms demonstrating homozygous mutated allele (Proband) showing the fused upstream (orange) and downstream (green) sequences.
Population screening
The identified frameshift variant was found in carrier state in 2 out of 48 healthy ethnically matched control samples in the probands’ village, i.e the carrier frequency is 1:24 (4.2% carriers). Accordingly, this variant was proposed and approved by the Israeli ministry of health for inclusion in “Health Basket” for population screening free of charge for this village.
Discussion
Our study describes ocular abnormalities that were not previously depicted in two sisters with a rare genetic disease—DA5D, caused by a pathogenic genetic variant in the ECEL1 gene. DA5D is characterized by joint contractures involving the upper and lower limbs, severe scoliosis and other orthopedic deformities, and ocular manifestations. To date 54 patients with AR DA5D are described in the literature. The most prominent ocular pathologies were ptosis (40/54 patients) and strabismus (14 patients) (see ).
Table 1. Clinical findings in our probands and in patients previously reported.
The two sisters reported here presented to us in their late 40‘s, already with scarred corneas, letting us learn about the natural history and the long-term ophthalmic and skeletal complications of DA5D.
Their detailed ophthalmology exam showed the DA5D ophthalmic phenotypic features that were considered part of the ECEL1 gene phenotype in the past: myogenic ptosis with poor levator palpebrae function, limitation in up gaze, lagophthalmos, and refractive errors. In addition, a significant lower lid retraction, and inferior scleral show were noted. We have noticed these findings in figures of patients from previous publications (Citation14,Citation16–18)), however they were never reported as part of the phenotypic spectrum of DA5D. Lower lid retraction is most probably due to lower eyelid retractors’ fibrosis. This fibrosis is probably progressive and thus is more pronounced as patients age. Looking at childhood photos of our index patients (), no inferior scleral show was seen. Furthermore, symptoms of dry eyes and tearing first appeared in their twenties, suggesting that the inferior scleral show developed over time.
Corneal scarring and vascularization due to prolonged exposure, were also seen. Lower eyelids retraction, and myogenic ptosis that further promotes corneal exposure during sleep are most probably the causes for the corneal scarring and vascularization seen in these patients, and parts of the refractive errors are possibly a consequence of scarred cornea. This decrease in corneal sensation might arise as a complication of the exposure itself as was previously shown (Citation29,Citation30). However, ECEL1 is clearly found in neural tissues (Citation18,Citation19), hence the pathogenic genetic variant might further promote the vulnerability of the fifth cranial nerve which is responsible for the corneal sensation.
Ptosis is the main characteristic of DA5D; however, its etiology is not well understood. Based on our clinical examination: the lack of eyelid crease and the poor levator function, we assume that this ptosis might be congenital and of myogenic nature, possibly due to fibrosis of the levator palpebrae muscle. This hypothesis conflicts the possible neurogenic etiology of DA5D syndrome (Citation31), and more specifically the possible neurogenic basis of the ptosis seen in our patients. Other than poor levator function we also noticed limitation in up gaze which was previously described (Citation5). We assumed that this is due to fibrosis of the inferior rectus muscle as seen in the retractors of the lower eyelid. In order to test this hypothesis, we performed force duction test and noticed restriction in elevation, suggesting inferior rectus fibrosis. Nagata knocked out mice showed axon guidance defects of the sixth cranial nerve, while other ocular motor nerves did not show this difference (Citation32). However, different phenotypes of DA5D exist. Whether the guidance defect is the reason for the possible fibrosis of the inferior rectus remains unclear. We suggest that fibrosis continues to evolve as patient ages, and should be further investigated by direct quantification of these features in other patients in childhood and as they grow up. We obviously weren’t able to perform such examinations in our patients due to their late presentation.”
Further investigations including levator palpebrae muscle and rectus muscles biopsy or EMG be conducted in order to confirm the mechanism. EMG was planned for the probands but was not carried out due to technical difficulties.
The ocular natural history of DA5D has been little described. Ulmann et al. described a 21 years old patient that had incomplete eye closure and convergent strabismus that were not present at a younger age (Citation20). Dieterich et al. described two patients, 41 and 32 years old, that presented with lagophthalmos (Citation13). The oldest patient with DA5D that was previously described in literature was 41 years old. He had pseudo-exophthalmos, lagophthalmos, and ptosis. No records regarding ophthalmic complications are available for this patient. Our patients presented to our ophthalmology and genetic clinics at a relatively old age, with late ophthalmic and skeletal sequels.
Both sisters were operated to address the corneal exposure. Because of very severe restrictive lung disease they could not tolerate general anesthesia. Due to extreme scoliosis, they could not lie down long enough to enable performing complicated eyelid surgeries. Therefore, under local anesthesia, relatively minor procedures were executed. In order to minimize corneal exposure by narrowing the palpebral fissure, both patients had permanent tarsorrhaphy in both eyes. The second sister had right eye lower lid retractors recession and elongation of posterior lamella using ear cartilage as well. Ptosis was not corrected since it did not block the visual axis.
It is important to recognize that the most dreadful long-term ophthalmic complication of this syndrome is the exposed cornea, that other than dry eyes and scarring may lead to corneal perforation and blindness. Measures should be taken to address the corneal exposure beforehand.
The identified frameshift variant p.Phe37Cysfs *151 was reported in ClinVar in homozygous state in three additional patients and in nine heterozygous carriers in GenomAD (Exomes & Genomes frequency = 0.0001726). This relatively high abundancy may be explained by the presence of 7 identical base pairs flanking the deleted region from both sides, which makes this region a hot spot for deletions by unequal crossing over.
Initially, it was believed that there aren’t any ocular motility issues associated with the AR ECEL1 gene mutations (Citation14). The first ocular motility limitation that was described was Duane syndrome (Citation13). Later on, other gaze limitations were described (Citation16,Citation19).
It has been pointed out that ECEL1 could be considered as a causal gene of another congenital contracture disorder termed Congenital Cranial Dis-innervation Disorder (CCDD) (Citation18,Citation19). This is a heterogeneous group of disorders resulting from aberrant wiring of cranial motor nerves. Nagata et al. show that ECEL1 gene knocked out mice had wandering or stalled phenotypes of abducens nerve on its pathway toward the lateral rectus muscle. This could possibly be an evidence of axon guidance defects as part of CCDD related to ECEL1 mutations (Citation32).
In contrast to most causal genes of DA, encoding proteins that relate to the muscle contraction apparatus, ECEL1 is predominantly expressed in neuronal tissue from embryonic stages (Citation33).
Interestingly, ECEL1 induction might be a part of the retinal neuroprotective response to an axonal injury in mice (Citation31). ECEL1 gene expression has been shown to be induced in mice retinal ganglion cells (RGCs), in a model causing optic nerve injury.
Knockdown of ECEL1 promoted the loss of RGCs after optic nerve crush (Citation31). Although our patients did not experience optic nerve trauma and showed no optic nerve disorder clinically, these findings suggest the necessity to further evaluate the optic nerve and the ganglion cells layer of the retina in patients known to have an ECEL1 mutation.
Such evaluation should include optical coherence tomography (OCT) of the retinal nerve fiber layer and of the ganglion cell layer and visual field testing.
Summary
This study delineates ocular abnormalities in two DA5D patients, studied both genetically and ophthalmologically, thus expanding the related clinical phenotype.
As the two sisters presented to our clinic at their fifth decade of life, we were able to learn about the natural history of the disease and the long-term ophthalmic and skeletal complications relevant to DA5D.
Extending our knowledge about rare genetic diseases such as DA5D and their natural history, may help us diagnose them more accurately and prevent expected later complications. Understanding the genetic basis of this rare disorder, might enable better care, accurate genetic counseling and possible prevention.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Hall JG. Arthrogryposis multiplex congenita: etiology, genetics, classification, diagnostic approach, and general aspects. J Pediatr Orthop. 1997;6(3):159–66. doi:10.1097/01202412-199707000-00002.
- Obi PWEE. Ocular findings in arthrogryposis congenita. Invest Ophthalmol. 2007;48(13):4839.
- Kimber E, Tajsharghi H, Kroksmark A-K, Oldfors A, Tulinius M. Distal arthrogryposis: clinical and genetic findings. Acta Paediatr. 2012 Aug;101(8):877–87. doi:10.1111/j.1651-2227.2012.02708.x.
- Beals RK, Weleber RG. Distal arthrogryposis 5: a dominant syndrome of peripheral contractures and ophthalmoplegia. Am J Med Genet. 2004 Nov;131A(1):67–70. doi:10.1002/ajmg.a.30289.
- Castori M, Rinaldi R, Barboni L, Tanzilli P, Bamshad M, Grammatico P. Juvenile macular dystrophy and forearm pronation-supination restriction presenting with features of distal arthrogryposis type 5. Am J Med Genet Part A. 2009 Mar;149A(3):482–86. doi:10.1002/ajmg.a.32668.
- Navti OB, Kinning E, Vasudevan P, Barrow M, Porter H, Howarth E, Konje J, Khare M. Review of perinatal management of arthrogryposis at a large UK teaching hospital serving a multiethnic population. Prenat Diagn. 2010 Jan;30(1):49–56. doi:10.1002/pd.2411.
- Kalampokas E, Kalampokas T, Sofoudis C, Deligeoroglou E, Botsis D. Diagnosing arthrogryposis multiplex congenita: a review. ISRN Obstet Gynecol. 2012;2012:1–6. doi:10.5402/2012/264918.
- Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: a review and update. J Bone Jt Surg-Am Vol. 2009 July;91(Suppl 4):40–46. doi:10.2106/JBJS.I.00281.
- Bamshad M, Jorde LB, Carey JC. A revised and extended classification of the distal arthrogryposes. Am J Med Genet. 1996 Nov;65(4):277–81. doi:10.1002/SICI1096-86281996111165:4<277:AID-AJMG6>3.0.CO;2-M.
- Hall JG, Reed SD, Greene G. The distal arthrogryposes: delineation of new entities - review and nosologic discussion. Am J Med Genet. 1982 Feb;11(2):185–239. doi:10.1002/ajmg.1320110208.
- Haliloglu G, Becker K, Temucin C, Talim B, Küçükşahin N, Pergande M, Motameny S, Nürnberg P, Aydingoz U, Topaloglu H, et al. Recessive PIEZO2 stop mutation causes distal arthrogryposis with distal muscle weakness, scoliosis and proprioception defects J Hum Genet. 2017 Apr;62(4):497–501. doi:10.1038/jhg.2016.153.
- Jungbluth H, Treves S, Zorzato F, Sarkozy A, Ochala J, Sewry C, Phadke R, Gautel M, Muntoni F. Congenital myopathies: disorders of excitation–contraction coupling and muscle contraction. Nat Rev Neurol. 2018 Mar;14(3):151–67. doi:10.1038/nrneurol.2017.191.
- Dieterich K, Quijano-Roy S, Monnier N, Zhou J, Fauré J, Smirnow DA, Carlier R, Laroche C, Marcorelles P, Mercier S, et al. The neuronal endopeptidase ECEL1 is associated with a distinct form of recessive distal arthrogryposis. Hum Mol Genet. 2013 Apr;22(8):1483–92. doi:10.1093/hmg/dds514.
- McMillin MJ, Below J, Shively K, Beck A, Gildersleeve H, Pinner J, Gogola G, Hecht J, Grange D, Harris D, et al. Mutations in ECEL1 cause distal arthrogryposis type 5D. Am J Hum Genet. 2013 Jan;92(1):150–56. doi:10.1016/j.ajhg.2012.11.014.
- Todd EJ, Yau KS, Ong R, Slee J, McGillivray G, Barnett CP, Haliloglu G, Talim B, Akcoren Z, Kariminejad A, et al. Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J Rare Dis. 2015 Dec;10(1):148. doi:10.1186/s13023-015-0364-0.
- Shaheen R, Al-Owain M, Khan AO, Zaki MS, Hossni HAA, Al-Tassan R, Eyaid W, Alkuraya FS. Identification of three novel ECEL1 mutations in three families with distal arthrogryposis type 5D. Clin Genet. 2014 June;85(6):568–72. doi:10.1111/cge.12226.
- Barnett CP, Todd EJ, Ong R, Davis MR, Atkinson V, Allcock R, Laing N, Ravenscroft G. Distal arthrogryposis type 5D with novel clinical features and compound heterozygous mutations in ECEL1. Am J Med Genet Part A. 2014 July;164(7):1846–49. doi:10.1002/ajmg.a.36342.
- Khan AO, Shaheen R, Alkuraya FS. The ECEL1-related strabismus phenotype is consistent with congenital cranial dysinnervation disorder. J Am Assoc Pediatr Ophthalmol Strabismus. 2014 Aug;18(4):362–67. doi:10.1016/j.jaapos.2014.03.005.
- Shaaban S, Duzcan F, Yildirim C, Chan W-M, Andrews C, Akarsu NA, Engle EC. Expanding the phenotypic spectrum of ECEL1 -related congenital contracture syndromes. Clin Genet. 2014 June;85(6):562–67. doi:10.1111/cge.12224.
- Ullmann U, D’Argenzio L, Mathur S, Whyte T, Quinlivan R, Longman C, Farrugia ME, Manzur A, Willis T, Jungbluth H, et al. ECEL1 gene related contractural syndrome: long-term follow-up and update on clinical and pathological aspects. Neuromuscul Disord. 2018 Sept;28(9):741–49. doi:10.1016/j.nmd.2018.05.012.
- Stattin E-L, Johansson J, Gudmundsson S, Ameur A, Lundberg S, Bondeson M-L, Wilbe M. A novel ECEL1 mutation expands the phenotype of distal arthrogryposis multiplex congenita type 5D to include pretibial vertical skin creases. Am J Med Genet Part A, 2018 June;176(6):1405–10. doi:10.1002/ajmg.a.38691.
- Bayram Y, Karaca E, Coban Akdemir Z, Yilmaz EO, Tayfun GA, Aydin H, Torun D, Bozdogan ST, Gezdirici A, Isikay S, et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest. 2016 Jan;126(2):762–78. doi:10.1172/JCI84457.
- Huddar A, Polavarapu K, Preethish-Kumar V, Bardhan M, Unnikrishnan G, Nashi S, Vengalil S, Priyadarshini P, Kulanthaivelu K, Arunachal G, et al. Expanding the phenotypic spectrum of ECEL1-associated distal arthrogryposis. Child (Basel, Switzerland). 2021 Oct;8(10). doi:10.3390/children8100909.
- Patil SJ, Rai GK, Bhat V, Ramesh VA, Nagarajaram HA, Matalia J, Phadke SR. Distal arthrogryposis type 5D with a novel ECEL1 gene mutation. Am J Med Genet Part A. 2014 Nov;164(11):2857–62. doi: 10.1002/ajmg.a.36702.
- Umair M, Khan A, Hayat A, Abbas S, Asiri A, Younus M, Amin W, Nawaz S, Khan S, Malik E, et al. Biallelic missense mutation in the ECEL1 underlies distal arthrogryposis type 5 (DA5D). Front Pediatr. 2019 Aug;7. doi:10.3389/fped.2019.00343.
- Jin J-Y, Liu D-Y, Jiao Z-J, Dong Y, Li J, Xiang R. The novel compound Heterozygous Mutations of ECEL1 Identified in a family with distal arthrogryposis type 5D. Biomed Res Int. 2020 May;2020;1–6. doi:10.1155/2020/2149342.
- Rai A, Puri RD, Phadke SR. Extending the phenotype and an ECEL1 gene mutation in distal arthrogryposis type 5D. Clin Dysmorphol. 2018 Oct;27(4):130–34. doi:10.1097/MCD.0000000000000236.
- Hamzeh AR, Nair P, Mohamed M, Saif F, Tawfiq N, Khalifa M, Al-Ali MT, Bastaki F. A novel variant in the endothelin-converting enzyme-like 1 (ECEL1) gene in an Emirati child. Med Princ Pract. 2017;26(2):195–98. doi:10.1159/000456034.
- Hoşal BM, Ornek N, Zilelioğlu G, Elhan AH. Morphology of corneal nerves and corneal sensation in dry eye: a preliminary study. Eye (Lond). 2005 Dec;19(12):1276–79. doi:10.1038/sj.eye.6701760.
- Bourcier T, Acosta MC, Borderie V, Borra´s F, Gallar J, Bury T, Laroche L, Belmonte C. Decreased corneal sensitivity in patients with dry eye. Invest Ophthalmol Vis Sci. 2005 July;46(7):2341–45. doi:10.1167/iovs.04-1426.
- Sato K, Shiga Y, Nakagawa Y, Fujita K, Nishiguchi KM, Tawarayama H, Murayama N, Maekawa S, Yabana T, Omodaka K, et al. Ecel1 knockdown with an AAV2-mediated CRISPR/Cas9 system promotes optic nerve damage-induced RGC death in the mouse retina. Investig Ophthalmol Vis Sci. 2018;59(10):3943–51. doi:10.1167/iovs.18-23784.
- Nagata K, Takahashi M, Kiryu-Seo S, Kiyama H, Saido TC. Distinct functional consequences of ECEL1/DINE missense mutations in the pathogenesis of congenital contracture disorders. Acta Neuropathol Commun. 2017 Dec;5(1):83. doi:10.1186/s40478-017-0486-9.
- Valdenaire O, Richards JG, Faull RL, Schweizer A. XCE, a new member of the endothelin-converting enzyme and neutral endopeptidase family, is preferentially expressed in the CNS. Mol Brain Res. 1999 Feb;64(2):211–21. doi:10.1016/S0169-328X(98)00321-0.