
ABSTRACT
Background
Anophthalmia, microphthalmia and coloboma are a genetically heterogenous spectrum of developmental eye disorders. Recently, variants in the Wnt-pathway gene Frizzled Class Receptor 5 (FZD5) have been identified in individuals with coloboma and rarely microphthalmia, sometimes with additional phenotypes and variable penetrance.
Materials and Methods
We identified variants in FZD5 in individuals with developmental eye disorders from the UK (including the DDD Study [www.ddduk.org/access.html]), France and Spain using whole genome/exome sequencing or customized NGS panels of ocular development genes.
Results
We report eight new families with FZD5 variants and ocular coloboma. Three individuals presented with additional syndromic features, two explicable by additional variants in other genes (SLC12A2 and DDX3X). In two families initially showing incomplete penetrance, re-examination of apparently unaffected carrier individuals revealed subtle ocular colobomatous phenotypes. Finally, we report two families with microphthalmia in addition to coloboma, representing the second and third reported cases of this phenotype in conjunction with FZD5 variants.
Conclusions
Our findings indicate FZD5 variants are typically associated with isolated ocular coloboma, occasionally microphthalmia, and that extraocular phenotypes are likely to be explained by other gene alterations.
Introduction
Anophthalmia, microphthalmia and coloboma (AMC) form a phenotypic spectrum of developmental eye disorders affecting 11.9 per 100,000 live births (Citation1). These conditions are genetically heterogeneous with variants in 112 genes currently considered diagnostic (https://panelapp.genomicsengland.co.uk/panels/509/), with 350–400 further genes implicated. However, approximately 40% of severely affected AMC individuals, and >75% with coloboma, remain genetically undiagnosed (Citation2–4).
Frizzled Class Receptor 5 (FZD5, OMIM *601723) is a Wnt-pathway gene required for Sox2 expression in Xenopus optic vesicles, with mice carrying a homozygous conditional loss-of-function allele exhibiting microphthalmia and coloboma, germline knockout models being embryonic lethal (Citation5–8). Recently, 13 families with heterozygous variants in this gene have been described with eye phenotypes including coloboma, microphthalmia and myopia. Both syndromic and non-syndromic affected individuals are reported, with incomplete penetrance also described (Citation9–11). Therefore, the phenotypic spectrum associated with FZD5 variants remains unclear. Here, we report eight new families with developmental eye anomalies and heterozygous FZD5 variants, clarifying our understanding of the associated phenotypic spectrum and penetrance.
Materials and methods
Informed consent was obtained according to the tenets of the Declaration of Helsinki for all participants. Full history, pedigree, ophthalmic and systemic evaluation were performed, including orthoptic and optometric assessment, slit lamp and dilated fundus examination. In some cases biometry and/or examination under anaesthesia were used. Diagnostic consents were obtained for families 2, 3, 4, 5 and 8. Research consents were obtained for Family 1 (Fundación Jimenez Díaz University Hospital [ID PIC015-18]), Family 6 (Deciphering Developmental Disorders (DDD) Study [Cambridge South Research Ethics Committee (10/H0305/83)], Republic of Ireland [GEN/284/12]); and Family 7 (UK “Genetics of Eye and Brain anomalies” study [Cambridge East Ethics Committee (04/Q0104/12)]) with additional diagnostic consent. Informed consent was obtained for the inclusion of all patient photographs. Variants were identified using whole genome/exome sequencing (WGS/WES) or customized NGS panels of ocular development genes in individuals from the UK (including the DDD Study [www.ddduk.org/access.html]), France and Spain. Individual 1 was analysed using a custom targeted NGS panel of 260 genes for Eye developmental diseases (SureSelect QXT capture custom design, Agilent Technologies, Santa Clara, California, USA) (Citation12). Libraries were sequenced on an Illumina NextSeq 500 platform. Mapping, calling and annotation for SNVs and CNVs, were performed using a custom in-house bioinformatic pipeline. Annotated VCF files were filtered by prioritization of potentially causal variants of canonical transcripts (QUAL >100, DP > 10, AF (ExAC, 1000 G, and GnomAD, Kaviar, SpanishFreq (CSVS) < 0.01), (CADD-Phred)>15 or NA). Individuals 2, 4, 6 and 8 underwent WES as part of the DDD study. Details of variant analysis have been published (Citation13). Briefly, files were filtered for coding region variants with a MAF < = 0.01% in the ExAC database, and which fitted expected inheritance models. Individuals 3 and 5 were screened using a customized panel of 119 ocular development genes, including FZD5 (NM_003468.3), as described previously, with all variants assessed by American College of Medical Genetics and Genomics (ACMG) criteria (Citation10). Individual 7 was screened using WGS, with variants annotated for, among others, presence in developmental eye disorder diagnostic panels (https://panelapp.genomicsengland.co.uk/panels/509/), RefSeq status, coding effect, gnomAD frequency, and ClinVar reports. Variants were filtered for those with a MAF ≤1% in gnomAD, and which were exonic and protein altering. Variants were validated by Sanger sequencing where appropriate. Screening for copy number variants was not routinely performed, but relevant data included where available.
Results
We identified eight individuals with ocular coloboma and FZD5 variants. Reported variants in individuals 1, 2, 3, 5, 7 and 8 are absent from the gnomAD database, while those in individuals 4 and 6 have a variant allele count of 1 (, ). Individuals carried no other potentially diagnostic variants unless stated. Variant locations are given according to NM_003468.4/NP_003459.2 and GRCh37/hg19.
Figure 1. Schematic representation of the location of FZD5 variants reported in individuals with ocular anomalies. Variants from previous studies are in black (Citation9–11), those from our data are indicated in red.
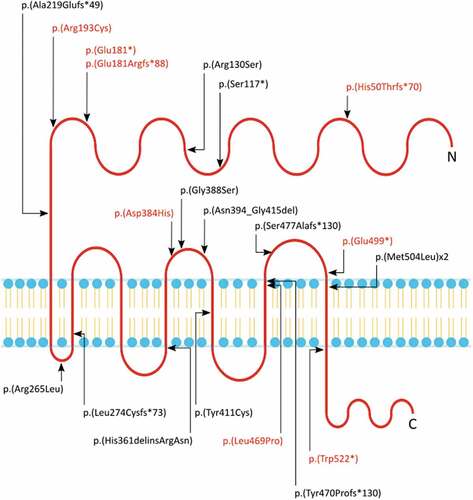
Figure 2. Pedigrees of the eight variants identified in the present study. Probands are indicated by an arrow, unknown genotypes are indicated by “?“.
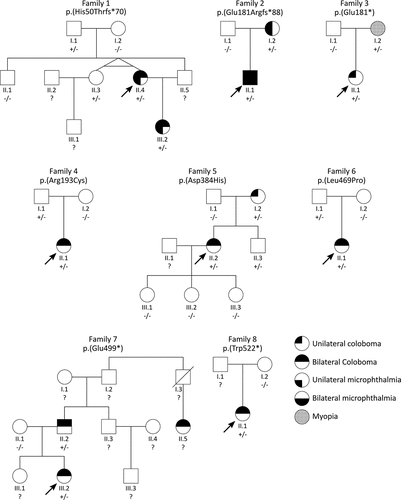
Figure 3. Phenotypic images for individuals from families 1, 2, 5, 6, 7, and 8. (a) images of Individual 1 (Family 1 II.4) showing left microphthalmic eye and iris coloboma (top), and fundoscopy of the right eye (bottom) (b) photograph of Individual 2 (Family 2 II.1) showing bilateral microphthalmia and iris coloboma. (c) images of both eyes of Individual 5 (Family 5 II.2) showing bilateral iris colobomas. (d) photograph of Individual 6 (Family 6 II.1) showing bilateral iris colobomas. (e) images of both eyes of Individual 7 (Family 7 III.2) showing bilateral iris colobomas. (f) fundoscopy images of the father of Individual 7 (Family 7 II.2) with bilateral cavernous disc anomalies indicated by an arrow (top—right eye, bottom—left eye). (g) images of both eyes of Individual 8 (Family 8 II.1) showing bilateral iris colobomas.
Table 1. Phenotypic summary of individuals 1–8. Variants given according to NM_003468.4/NP_003459.2 and GRCh37/hg19.
Individual 1
A female (, Family 1, II.4) presented with right colobomatous dysplastic optic disc, and left microphthalmia, coloboma of the iris and choroid (affecting the macula and optic nerve), and normal development. She has corneal diameters of right eye 12.3 mm and left eye 12 mm (). She carries a heterozygous FZD5 frameshift c.147delG;p.(His50Thrfs *70) variant, inherited from her asymptomatic father (I.1), who was unavailable for re-examination. Her affected daughter (III.2) carries the same variant and exhibited microphthalmia and extensive coloboma of the right eye, and a colobomatous dysplastic left optic disc. Individual 1’s monozygotic twin sister (II.3) carries the variant, but was asymptomatic and had a normal examination, including fundoscopy and biometry. The 5-month-old son of the twin sister (nephew of Individual 1) (III.1) had a dysplastic left optic disc, but genetic data is unavailable.
Individual 2
A male (, Family 2, II.1) presented with bilateral microphthalmia, iris and chorioretinal coloboma, atrial and ventricular septal defect, absent corpus callosum, cortical dysplasia, microcephaly, seizures, sensorineural deafness, and tracheoesophageal and anorectal anomalies (). He carries a heterozygous FZD5 frameshift c.539_540insG;p.(Glu181Argfs *88) variant, inherited from his mother (I.2) who had unilateral colobomatous microphthalmia and phthisis of the same eye. Individual 2 also carries a de novo likely pathogenic SLC12A2 (OMIM *600840) missense variant (NM_001046, c.980C>T; p.Ala327Val), previously published (Citation14). Individuals with SLC12A2 variants have neurodevelopmental, hearing and brain anomalies (Citation14–17), but to our knowledge variants in this gene have not been associated with coloboma.
Individual 3
A female (, Family 3, II.1) presented with isolated unilateral iris and chorioretinal coloboma. She carries a heterozygous FZD5 nonsense c.541 G>T;p.(Glu181*) variant, maternally inherited. The mother (I.2) has myopic astigmatism and was followed for mild childhood strabismus, but has no ocular coloboma.
Individual 4
A female (, Family 4, II.1) presented with bilateral iris and retinal coloboma, patent ductus arteriosus, atrial septal defect, and volvulus. Her MRI scan was normal. She carries a heterozygous FZD5 missense c.577C>T;p.(Arg193Cys) variant, inherited from her asymptomatic father (I.1). Her mother (I.2) has a history of epilepsy for which sodium valproate was taken during pregnancy, leading to an initial diagnosis of fetal valproate syndrome, which has been reported to include ocular coloboma (Citation18).
Individual 5
A female (, Family 5, II.2) presented with bilateral iris and chorioretinal coloboma, early onset cataract and mild episodic horizontal nystagmus (). She developed scoliosis in adolescence. She is an adult with normal cognitive skills. She carries a maternally inherited heterozygous FZD5 missense c.1150 G>C;p.(Asp384His) variant. On re-evaluation at 65 years-of-age her mother (I.2) was diagnosed with an asymptomatic left chorioretinal coloboma inferior to the optic nerve. Individual 5 has three asymptomatic daughters (III.1, III.2, III.3), none of whom carry the FZD5 variant. She has an asymptomatic brother (II.3) who carries the variant, but has not had fundoscopy performed.
Individual 6
A female (, Family 6, II.1) presented with bilateral iris, choroid and retinal colobomas affecting the disc and macula, with mild inferiorly displaced lenses thought to be secondary to lens colobomas (). She is registered severely visually impaired. She also has hypotonia, and delayed speech and motor development (walked at 18 months). There is no family history of similar ophthalmic phenotypes. At age 5 years her height and weight were on the 25th centile with a head circumference on the 0.4th centile with plagiocephaly. Microarray analysis did not reveal any pathogenic variants. She has a heterozygous FZD5 missense c.1406T>C;p.(Leu469Pro) variant inherited from her unaffected father (I.1). She also has a de novo pathogenic DDX3X (OMIM *300160) variant (NM_001193416.3, c.1171-2A>G), likely to account for her developmental phenotype. Variants in this gene are rarely associated with ocular colobomas (4/92 individuals) (Citation19).
Individual 7
A 5-month-old female (, Family 7, II.1) was referred with bilateral colobomas diagnosed in the first 2 weeks’ of life. There was a family history of colobomas in a paternal cousin (II.5), but her parents and sister did not report any ocular problems. She had manifest horizontal nystagmus, a small left/alternating exotropia and a visual acuity of each eye 0.64 cycl/cm (Teller). She had bilateral iris colobomas and large chorioretinal colobomas involving both discs, with a refraction of −1.50 DS in both eyes (). Her mother (II.1) had a normal ocular examination. Her father (II.2) had reduced right visual acuity, normal on the left (RE 0.16, N8, LE 0.1, N5). He had mild blue dot lens opacities, more evident on the right than the left, a right cavernous disc anomaly with a possible pit, and an increased optic cup with possible cavernous anomaly on the left (). On subsequent evaluation at 8.5 years of age, the proband’s development was normal. She had an extra row of teeth on the upper dentition. Her height was 50th centile, weight 50-75th centile and head circumference 9-25th centile. She carries a paternally inherited heterozygous FZD5 nonsense c.1495 G>T;p.(Glu499*) variant. The proband carries a previously published de novo heterozygous 1.3Mb chromosomal deletion including GJA8 (arr[hg19] 1q21.1q21.2 (146497694_147825519)x1 dn). While single nucleotide variants in GJA8 are associated with cataract and microphthalmia, the role of heterozygous deletions remains uncertain (Citation20) and the de novo nature of this deletion did not explain the paternal eye anomalies and colobomas in the paternal cousin.
Individual 8
A female (, Family 8, II.1) presented with bilateral iris and optic nerve colobomas, mild facial asymmetry with hypertelorism and downslanting palpebral fissures (). She also had a left upper visual field defect, mild asthma, arthritis affecting the knees and spine, and required some learning support at school. She carries a heterozygous FZD5 nonsense c.1566 G>A;p.(Trp522*) variant. Microarray testing was normal. The variant is absent in the mother (I.2). Paternal DNA was unavailable for testing.
Discussion
We present eight new families with ocular colobomas and FZD5 variants, including two families with microphthalmia. This brings the total number of reported independent families to 19 with ocular coloboma, three with additional microphthalmia (Citation9–11).
Seven of our 13 affected individuals have isolated eye phenotypes. In two studies linking FZD5 variants to ocular anomalies, there was no description of extraocular features (Citation9,Citation11). However, Aubert-Mucca et al. reported one of three patients with syndromic features, including bilateral deafness with cochlear malformations, and neurocognitive difficulties (Citation10). While Individual 2 also manifests sensorineural deafness, the FZD5 variant is inherited from their mother who has isolated unilateral colobomatous microphthalmia. Furthermore, Individual 2’s deafness and neurodevelopmental features are explicable by their additional de novo SLC12A2 missense variant (Citation14). Similarly, while Individual 6 manifests coloboma and developmental features, the latter is explicable by their de novo pathogenic DDX3X variant. However, in a recent study 4/92 individuals with DDX3X de novo missense or inframe deletion variants also had ocular colobomas (Citation19). Given that the FZD5 variant in Individual 6 is classified as of uncertain significance according to the ACMG guidelines () it is possible that their splicing variant in DDX3X contributes to their eye phenotype. Therefore, FZD5 variants appear to be associated with isolated ocular phenotypes, with extraocular features likely attributable to additional genetic findings.
Our data also detail the ocular phenotypic spectrum and penetrance associated with FZD5 variants. In their original pedigree, Liu et al. reported two unaffected individuals carrying the variant of interest, postulating incomplete penetrance (Citation9). Our study supports such incomplete penetrance and variable expressivity. Variants in Individuals 4 and 6 are inherited from apparently asymptomatic fathers. However, the mother of Individual 4 received sodium valproate during pregnancy, which has been historically linked to ocular coloboma (Citation18). However, the families described therein are unlikely to have received a full genetic workup. Therefore the contribution of this genetic change to Individual 4’s phenotype remains difficult to ascertain. In contrast, the FZD5 variant present in Individual 5 was initially thought to be inherited from an unaffected mother. However, upon re-examination the mother displayed mild coloboma-spectrum phenotypes. Interestingly, the variant in Individual 1 is inherited from a parent reported as unaffected, but who is unavailable for re-examination. Her monozygotic twin sister was unaffected, confirmed on examination. However, Individual 1’s daughter, who also carries the variant has microphthalmia and coloboma, showing remarkable variable penetrance and expressivity. Furthermore, the variant in Individual 3 is inherited from her mother who has myopia, a phenotype which has been associated with FZD5 variants (Citation11). However, as the myopia in Individual 3’s mother is mild, as seen in the general population, no genetic inferences can be made. Therefore, unaffected carriers appear to represent one end of a spectrum of ocular disorders of varying severity caused by FZD5 variants.
In addition to the previous reports of two carriers as unaffected, there are other descriptions of milder forms, including one carrier with unilateral optic disc/nerve pigmentary abnormality (Citation9), and three families with inferior chorioretinal hypoplasia and/or optic disc hypoplasia (Citation11), phenotypes which represent mild forms of uveal coloboma (Citation11,Citation21,Citation22). At the other end of the spectrum, we identified microphthalmia in Individual 1 and her daughter, and Individual 2 and his mother, being only the second and third reports of this phenotype in relation to FZD5 variants in humans (Citation11). However, there currently appears to be no correlation between the location or class of variant within FZD5 and the associated phenotype, with lack of penetrance associated with both missense and frameshift changes (Citation9–11), including in our own findings. Therefore, FZD5 variants appear to be associated with a spectrum of ocular coloboma and microphthalmia phenotypes, meaning that apparently unaffected carriers should be carefully examined for subtle signs, with genetic counselling provided to raise awareness of the variable phenotype and incomplete penetrance, and testing offered.
In conclusion, we present eight new families with FZD5 variants and ocular colobomas. Our data indicate that genetic alterations in this gene are associated with isolated coloboma with occasional microphthalmia, additional extra-ocular phenotypes being explicable by variants in other genes. Finally, our study identifies several cases of incomplete penetrance and variable expression, demonstrating the importance of reassessing asymptomatic carriers for subtle coloboma phenotypes, including cavernous disc anomalies or optic disc hypoplasia, mild inferior chorioretinal dysplasia and unilateral signs. Therefore, these data aid clinical diagnostic genetic testing and counselling.
Acknowledgments
We would like to thank the patients and their families for participating in this study. This work was supported by grants from Baillie Gifford, and MACS (Microphthalmia, Anophthalmia, Coloboma Support). The Deciphering Developmental Disorders (DDD) Study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003]. This study makes use of DECIPHER (http://decipher.sanger.ac.uk), which is funded by Wellcome. See Nature PMID: 25533962 or www.ddduk.org/access.html for full acknowledgement.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Shah SP, Taylor AE, Sowden JC, Ragge NK, Russell-Eggitt I, Rahi JS, Gilbert CE; Surveillance of Eye Anomalies (SEA-UK) Special Interest Group. Anophthalmos, microphthalmos, and typical coloboma in the United Kingdom: a prospective study of incidence and risk. Invest Ophthalmol Vis Sci. 2011;52(1):558–64. doi:10.1167/iovs.10-5263. PMID: 20574025
- Patel N, Khan AO, Alsahli S, Abdel-Salam G, Nowilaty SR, Mansour AM, Nabil A, Al-Owain M, Sogati S, Salih MA, et al. Genetic investigation of 93 families with microphthalmia or posterior microphthalmos. Clin Genet. 2018;93(6):1210–22. doi:10.1111/cge.13239. PMID: 29450879.
- Plaisancié J, Ceroni F, Holt R, Zazo Seco C, Calvas P, Chassaing N, Ragge NK. Genetics of anophthalmia and microphthalmia. Part 1: non-syndromic anophthalmia/microphthalmia. Hum Genet. 2019;138(8–9):799–830. doi:10.1007/s00439-019-01977-y. PMID: 30762128.
- Yoon KH, Fox SC, Dicipulo R, Lehmann OJ, Waskiewicz AJ. Ocular coloboma: genetic variants reveal a dynamic model of eye development. Am J Med Genet C Semin Med Genet. 2020;184(3):590–610. doi:10.1002/ajmg.c.31831. PMID: 32852110.
- Van Raay TJ, Moore KB, Iordanova I, Steele M, Jamrich M, Harris WA, Vetter ML. Frizzled 5 signaling governs the neural potential of progenitors in the developing Xenopus retina. Neuron. 2005;46(1):23–36. doi:10.1016/j.neuron.2005.02.023. PMID: 15820691.
- Liu C, Nathans J. An essential role for frizzled 5 in mammalian ocular development. Development. 2008;135(21):3567–76. doi:10.1242/dev.028076. PMID: 18832390.
- Liu C, Bakeri H, Li T, Swaroop A. Regulation of retinal progenitor expansion by Frizzled receptors: implications for microphthalmia and retinal coloboma. Hum Mol Genet. 2012;21(8):1848–60. doi:10.1093/hmg/ddr616. PMID: 22228100.
- Dijksterhuis JP, Baljinnyam B, Stanger SH, Ji Y, Andres O, Rubin JS, Hannoush RN, Schulte G. Systematic mapping of WNT-FZD protein interactions reveals functional selectivity by distinct WNT-FZD pairs. J Biol Chem. 2015;290(11):6789–98. doi:10.1074/jbc.M114.612648. PMID: 25605717.
- Liu C, Widen SA, Williamson KA, Ratnapriya R, Gerth-Kahlert C, Rainger J, Alur RP, Strachan E, Manjunath SH, Balakrishnan A, et al. A secreted WNT-ligand-binding domain of FZD5 generated by a frameshift mutation causes autosomal dominant coloboma. Hum Mol Genet. 2016;25(7):1382–91. doi:10.1093/hmg/ddw020. PMID: 26908622.
- Aubert-Mucca M, Pernin-Grandjean J, Marchasson S, Gaston V, Habib C, Meunier I, Sigaudy S, Kaplan J, Roche O, Denis D, et al. Confirmation of FZD5 implication in a cohort of 50 patients with ocular coloboma. Eur J Hum Genet. 2021;29(1):131–40. doi:10.1038/s41431-020-0695-8. PMID: 32737437.
- Jiang Y, Ouyang J, Li S, Xiao X, Sun W, Zhang Q. Confirming and expanding the phenotypes of FZD5 variants: coloboma, inferior chorioretinal hypoplasia, and high myopia. Mol Vis. 2021;27:50–60. PMID: 33633439.
- Tarilonte M, Ramos P, Moya J, Fernandez-Sanz G, Blanco-Kelly F, Swafiri ST, Villaverde C, Romero R, Tamayo A, Gener B, et al. Activation of cryptic donor splice sites by non-coding and coding PAX6 variants contributes to congenital aniridia. J Med Genet. 2022;59(5):428–37. doi:10.1136/jmedgenet-2020-106932.
- Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, King DA, Ambridge K, Barrett DM, Bayzetinova T, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385(9975):1305–14. doi:10.1016/S0140-6736(14)61705-0.
- McNeill A, Iovino E, Mansard L, Vache C, Baux D, Bedoukian E, Cox H, Dean J, Goudie D, Kumar A, et al. SLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect. Brain. 2020;143(8):2380–87. doi:10.1093/brain/awaa176. PMID : 32658972.
- Delpire E, Wolfe L, Flores B, Koumangoye R, Schornak CC, Omer S, Pusey B, Lau C, Markello T, Adams DR. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2cl cotransporter, NKCC1. Cold Spring Harb Mol Case Stud. 2016;2(6):a001289. doi:10.1101/mcs.a001289. PMID: 27900370.
- Mutai H, Wasano K, Momozawa Y, Kamatani Y, Miya F, Masuda S, Morimoto N, Nara K, Takahashi S, Tsunoda T, et al. Variants encoding a restricted carboxy-terminal domain of SLC12A2 cause hereditary hearing loss in humans. PLoS Genet. 2020;16(4):e1008643. doi:10.1371/journal.pgen.1008643. PMID: 32294086.
- Stödberg T, Magnusson M, Lesko N, Wredenberg A, Munoz DM, Stranneheim H, Wedell A. SLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia. Neurol Genet. 2020;6(4):e478. doi:10.1212/NXG.0000000000000478. PMID: 32754646.
- Jackson A, Fryer A, Clowes V, Clayton-Smith J. 2014. Ocular coloboma and foetal valproate syndrome: four further cases and a hypothesis for aetiology. Clin Dysmorphol. 23(2):74–75. doi:10.1097/MCD.0000000000000028
- Lennox AL, Hoye ML, Jiang R, Johnson-Kerner BL, Suit LA, Venkataramanan S, Sheehan CJ, Alsina FC, Fregeau B, Aldinger KA, et al. Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis during fetal cortical development. Neuron. 2020;106(3):404–20. doi:10.1016/j.neuron.2020.01.042. PMID: 32135084.
- Ceroni F, Aguilera-Garcia D, Chassaing N, Bax DA, Blanco-Kelly F, Ramos P, Tarilonte M, Villaverde C, da Silva LRJ, Ballesta-Martínez LRJ, et al. New GJA8 variants and phenotypes highlight its critical role in a broad spectrum of eye anomalies. Hum Genet. 2019;138(8–9):1027–42. doi:10.1007/s00439-018-1875-2. PMID: 29464339.
- Ravine D, Ragge NK, Stephens D, Oldridge M, Wilkie AOM. Dominant coloboma-microphthalmos syndrome associated with sensorineural hearing loss, hematuria, and cleft-lip/palate. Am J Med Genet. 1997;72(2):227–36. doi:10.1002/(sici)1096-8628(19971017)72:2<227:aid-ajmg19>3.0.co;2-p. PMID: 9382148.
- Ragge NK, Ravine D, Wilkie AO. Dominant inheritance of optic pits. Am J Ophthalmol. 1998;125(1):124–25. doi:10.1016/s0002-9394(99)80255-4. PMID: 9437334.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–24. doi:10.1038/gim.2015.30. PMID: 25741868.