
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Context
Peucedanol is a major extract of Peucedanum japonicum Thunb. (Apiaceae) roots, which is a commonly used herb in paediatrics. Its interaction with cytochrome P450 enzymes (CYP450s) would lead to adverse effects or even failure of therapy.
Objective
The interaction between peucedanol and CYP450s was investigated.
Materials and methods
Peucedanol (0, 2.5, 5, 10, 25, 50, and 100 μM) was incubated with eight human liver CYP isoforms (CYP1A2, 2A6, 3A4, 2C8, 2C9, 2C19, 2D6, and 2E1), in pooled human liver microsomes (HLMs) for 30 min with specific inhibitors as positive controls and untreated HLMs as negative controls. The enzyme kinetics and time-dependent study (0, 5, 10, 15, and 30 min) were performed to obtain corresponding parameters in vitro.
Results
Peucedanol significantly inhibited the activity of CYP1A2, 2D6, and 3A4 in a dose-dependent manner with IC50 values of 6.03, 13.57, and 7.58 μM, respectively. Peucedanol served as a non-competitive inhibitor of CYP3A4 with a Ki value of 4.07 μM and a competitive inhibitor of CYP1A2 and 2D6 with a Ki values of 3.39 and 6.77 μM, respectively. Moreover, the inhibition of CYP3A4 was time-dependent with the Ki/Kinact value of 5.44/0.046 min/μM.
Discussion and conclusions
In vitro inhibitory effect of peucedanol on the activity of CYP1A2, 2A6, and 3A4 was reported in this study. As these CYPs are involved in the metabolism of various drugs, these results implied potential drug-drug interactions between peucedanol and drugs metabolized by CYP1A2, 2D6, and 3A4, which needs further in vivo validation.
Keywords:
Introduction
Herbal medicine is one of the most common treatment methods in paediatrics in traditional Chinese medicine, which plays a crucial role in the Chinese healthcare system and has been used as alternative medicine worldwide. Unlike Western therapeutics, herbal medicines are complex mixtures, and these complex mixtures may interact with each other (Ge Citation2019). Besides the known pharmacological activities of Peucedanum japonicum Thunb. (Apiaceae), including antiallergic effects, cardiopulmonary protection, and antitumor activity (Ren et al. Citation2013), Peucedanum japonicum roots are often prescribed against cough, colds, headaches, and antifebrile in paediatrics (Sarkhail Citation2014). Peucedanol mainly exists in the root of Peucedanum japonicum, which is a commonly used herb in paediatrics (Hong and Kim Citation2017). The effect of peucedanol on the activity of cytochrome P450 (CYP450) enzymes is critical to guide the clinical prescription of peucedanol.
CYP450 enzymes are membrane-bound hemoproteins, which are essential for the metabolism of medications, detoxification of xenobiotics, and cellular metabolism (Guengerich et al. Citation2016). Induction or inhibition of CYP450 enzymes is a major mechanism that results in drug-drug interaction (Lynch and Price Citation2007). CYP450 enzymes can be transcriptionally activated or inhibited by various xenobiotics and endogenous substrates. Previously, a number of herbs have been reported to exert inhibitory effects on the activity of CYP450 enzymes, such as bergenin, pulvinic acid, and natural furanocoumarins (Guo et al. Citation2000; Huang et al. Citation2009; Dong et al. Citation2018). Drug-drug interactions induced by changes of CYP450 activity were also assessed in many studies (Yamamoto et al. Citation2017; Giri et al. Citation2018; Nishihara et al. Citation2019). Therefore, it is necessary to investigate the interaction between peucedanol and CYP450 enzymes to state the effect of peucedanol on the activity of CYPs, which can provide more guidance for the clinical application and prescription of peucedanol.
Peucedanol was incubated with eight major CYP isoforms (CYP1A2, 2A6, 3A4, 2C8, 2C9, 2C19, 2D6, and 2E1) in pooled human liver microsomes (HLMs). The effect of peucedanol on the activity of CYPs was assessed by a series of probe substrates and reactions. Moreover, enzyme kinetic studies and time-dependent studies were performed to obtain the potential mode of peucedanol on CYP enzymes.
Materials and methods
Chemicals
Peucedanol (≥98%, ) and testosterone (≥98%) were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). d-Glucose-6-phosphate, glucose-6-phosphate dehydrogenase, corticosterone (≥98%), NADP+, phenacetin (≥98%), acetaminophen (≥98%), 4-hydroxymephenytoin (≥98%), 7-hydroxycoumarin (≥98%), 4′-hydroxydiclofenac (≥98%), sulfaphenazole (≥98%), quinidine (≥98%), tranylcypromine (≥98%), chlorzoxazone (≥98%), 6-hydroxychlorzoxazone (≥98%), paclitaxel (≥98%), 6β-hydroxytestosterone (≥98%), clomethiazole (≥98%), and furafylline (≥98%) were obtained from Sigma Chemical Co. Montelukast (≥98%) was obtained from Beijing Aleznova Pharmaceutical (Beijing, China). Coumarin (≥98%), diclofenac (≥98%), dextromethorphan (≥98%), and ketoconazole (≥98%) were purchased from ICN Biomedicals. Pooled HLMs were purchased from BD Biosciences Discovery Labware. All other reagents and solvents were of analytical reagent grade.
Figure 1. The chemical structure of peucedanol.
Assay with human liver microsomes
The effect of peucedanol on the activity of CYP isoforms was investigated in HLMs with specific probe reactions summarized in , based on previously reported studies (Zhang et al. Citation2007; Qi et al. Citation2013; Zhang et al. Citation2017) phenacetin O-deethylation for CYP1A2, coumarin 7-hydroxylation for CYP2A6, testosterone 6β-hydroxylation for CYP3A4, paclitaxel 6α-hydroxylation for CYP2C8, diclofenac 4′-hydroxylation for CYP2C9, S-mephenytoin 4-hydroxylation for CYP2C19, chlorzoxazone dextromethorphan O-demethylation for CYP2D6, and 6-hydroxylation for CYP2E1. The incubation systems with the final volume of 200 μL were composed of 100 mM potassium phosphate buffer (pH 7.4), the appropriate concentration of HLMs, a corresponding probe substrate and peucedanol (or positive inhibitor for different probe reactions) and an NADPH generating system, including 1 mM NADP+, 10 mM glucose-6-phosphate, 1 U/mL of glucose-6-phosphate dehydrogenase, and 4 mM MgCl2. All incubations were performed in triplicate, and the mean values were used.
Table 1. CYP450 isoforms tested, maker reactions, incubation conditions and Km used in the inhibition study.
Probe substrates and positive inhibitors (except for dextromethorphan and quinidine which were dissolved in water) and peucedanol (100 μM) were dissolved in methanol, with a final concentration of 1% (v/v), and 1% neat methanol was added to the incubations without inhibitor. The positive inhibitor concentrations were as follows: 10 μM furafylline for CYP1A2, 10 μM tranylcypromine for CYP2A6, 1 μM ketoconazole for CYP3A4, 5 μM montelukast for CYP2C8, 10 μM sulfaphenazole for CYP2C9, 50 μM tranylcypromine for CYP2C19, 10 μM quinidine for CYP2D6, 50 μM clomethiazole for CYP2E1. The final microsomal protein concentration and incubation time for the different probe reactions are shown in .
After preincubating for 3 min at 37 °C, a NADPH-generating system was added to start the reaction. The reaction was terminated after 30 min by adding a 100 μL acetonitrile (10% trichloroacetic acid for CYP2A6) internal standard mix, and the solution was placed on ice. Then, the mixture was centrifuged at 12,000 rpm for 10 min, and an aliquot (50 μL) of supernatant was transferred for HPLC analysis. The instrument used in this study was Agilent 1260 series instrument with DAD and FLD detector, and the quantitative assay for the corresponding metabolites was performed as previously reported (Lang et al. Citation2017; Zhang et al. Citation2017). The corresponding conditions are summarized in , and the HPLC profile results are shown in Supplementary materials.
Table 2. HPLC conditions for the analyses of CYP450s corresponding metabolics.
Dose-dependent study and kinetic studies of peucedanol
Peucedanol (100 μM) was incubated with eight CYP isoforms to screen CYPs activity of which was inhibited by peucedanol significantly. Then, the kinetic studies were conducted with 0, 2.5, 5, 10, 25, 50, and 100 μM peucedanol and different concentration of probe substrates (20–100 μM of phenacetin for CYP1A2, 20–100 μM of testosterone for CYP3A4, and 10–50 μM of dextromethorphan for CYP2D6) to obtain the half inhibition concentration (IC50) and Ki values of the CYPs inhibited by peucedanol.
Time-dependent inhibition study of peucedanol
A time-dependent study was performed to investigate the effect of incubation time on the inhibition by peucedanol. Briefly, 20 μM peucedanol was pre-incubated with HLMs (1 mg/mL) in the presence of an NADPH-generating system for 30 min at 37 °C. Then, an aliquot (20 μL) was transferred to another incubation tube (final volume 200 μL) containing an NADPH-generating system and probe substrates whose final concentrations were approximate to Km and continued incubation to determine the residual activity. After incubating for 0, 5, 10, 15, and 30 min, the reactions were terminated by adding a 100 μL acetonitrile internal standard mix and then placed on ice; the corresponding metabolites were determined by HPLC ().
To determine the Ki and Kinact values for the inactivation of CYP3A4, the incubations were conducted using higher probe substrate concentrations (approximately 4-fold Km values) and various concentrations of peucedanol (0–50 μM) after different preincubation times (0–30 min), with a two-step incubation scheme, as described above.
Statistical analysis
The enzyme kinetic parameters for the probe reaction were estimated from the best fit line, using least-squares linear regression of the inverse substrate concentration versus the inverse velocity (Lineweaver-Burk plots), and the mean values were used to calculate Vmax and Km. Inhibition data from the experiments that were conducted using multiple compound concentrations were represented by Dixon plots, and inhibition constant (Ki) values were calculated using non-linear regression according to the following equations:
where I is the concentration of the compound, Ki is the inhibition constant, S is the concentration of the substrate, and Km is the substrate concentration at half the maximum velocity (Vmax) of the reaction. For the time-dependent inhibition, the Ki and Kinact of CYP3A4 was calculated by the following equation:
where Kobs is the pseudo-first-order rate constant of inactivation at inactivated concentration [I], Kinact is the maximum inactivation rate (a theoretical value that cannot be experimentally observed), and Ki is the inactivated concentration when the rate of inactivation reaches half of Kinact. The mechanism of the inhibition was inspected using the Lineweaver-Burk plots and the enzyme inhibition models. All experiments were performed in triplicate and the data comparison was performed using Student’s t-test or ANOVA and performed using IBM SPSS statistics 20 (SPSS Inc.).
Results
Peucedanol inhibits the activity of CYP1A2, 2D6, and 3A4
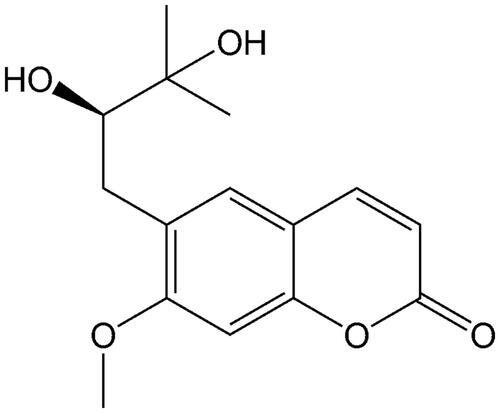
The effect of peucedanol and specific inhibitor on the activity of CYPs was investigated in pooled HLMs. The typical inhibitors significantly decreased the activity of all CYP isoforms compared with control (p < 0.05). Peucedanol showed a significant inhibitory effect on the activity of CYP1A2, 2D6, and 3A4, of which the activity decreased to 33.32%, 16.73%, and 12.23%, respectively, whereas other CYPs were not affected by peucedanol (p < 0.05, ). Additionally, the inhibition of CYP1A2, 3A4, and 2D6 was demonstrated in a dose-dependent manner and the IC50 values were obtained as 6.03, 7.58, and 13.57 μM ().
Figure 2. Effect of peucedanol on the activity of eight CYP isoforms, including CYP1A2, 2A6, 3A4, 2C8, 2C9, 2C19, 2D6, and 2E1. *p < 0.05. Negative control: without any treatment; Peucedanol: treated with 100 μM peucedanol; Positive control: treated with corresponding inhibitors.

Figure 3. The inhibition of CYP1A2 (A), 3A4 (B), and 2D6 (C) by peucedanol was in a dose-dependent manner, with the IC50 values of 6.03, 7.58, and 13.57 μM, respectively.
The competitive inhibition of CYP1A2 in Ki, and 2D6 and non-competitive inhibition of CYP3A4
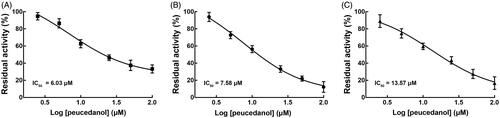
The Lineweaver-Burk plots of CYP1A2 and 2D6 inhibition showed that peucedanol was a competitive inhibitor of CYP1A2 and 2D6 ( and ). Additionally, the Ki values of CYP1A2 and 2D6 were obtained as 3.39 and 6.77 μM, respectively ( and ). In contrast, the inhibition of CYP3A4 was fitted in a non-competitive manner with the Ki value of 4.07 μM ().
Figure 4. The inhibition model fitting and corresponding parameters calculation of CYP1A2 catalysed reactions (phenacetin O-deethylation). (A) Lineweaver-Burk plots of inhibition of peucedanol on CYP1A2 with phenacetin (20–100 μM) in the absence or presence of peucedanol (0–30 μM). The inhibition of CYP1A2 was best fitting with competitive manner. (B) Secondary plot with the values of Km/Vmax in . All data represent the mean of the incubations (performed in triplicate).
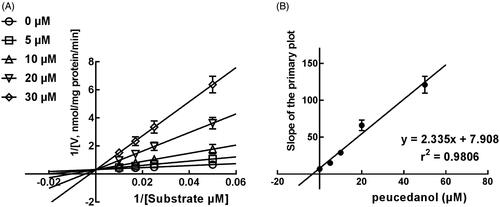
Figure 5. The inhibition model fitting and corresponding parameters calculation of CYP2D6 catalysed reactions (diclofenac 4′-hydroxylation) in pooled HLM. (A) Lineweaver-Burk plots of inhibition of peucedanol on CYP2D6 with dextromethorphan (10–50 μM) in the absence or presence of peucedanol (0–50 μM). The inhibition of CYP2D6 was best fitting with competitive manner. (B) Secondary plot with the values of Km/Vmax in . All data represent the mean of the incubations (performed in triplicate).
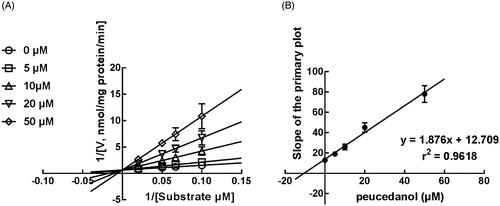
Figure 6. The inhibition model fitting and corresponding parameters calculation of CYP3A4 catalysed reactions (testosterone 6 b-hydroxylation) in pooled HLM. (A) Lineweaver-Burk plots of the of inhibition of peucedanol on CYP3A4 with testosterone (20–100 μM) in the absence or presence of peucedanol (0–30 μM). The inhibition of CYP3A4 was best fitting with non-competitive manner. (B) Secondary plot with the value of Km/Vmax in . All data represent the mean of the incubations (performed in triplicate).
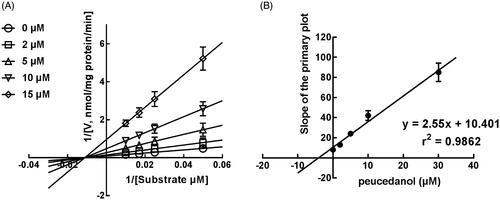
The time-dependent inhibition of CYP3A4
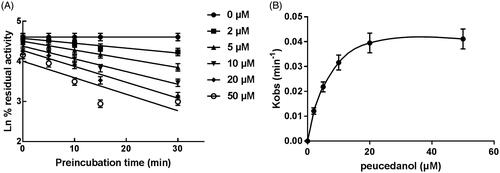
CYP1A2, 2D6, and 3A4 were incubated with peucedanol for different time. It was found that the activity of CYP3A4 significantly decreased with the incubation time, whereas CYP1A2 and 2D6 were not affected indicating the inhibition of CYP3A4 was time-dependent. Moreover, the corresponding parameters of the time-dependent inhibition of CYP3A4 were obtained by the non-linear regression analysis (). The value of Ki/Kinact of CYP3A4 was 5.44/0.046 min/μM means approximately 4.6% CYP3A4 was inactivated each minute when incubating with a saturating concentration of peucedanol in HLMs.
Figure 7. Time and concentration-inactivation of microsomal CYP3A4 activity by peucedanol in the presence of NADPH. (A) The initial rate constant of inactivation of CYP3A4 by each concentration (Kobs) was determined through linear regression analysis of the natural logarithm of the percentage of remaining activity versus pre-incubation time. (B) The Ki and Kinact values were determined through non-linear analysis of the Kobs versus the peucedanol concentration.
Discussion
In Chinese traditional medicine, the concomitant use of herbal medicines may induce clinically relevant herb-drug interaction or adverse reaction or even metabolic disorders, when some herbs are co-administrated with those drugs with narrow therapeutic indices (Chua et al. Citation2015; Ma and Ma Citation2016; Li et al. Citation2019). Therefore, because of the centrally important role of CYPs in drug-drug interaction and toxicology studies, it is necessary to assess the CYPs inhibition or induction potential of commonly used drugs or herbs.
CYP1A2, 2D6, and 3A4 are major isoforms of CYP involved in the metabolism of a large number of drugs, such as mexiletine, tamoxifen, and amlodipine (Nakajima et al. Citation1998; Zhu et al. Citation2014; MacLeod et al. Citation2017). Previously, CYP3A4 has been reported to mediate the drug-drug interaction between verapamil and oridonin result in the increase of oridonin exposure (Liu et al. Citation2019). Peucedanol was found to be a non-competitive inhibitor of CYP3A4 in this study and the inhibitory effect of peucedanol was demonstrated to be time-dependent indicating the incubation time is an important factor in the interaction between peucedanol and CYP3A4. CYP3A4 metabolizes a large variety of paediatric drugs, which makes it possible that peucedanol may co-administrated with CYP3A4 substrates (Rafaniello et al. Citation2018; Faria et al. Citation2019).
In addition, peucedanol also exerted a significant inhibitory effect on the activity of CYP1A2 and 2D6 in a competitive manner. The different inhibitory manners of peucedanol in CYP1A2, 2D6, and 3A4 may be a result of the chemical structure of peucedanol. Although these two CYP isoforms correspond to a minor part of CYPs, they are also involved in the metabolism of a variety of drugs, which significantly increase the potential for drug-drug interaction (Urichuk et al. Citation2008).
Besides the CYPs in the liver, the intestinal CYPs also play vital roles in the metabolism of drugs (Thelen and Dressman Citation2009). These studies investigated the interaction between peucedanol and CYP enzymes in pooled HLMs, the effect of peucedanol on the activity of intestinal CYPs should be paid attention in further study, which can provide more clinical guidance for the use of peucedanol. Moreover, the effect of peucedanol on the activity of CYPs was studied in vitro in this research. Further in vivo studies are needed to assess the risk of potential interaction and verify the in vitro results obtained in this study.
Conclusions
The significant inhibitory effect of peucedanol on the activity of CYP1A2, 2D6, and 3A4 was found in the present study. The inhibition of CYP1A2, 2D6, and 3A4 was found to perform in a dose-dependent manner with various IC50 values. Moreover, the inhibition of CYP3A4 was demonstrated to be time-dependent, which indicated that the incubation time is an important factor in the interaction between peucedanol and CYP3A4. These results implied the potential herb-drug or herb-herb interaction might occur during the co-administration between peucedanol and other drugs metabolized by CYP1A2, 2D6, or 3A4, which needs further in vivo validation.
SI_Figure_8.tif
Download ()SI_Figure_7.tif
Download ()SI_Figure_6.tif
Download ()SI_Figure_5.tif
Download ()SI_Figure_4.tif
Download ()SI_Figure_3.tif
Download ()SI_Figure_2.tif
Download ()SI_Figure_1.tif
Download ()Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Chua YT, Ang XL, Zhong XM, Khoo KS. 2015. Interaction between warfarin and Chinese herbal medicines. Singapore Med J. 56(1):11–18.
- Dong G, Zhou Y, Song X. 2018. In vitro inhibitory effects of bergenin on human liver cytochrome P450 enzymes. Pharm Biol. 56(1):620–625.
- Faria J, Solverson M, Faria M, Benoit M, McCormick M. 2019. Potential cytochrome P450 drug-drug interactions among pediatric patients undergoing tonsillectomy. Otolaryngol Head Neck Surg. 160(1):145–149.
- Ge GB. 2019. Deciphering the metabolic fates of herbal constituents and the interactions of herbs with human metabolic system. Chin J Nat Med. 17(11):801–802.
- Giri P, Gupta L, Naidu S, Joshi V, Patel N, Giri S, Srinivas NR. 2018. In vitro drug-drug interaction potential of sulfoxide and/or sulfone metabolites of albendazole, Triclabendazole, Aldicarb, Methiocarb, Montelukast and Ziprasidone. Drug Metab Lett. 12(2):101–116.
- Guengerich FP, Waterman MR, Egli M. 2016. Recent structural insights into cytochrome P450 function. Trends Pharmacol Sci. 37(8):625–640.
- Guo LQ, Taniguchi M, Xiao YQ, Baba K, Ohta T, Yamazoe Y. 2000. Inhibitory effect of natural furanocoumarins on human microsomal cytochrome P450 3A activity. Jpn J Pharmacol. 82(2):122–129.
- Hong MJ, Kim J. 2017. Determination of the absolute configuration of khellactone esters from Peucedanum japonicum roots. J Nat Prod. 80(5):1354–1360.
- Huang YT, Onose J, Abe N, Yoshikawa K. 2009. In vitro inhibitory effects of pulvinic acid derivatives isolated from Chinese edible mushrooms, Boletus calopus and Suillus bovinus, on cytochrome P450 activity. Biosci Biotechnol Biochem. 73(4):855–860.
- Lang J, Li W, Zhao J, Wang K, Chen D. 2017. Inhibitory effects of curculigoside on human liver cytochrome P450 enzymes. Xenobiotica. 47(10):849–855.
- Li J, Liang Q, Sun G. 2019. Interaction between traditional Chinese medicine and anticoagulant/antiplatelet drugs. Curr Drug Metab. 20(9):701–713.
- Liu J, Zhang N, Li N, Fan X, Li Y. 2019. Influence of verapamil on the pharmacokinetics of oridonin in rats. Pharm Biol. 57(1):787–791.
- Lynch T, Price A. 2007. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 76(3):391–396.
- Ma BL, Ma YM. 2016. Pharmacokinetic herb-drug interactions with traditional Chinese medicine: progress, causes of conflicting results and suggestions for future research. Drug Metab Rev. 48(1):1–26.
- MacLeod AK, McLaughlin LA, Henderson CJ, Wolf CR. 2017. Application of mice humanized for CYP2D6 to the study of tamoxifen metabolism and drug-drug interaction with antidepressants. Drug Metab Dispos. 45(1):17–22.
- Nakajima M, Kobayashi K, Shimada N, Tokudome S, Yamamoto T, Kuroiwa Y. 1998. Involvement of CYP1A2 in mexiletine metabolism. Br J Clin Pharmacol. 46(1):55–62.
- Nishihara M, Yamasaki H, Czerniak R, Jenkins H. 2019. In vitro assessment of potential for CYP-Inhibition-based drug-drug interaction between vonoprazan and clopidogrel. Eur J Drug Metab Pharmacokinet. 44(2):217–227.
- Qi XY, Liang SC, Ge GB, Liu Y, Dong PP, Zhang JW, Wang AX, Hou J, Zhu LL, Yang L, et al. 2013. Inhibitory effects of sanguinarine on human liver cytochrome P450 enzymes. Food Chem Toxicol. 56:392–397.
- Rafaniello C, Sessa M, Bernardi FF, Pozzi M, Cheli S, Cattaneo D, Baldelli S, Molteni M, Bernardini R, Rossi F, et al. 2018. The predictive value of ABCB1, ABCG2, CYP3A4/5 and CYP2D6 polymorphisms for risperidone and aripiprazole plasma concentrations and the occurrence of adverse drug reactions. Pharmacogenomics J. 18(3):422–430.
- Ren L, Du X, Hu M, Yan C, Liang T, Li Q. 2013. Design, synthesis and antitumor activity of novel 4-methyl-(3′S,4′S)-cis-khellactone derivatives. Molecules. 18(4):4158–4169.
- Sarkhail P. 2014. Traditional uses, phytochemistry and pharmacological properties of the genus Peucedanum: a review. J Ethnopharmacol. 156:235–270.
- Thelen K, Dressman JB. 2009. Cytochrome P450-mediated metabolism in the human gut wall. J Pharm Pharmacol. 61(5):541–558.
- Urichuk L, Prior TI, Dursun S, Baker G. 2008. Metabolism of atypical antipsychotics: involvement of cytochrome p450 enzymes and relevance for drug-drug interactions. Curr Drug Metab. 9(5):410–418.
- Yamamoto T, Furihata K, Hisaka A, Moritoyo T, Ogoe K, Kusayama S, Motohashi K, Mori A, Iwatsubo T, Suzuki H. 2017. Notable drug-drug interaction between etizolam and itraconazole in poor metabolizers of cytochrome P450 2C19. J Clin Pharmacol. 57(11):1491–1499.
- Zhang JW, Liu Y, Cheng J, Li W, Ma H, Liu HT, Sun J, Wang LM, He YQ, Wang Y, et al. 2007. Inhibition of human liver cytochrome P450 by star fruit juice. J Pharm Pharm Sci. 10(4):496–503.
- Zhang H, Ya G, Rui H. 2017. Inhibitory effects of triptolide on human liver cytochrome P450 enzymes and P-glycoprotein. Eur J Drug Metab Pharmacokinet. 42(1):89–98.
- Zhu Y, Wang F, Li Q, Zhu M, Du A, Tang W, Chen W. 2014. Amlodipine metabolism in human liver microsomes and roles of CYP3A4/5 in the dihydropyridine dehydrogenation. Drug Metab Dispos. 42(2):245–249.