
Abstract
Context
Poziotinib and vonoprazan are two drugs mainly metabolized by CYP3A4. However, the drug-drug interaction between them is unknown.
Objective
To study the interaction mechanism and pharmacokinetics of poziotinib on vonoprazan.
Materials and methods
In vitro experiments were performed with rat liver microsomes (RLMs) and the contents of vonoprazan and its metabolite were then determined with UPLC-MS/MS after incubation of RLMs with vonoprazan and gradient concentrations of poziotinib. For the in vivo experiment, rats in the poziotinib treated group were given 5 mg/kg poziotinib by gavage once daily for 7 days, and the control group was only given 0.5% CMC-Na. On Day 8, tail venous blood was collected at different time points after the gavage administration of 10 mg/kg vonoprazan, and used for the quantification of vonoprazan and its metabolite. DAS and SPSS software were used for the pharmacokinetic and statistical analyses.
Results
In vitro experimental data indicated that poziotinib inhibited the metabolism of vonoprazan (IC50 = 10.6 μM) in a mixed model of noncompetitive and uncompetitive inhibition. The inhibitory constant Ki was 0.574 μM and the binding constant αKi was 2.77 μM. In vivo experiments revealed that the AUC(0-T) (15.05 vs. 90.95 μg/mL·h) and AUC(0-∞) (15.05 vs. 91.99 μg/mL·h) of vonoprazan increased significantly with poziotinib pretreatment. The MRT(0-∞) of vonoprazan increased from 2.29 to 5.51 h, while the CLz/F value decreased from 162.67 to 25.84 L/kg·h after pretreatment with poziotinib.
Conclusions
Poziotinib could significantly inhibit the metabolism of vonoprazan and more care may be taken when co-administered in the clinic.
Introduction
Acid-related diseases (ARDs) are common diseases worldwide, which mainly include the peptic ulcer disease (PUD) and the gastroesophageal reflux disease (GERD). For a long period, PUD has been one of the major scourges to humanity, and is associated with a high incidence of morbidity and mortality. However, in the past two decades GERD has replaced PUD as the major reason for physician consultation, due to foregut-related symptoms (Sachs et al. Citation2014). It was considered that PUD only occurred in the presence of gastric acid, leading to the pronouncement ‘no acid, no ulcer’ (Prout Citation1974). Obtaining a continuous and stable anti-acid effect within 24 h is the key point for the treatment of acid-related diseases (Mori and Suzuki Citation2019). At present, proton pump inhibitors (PPIs) and potassium competitive acid blockers (P-CABs) are the main drugs for the treatment of gastric acid-related diseases (Oshima and Miwa Citation2018). Among them, PPIs are prodrugs and need to be rearranged into their active form with the aid of gastric acid to irreversibly inhibit proton pumps and gastric acid secretion (Marabotto et al. Citation2020). Therefore, the effect of PPIs is relatively time-delayed and has a short half-life (Martinucci et al. Citation2017). Compared with PPIs, P-CABs have a higher acid inhibition intensity and longer acid inhibition duration, which makes them have wider clinical application prospects.
Vonoprazan fumarate, a potassium ion competitive acid blocker, is a novel acid suppressor (Garnock-Jones Citation2015). Clinical and animal experiments (Murakami et al. Citation2016; Kogame et al. Citation2017) showed that the prototype of vonoprazan can rapidly relieve gastrointestinal symptoms and has the advantages of more rapid onset, longer half-life, and stronger acid inhibition effect relative to those of the traditional PPIs prazole drugs (Shin and Kim Citation2013; Sugano Citation2018). In addition, traditional PPIs are mainly metabolized by CYP2C19 (Kogame et al. Citation2017; Mori and Suzuki Citation2019), while vonoprazan is not (Sugimoto et al. Citation2020; Wang Y et al. Citation2020). Thus, the highly polymorphic CYP2C19 has little impact on gastric acid inhibition after vonoprazan administration, and the efficacy and effective dose of vonoprazan does not differ significantly among different individuals, especially in the East Asian populations that have a relatively higher proportion of the defective allele CYP2C19*2 or CYP2C19*3 than other races (Hu et al. Citation2012).
Previous studies reported that vonoprazan can be metabolized by various metabolic enzymes in human hepatocytes, such as CYP3A4, CYP2B6, CYP2D6, and the non-CYP enzyme SULT2A1, producing the main metabolite M-I and other metabolites such as M-II, M-III, M-IV-SUL, N-demethylated and N-sulfated vonoprazan (Yoneyama et al. Citation2016; Kogame et al. Citation2017; Yamasaki et al. Citation2017; Sugimoto et al. Citation2020). It has been reported that M-I, mainly metabolized by CYP3A4, is the most important metabolite and drug-drug interactions might occur when vonoprazan is coadministered with other CYP3A4 metabolized drugs (Yamasaki et al. Citation2017). For example, voriconazole, a broad-spectrum antifungal drug and a well-known inhibitor of CYP3A4 could significantly inhibit the metabolism of vonoprazan if coadministered in the clinic (Shen et al. Citation2020).
Poziotinib is a novel tyrosine kinase small molecule inhibitor that targets the rare exon 20 insertion mutations in epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) (Kim et al. Citation2013). Previous studies revealed that poziotinib was beneficial for the treatment of acquired mutations (EGFR) caused by clinically localized non-small cell lung (Rosell and Cardona Zorrilla Citation2021), breast (Kim et al. Citation2020), and gastric cancer (Kim et al. Citation2019). In the clinic, many cancer patients need to take antineoplastic drugs for a long time, and some of them have to take acid suppressive drugs at the same time to cure the gastrointestinal side effects of antitumor drugs. Patients with digestive system tumors, such as gastric cancer, are required to take drugs for both antineoplastic therapy and acid suppression. A previous study reported that poziotinib was mainly metabolized to M1 by CYP3A4 and partly metabolized to M2 by CYP2D6 (Ji et al. Citation2021). Considering that poziotinib and vonoprazan are both newly developed drugs mainly metabolized by CYP3A4 and have a high possibility of coadministration, it is very important to investigate the potential interactions between them to provide a theoretical basis for their coadministration and dose adjustment in the clinic.
Materials and methods
Chemicals and reagents
Poziotinib (purity > 98%) and vonoprazan (purity > 98%) were purchased from Sunflower Technology Development Co. (Beijing, China). The vonoprazan metabolite M-I was obtained from Wuxi Medical Technology Co. (Wuxi, China). Diazepam [internal standard (IS); purity > 98%] was purchased from Tianjin Golden York Pharmaceutical Co. (Tianjin China). Methanol and acetonitrile (chromatographic grade) were obtained from Merck GmbH (Darmstadt, Germany). Formic acid (chromatographic grade) was obtained from Sigma-Aldrich (St. Louis, MO, USA). Ultrapure water was obtained from a Milli-Q water purification system (Millipore, Billerica, MA, USA). Rat liver microsomes (RLMs) were prepared in our laboratory. All other chemicals and biologicals were of analytical grade or higher.
Animals and treatment
Male Sprague-Dawley (SD) rats, weighting 300 ± 20 g, were provided by the Experimental Animal Centre of Wenzhou Medical University and fed under standard conditions at a temperature of 23 ± 2 °C, a relative humidity of 50 ± 10%, and light for 12 h per day. Before the experiment, no other drugs were administered, and the animals were fasted for 12 h. The experimental procedures and protocols conformed to the animal ethics standards and were approved by the ethics committee of Wenzhou Medical University (Animal Ethics Approval Number: wydw2019-650).
In vitro experiments
Rat liver microsomes (RLMs) were prepared according to the previously reported differential centrifugation method (Eriksson Citation1978; Wang et al. Citation2018), and the concentration of total protein in the RLM was determined to be 28 mg/mL by the BCA protein quantitative method. Then, a 200 μL incubation system was established, which consisted of 5 μg RLM, 100 mM phosphate buffer (pH 7.4), 10 μM vonoprazan (close to its Km), and a series gradient concentration of poziotinib (100, 50, 10, 5, 1, 0.1, 0.01 μM). The mixture was preincubated at 37 °C for 5 min, and NADPH (1 mM) was then added to start the reaction after vortexing. The mixture was placed at 37 °C in a water bath for 30 min with shaking, and 200 μL acetonitrile was added to terminate the reaction and precipitate the protein. Diazepam-acetonitrile solution was then added accurately to serve as the internal standard. After vortexing, the mixture was centrifuged at 12,000 rpm for 10 min and the supernatant was transferred to the sample bottle for further separation. Vonoprazan and its metabolite M-I were detected by UPLC-MS/MS according to the previously reported method (Shen et al. Citation2020). The nonlinear regression equation was fitted with GraphPad Prism 9.0 software to calculate the IC50 value of poziotinib.
To better understand the inhibitory mechanism of poziotinib on vonoprazan, a series of gradient concentrations of vonoprazan were used in the reaction, which included 1/4-, 1/2-, 1-, and 2-times the Km value. Similarly, concentrations of 0, 1/4-, 1/2-, 1-, and 2-times the IC50 of poziotinib were also included in the experiment. The above mentioned UPLC-MS/MS method was used for the quantification of the vonoprazan metabolite M-I. Ki and αKi values were calculated, and the inhibitory mechanism was analyzed using a primary Lineweaver-Burk diagram, Dixon diagram, and secondary Lineweaver-Burk diagram by GraphPad Prism 9.0.
Pharmacokinetic experiments in vivo
Twelve healthy male SD rats were randomly divided into two groups. The poziotinib group was given 5 mg/kg poziotinib by gavage every day for 7 days, and rats in the control group only received equal amounts of 0.5% CMC-Na solution in the pre-treatment time. Then animals in the poziotinib group and the control group were fasted for 12 h. On Day 8, the poziotinib group was given poziotinib (5 mg/kg) by gavage, and vonoprazan (10 mg/kg) was given 30 min later, while the control group was only given vonoprazan (10 mg/kg) by gavage. Tail venous blood (300 µL) was collected at 0.083, 0.25, 0.5, 1, 2, 3, 4, 6, 12, and 24 h after the administration of vonoprazan, and then the blood was centrifuged at 4,000 rpm, and 4 °C for 10 min. The serum was collected and stored at −80 °C. After thawing at room temperature, 100 μL of the serum sample was removed and 200 μL of acetonitrile containing diazepam (500 ng/mL) was then added to precipitate proteins. After vortexing, 150 μL of supernatant was removed and centrifuged at 13,000 rpm for 5 min. Then, 2 μL of sample was used for the detection of vonoprazan and its main metabolite M-I by UPLC-MS/MS.
DAS (version 3.2.8; The People’s Hospital of Lishui) was used to fit and calculate the pharmacokinetic parameters of vonoprazan and its metabolite M-I, which include peak time (Tmax), maximum plasma concentration (Cmax), elimination half-life (T1/2), area under the drug-time curve (AUC) and clearance rate (CLz/F). SPSS (version 25.0, IBM, USA) was used for the data processing. Student’s t-test was used to compare the normal distribution of the data, and the Mann–Whitney U test was used for the nonnormal distribution of the data. *p < 0.05 indicates a significant difference compared to the control group and **p < 0.001 indicates an extremely significant difference compared to the control group.
Liquid chromatography and mass spectrometry conditions of vonoprazan, metabolite M-I, and internal standard
ACQUITY I-Class UPLC and Waters XEVO TQD MS (Milford, MA, USA) were used for the detection of vonoprazan and M-I with an ACQUITY UPLC BEH C18 chromatographic column (50 × 2.1 mm, 1.7 μm) at 40 °C. The mobile phase consisted of acetonitrile and 0.1% formic acid in water in gradient proportions, as shown in , with a flow rate of 0.4 mL/min. The injection volume of the sample was 2 μL. The scanning method was multiple reaction monitoring (MRM) with detection in positive ion mode and an ESI + ion source. Other mass spectrometry parameters were as follows: capillary voltage 2.0 kV, ion source temperature 150 °C, dissolvent temperature 500 °C, argon flow rate 0.15 mL/min, cone hole gas flow 50 L/h, solvent gas flow 1000 L/h. The precursor ion and product ion were m/z 346.04→314.97 for vonoprazan, m/z 347.50→205.00 for M-I, and m/z 285.10→193.10 for internal standard (IS), diazepam.
Table 1. Mobile phase ratio.
Results
In vitro effect of poziotinib on vonoprazan
As shown in , the IC50 value of poziotinib was 10.6 μM, indicating that poziotinib had an inhibitory effect on vonoprazan in vitro. The Lineweaver-Burk plot for poziotinib inhibition of vonoprazan metabolism in rat liver microsomes indicated that this inhibition effect belongs to a mixed uncompetitive and noncompetitive inhibition with Ki = 0.574 μM and αKi = 2.77 μM ().
Figure 1. Michaelis–Menten kinetics (A) and the IC50 value (B) of vonoprazan in rat liver microsomes.
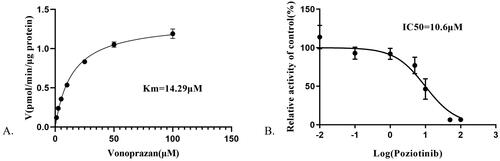
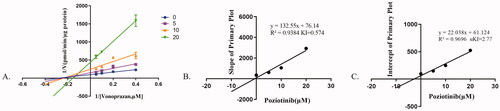
Figure 2. The inhibitory mechanism of poziotinib on vonoprazan. Data shown are the mean ± standard deviation of triplicate experiments. (A) Lineweaver–Burk plots for poziotinib (0, 2.5, 5, 10, 20 μM) inhibition of vonoprazan (0, 5, 10, 20 μΜ) in rat liver microsomes. (B) Slope of the primary plot. (C) Intercept of primary plot.
In vivo pharmacokinetics detection
The mean plasma concentration–time profiles of vonoprazan and M-I in rats are illustrated in , and detailed pharmacokinetic parameters are listed in and . Pretreatment with poziotinib led to a significant increase in the values of AUC(0-T), AUC(0-∞), MRT(0-T), MRT(0-∞), and Cmax of vonoprazan, whereas the values of Vz/F and CLz/F were significantly decreased compared to those of the control group.
Figure 3. Mean plasma concentration–time profiles of vonoprazan (A) and vonoprazan M-I (B) after an oral administration of vonoprazan (10 mg/kg) to rats in the presence and absence of poziotinib (5 mg/kg). n = 6 per group. Data are expressed as mean ± SD. The poziotinib group was pretreated with poziotinib (5 mg/kg, oral) before vonoprazan (10 mg/kg, oral) and the control group was administered vonoprazan (10 mg/kg, oral).
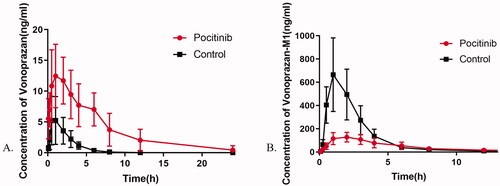
Table 2. Mean pharmacokinetic parameters of vonoprazan after oral administration (10 mg/kg) to rats in the presence and absence of poziotinib (5 mg/kg).
Table 3. Mean pharmacokinetic parameters of M-I after oral administration of vonoprazan (10 mg/kg) to rats in the presence and absence of poziotinib (5 mg/kg).
Compared with the control group, the AUC(0-T) and AUC(0-∞) of vonoprazan in the poziotinib group were increased to 6.02- and 6.11-fold, with MRT(0-T) and MRT(0-∞) increased to 2.30- and 2.41-fold, respectively. When pre-administered with poziotinib for 7 days, the Cmax, Tmax, and T1/2 values of vonoprazan were significantly increased to 2.7-, 3.0-, and 2.2-fold, while the values of Vz/F and CLz/F were reduced by 70 and 84%, respectively. In contrast, most pharmacokinetic parameters of the main metabolite of vonoprazan, M-I, were reduced when rats were pretreated with poziotinib. Comparing the poziotinib group with the control group, the AUC(0-T), AUC(0-∞), and Cmax values of M-I were reduced by 38, 33, and 71%, respectively. These results demonstrated that pretreatment with poziotinib could inhibit the metabolism of vonoprazan in rats.
Discussion
Drug-drug interaction (DDI) refers to the simultaneous or sequential administration of more than two drugs in which one drug alters the pharmacological effects of the other (Lee et al. Citation2015; Liu et al. Citation2017). When DDI occurs among drugs, administration of a regular dose may increase the drug toxicity or weaken the therapeutic effect of some drugs (Wang J et al. Citation2020), resulting in the occurrence of adverse drug reactions or treatment failure. Most clinically used drugs are metabolized by cytochrome P450 enzymes in the liver and the intestinal tract, and the inhibition or induction of P450 enzymatic activity is the main cause of metabolic DDIs in the clinic (Benet et al. Citation2019). According to the recommendation of the FDA, DDI studies should give more attention to seven major CYP isoenzymes: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 (Kato Citation2020; Wang J et al. Citation2020). Among them, CYP3A4 is one of the most important isozyme subfamilies, accounting for 40% of all CYP content and metabolizing ∼30–40% of clinically used drugs (Werk and Cascorbi Citation2014). It also has many active sites that can match substrates with different sizes and chemical properties (Tyzack and Kirchmair Citation2019; Kato Citation2020). Specifically, CYP3A4 can bind and oxidize some substrates in a synergistic way, leading to the induction or inactivation of its catalytic activity by these substrates or drugs (Sevrioukova and Poulos Citation2015). For example, macrolide antibiotics are reported to be a strong inhibitor of CYP3A4 and can cause serious adverse reactions when combined with calcium blockers (Gandhi et al. Citation2013).
As a new acid suppressor mainly metabolized by CYP3A4, vonoprazan has the advantages of strong acid suppressive ability and lasting effect compared with the traditional acid suppressor PPI (Echizen Citation2016; Yamasaki et al. Citation2017). More importantly, PPI is mainly metabolized by the CYP2C19 enzyme and some allelic mutations in the CYP2C19 gene can affect the metabolic activity of the enzyme, which may lead to different treatment outcomes when using the same drug dose for different individuals (El Rouby et al. Citation2018). In contrast, the drug curative effect of vonoprazan is not affected by the polymorphic variability of CYP2C19 (Shin et al. Citation2011), whereas more attention may be given to the potential drug-drug interactions when combined with other drugs. Suzuki et al. (Citation2003) and Nishihara et al. (Citation2019) proved that clarithromycin, metabolized by CYP3A4 and as a CYP3A4/5 inhibitor, can inhibit the metabolite of vonoprazan. Suzuki et al. (Citation2021) showed that the blood concentration of tacrolimus could significantly increase by 65.8% after switching from rabeprazole, a conventional PPI drug, to vonoprazan coadministration in kidney transplant recipients. These examples remind us that when vonoprazan is used in combination with other drugs, especially for drugs that are also metabolized by CYP3A4, it is necessary to guard against the occurrence of adverse drug reactions, and the dose should be adjusted, or drugs should be changed if necessary.
As a new third-generation tyrosine kinase small molecule inhibitor, poziotinib belongs to the quinazoline derivatives that also include afatinib, erlotinib, gefitinib, dacomitinib, etc. (Kim et al. Citation2013). It has been reported that quinazoline drugs are mainly metabolized by CYP2D6 and CYP3A4 but exhibit different effects on the metabolic activities of CYP enzymes (Wang J et al. Citation2020). Dungo and Keating (Citation2013) reported that afatinib had no impact on the catalytic activity of any CYP enzyme. Ma et al. (Citation2015) indicated that erlotinib could inhibit the activities of CYP2B1 and CYP3A1/2 in rats. Prommer (Citation2004) and Hiraide et al. (Citation2019) reported that gefitinib exhibited inhibitory effects on both CYP2C9 and CYP2D6 enzymes. Shirley (Citation2018) showed that dacomitinib was an effective inhibitor of CY2D6. Recently, Wang J et al. (Citation2020) reported that poziotinib could inhibit the activities of CYP2B1 and CYP2C11 in rats but induce the activities of CYP1A2 and CYP2E1. However, no reports have indicated the impact of poziotinib on the activity of CYP3A4 to date.
Considering that both vonoprazan and poziotinib can be metabolized by CYP3A4, some potential drug-drug interactions might occur between them. In this study, the in vitro data truly indicated that poziotinib could significantly inhibit the metabolism of vonoprazan through a mixed inhibitory mechanism. In addition, the pharmacokinetic parameters of vonoprazan in poziotinib pretreated rats were significantly increased compared with those in the control group. The serum concentration and peak time of metabolite M-I in the experimental group were much lower than those in the control group, indicating that the metabolite production of vonoprazan in rats was significantly reduced. These data revealed that poziotinib could significantly inhibit the metabolism of vonoprazan both in vitro and in vivo.
Although our data confirmed the DDIs between vonoprazan and poziotinib, some limitations still exist in this study. First, the poziotinib was regarded as the inhibitor, and vonoprazan was used as the substrate in all the experiments. We are not sure whether vonoprazan also has an inhibitory effect on the metabolism of poziotinib if we set poziotinib as the substrate. Second, we only focused on the quantification of the main metabolite of vonoprazan M-I in this experiment. Yamasaki et al. (Citation2017) pointed out that vonoprazan could be metabolized in two different pathways: one is the oxidative pathway via CYP450 such as CYP3A4, CYP2B6, CYP2C19, and CYP2D6, and the other is the nonoxidative pathway via SULT2A1. It is believed that CYP3A4 mainly contributed only to the metabolism of vonoprazan to M-I, M-III, and N-demethylated TAK-438. Thus, the measurement of other metabolites may be needed for a comprehensive evaluation of the impact of poziotinib on vonoprazan metabolism. Finally, all the experiments were performed in rats, and it has been reported that the metabolites of vonoprazan in rats and humans are not perfectly consistent. Thus, further investigations on humans must be performed to better understand the interactions of poziotinib and vonoprazan in the clinic.
Conclusions
Our data revealed that poziotinib could significantly inhibit the metabolism of vonoprazan and alter its pharmacokinetic parameters both in vivo and in vitro. These data indicated that more care may be taken when poziotinib and vonoprazan are coadministered in the clinic, although further clinical investigation is still needed to clarify these potential drug-drug interactions in humans.
Acknowledgments
The authors thank the members of Beijing Hospital and Lishui People’s Hospital for their constructive advice and full support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Benet LZ, Bowman CM, Koleske ML, Rinaldi CL, Sodhi JK. 2019. Understanding drug-drug interaction and pharmacogenomic changes in pharmacokinetics for metabolized drugs. J Pharmacokinet Pharmacodyn. 46(2):155–163.
- Dungo RT, Keating GM. 2013. Afatinib: first global approval. Drugs. 73(13):1503–1515.
- Echizen H. 2016. The first-in-class potassium-competitive acid blocker, vonoprazan fumarate: pharmacokinetic and pharmacodynamic considerations. Clin Pharmacokinet. 55(4):409–418.
- El Rouby N, Lima JJ, Johnson JA. 2018. Proton pump inhibitors: from CYP2C19 pharmacogenetics to precision medicine. Expert Opin Drug Metab Toxicol. 14(4):447–460.
- Eriksson LC. 1978. Preparation of liver microsomes with high recovery of endoplasmic reticulum and a low grade of contamination. Biochim Biophys Acta. 508(1):155–164.
- Gandhi S, Fleet JL, Bailey DG, McArthur E, Wald R, Rehman F, Garg AX. 2013. Calcium-channel blocker-clarithromycin drug interactions and acute kidney injury. JAMA. 310(23):2544–2553.
- Garnock-Jones KP. 2015. Vonoprazan: first global approval. Drugs. 75(4):439–443.
- Hiraide M, Minowa Y, Nakano Y, Suzuki K, Shiga T, Nishio M, Miyoshi J, Takahashi H, Hama T. 2019. Drug interactions between tyrosine kinase inhibitors (gefitinib and erlotinib) and warfarin: assessment of international normalized ratio elevation characteristics and in vitro CYP2C9 activity. J Oncol Pharm Pract. 25(7):1599–1607.
- Hu LM, Dai DP, Hu GX, Yang JF, Xu RA, Yang LP, Qian JC, Ge RS, Cai JP. 2012. Genetic polymorphisms and novel allelic variants of CYP2C19 in the Chinese Han population. Pharmacogenomics. 13(14):1571–1581.
- Ji W, Shen J, Wang B, Chen F, Meng D, Wang S, Dai D, Zhou Y, Wang C, Zhou Q. 2021. Effects of dacomitinib on the pharmacokinetics of poziotinib in vivo and in vitro. Pharm Biol. 59(1):457–464.
- Kato H. 2020. Computational prediction of cytochrome P450 inhibition and induction. Drug Metab Pharmacokinet. 35(1):30–44.
- Kim E, Kim H, Suh K, Kwon S, Lee G, Park NH, Hong J. 2013. Metabolite identification of a new tyrosine kinase inhibitor, HM781-36B, and a pharmacokinetic study by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 27(11):1183–1195.
- Kim JY, Park K, Im SA, Jung KH, Sohn J, Lee KS, Kim JH, Yang Y, Park YH. 2020. Clinical implications of HER2 mRNA expression and intrinsic subtype in refractory HER2-positive metastatic breast cancer treated with pan-HER inhibitor, poziotinib. Breast Cancer Res Treat. 184(3):743–753.
- Kim TY, Han HS, Lee KW, Zang DY, Rha SY, Park YI, Kim JS, Lee KH, Park SH, Song EK, et al. 2019. A phase I/II study of poziotinib combined with paclitaxel and trastuzumab in patients with HER2-positive advanced gastric cancer. Gastric Cancer. 22(6):1206–1214.
- Kogame A, Takeuchi T, Nonaka M, Yamasaki H, Kawaguchi N, Bernards A, Tagawa Y, Morohashi A, Kondo T, Moriwaki T, et al. 2017. Disposition and metabolism of TAK-438 (vonoprazan fumarate), a novel potassium-competitive acid blocker, in rats and dogs. Xenobiotica. 47(3):255–266.
- Lee B, Ji HK, Lee T, Liu KH. 2015. Simultaneous screening of activities of five cytochrome P450 and four uridine 5′-diphospho-glucuronosyltransferase enzymes in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Drug Metab Dispos. 43(7):1137–1146.
- Liu L, Sun S, Rui H, Li X. 2017. In vitro inhibitory effects of dihydromyricetin on human liver cytochrome P450 enzymes. Pharm Biol. 55(1):1868–1874.
- Ma J, Wang S, Zhang M, Zhang Q, Zhou Y, Lin C, Lin G, Wang X. 2015. Simultaneous determination of bupropion, metroprolol, midazolam, phenacetin, omeprazole and tolbutamide in rat plasma by UPLC-MS/MS and its application to cytochrome P450 activity study in rats. Biomed Chromatogr. 29(8):1203–1212.
- Marabotto E, Ziola S, Savarino V, Giannini EG, Furnari M, Bodini G, Zingone F, Ghisa M, Barberio B, Zentilin P, et al. 2020. Vonoprazan fumarate for the treatment of gastric ulcers: a short review on emerging data. Clin Exp Gastroenterol. 13:99–104.
- Martinucci I, Blandizzi C, Bodini G, Marabotto E, Savarino V, Marchi S, de Bortoli N, Savarino E. 2017. Vonoprazan fumarate for the management of acid-related diseases. Expert Opin Pharmacother. 18(11):1145–1152.
- Mori H, Suzuki H. 2019. Role of acid suppression in acid-related diseases: proton pump inhibitor and potassium-competitive acid blocker. J Neurogastroenterol Motil. 25(1):6–14.
- Murakami K, Sakurai Y, Shiino M, Funao N, Nishimura A, Asaka M. 2016. Vonoprazan, a novel potassium-competitive acid blocker, as a component of first-line and second-line triple therapy for Helicobacter pylori eradication: a phase III, randomised, double-blind study. Gut. 65(9):1439–1446.
- Nishihara M, Yamasaki H, Czerniak R, Jenkins H. 2019. In vitro assessment of potential for CYP-inhibition-based drug-drug interaction between vonoprazan and clopidogrel. Eur J Drug Metab Pharmacokinet. 44(2):217–227.
- Oshima T, Miwa H. 2018. Potent potassium-competitive acid blockers: a new era for the treatment of acid-related diseases. J Neurogastroenterol Motil. 24(3):334–344.
- Prommer E. 2004. Gefitinib: a new agent in palliative care. Am J Hosp Palliat Care. 21(3):222–227.
- Prout W. 1974. On the nature of the acid and saline matters usually existing in the stomachs of animals. Gut. 15(1):45.1–49.1.
- Rosell R, Cardona Zorrilla AF. 2021. Poziotinib treatment in intractable NSCLC: epidermal growth factor receptor and human epidermal growth factor receptor 2 exon 20 insertion mutation disease. Eur J Cancer. 149:233–234.
- Sachs G, Shin JM, Munson K, Scott DR. 2014. Gastric acid-dependent diseases: a twentieth-century revolution. Dig Dis Sci. 59(7):1358–1369.
- Sevrioukova IF, Poulos TL. 2015. Current approaches for investigating and predicting cytochrome P450 3A4-ligand interactions. Adv Exp Med Biol. 851:83–105.
- Shen J, Wang B, Wang S, Chen F, Meng D, Jiang H, Zhou Y, Geng P, Zhou Q, Liu B. 2020. Effects of voriconazole on the pharmacokinetics of vonoprazan in rats. Drug Des Dev Ther. 14:2199–2206.
- Shin JM, Inatomi N, Munson K, Strugatsky D, Tokhtaeva E, Vagin O, Sachs G. 2011. Characterization of a novel potassium-competitive acid blocker of the gastric H,K-ATPase, 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamin e monofumarate (TAK-438. J Pharmacol Exp Ther. 339(2):412–420.
- Shin JM, Kim N. 2013. Pharmacokinetics and pharmacodynamics of the proton pump inhibitors. J Neurogastroenterol Motil. 19(1):25–35.
- Shirley M. 2018. Dacomitinib: first global approval. Drugs. 78(18):1947–1953.
- Sugano K. 2018. Vonoprazan fumarate, a novel potassium-competitive acid blocker, in the management of gastroesophageal reflux disease: safety and clinical evidence to date. Therap Adv Gastroenterol. 11:1756283x17745776.
- Sugimoto M, Hira D, Murata M, Kawai T, Terada T. 2020. Effect of antibiotic susceptibility and CYP3A4/5 and CYP2C19 genotype on the outcome of vonoprazan-containing Helicobacter pylori eradication therapy. Antibiotics. 9(10):645.
- Suzuki A, Iida I, Hirota M, Akimoto M, Higuchi S, Suwa T, Tani M, Ishizaki T, Chiba K. 2003. CYP isoforms involved in the metabolism of clarithromycin in vitro: comparison between the identification from disappearance rate and that from formation rate of metabolites. Drug Metab Pharmacokinet. 18(2):104–113.
- Suzuki Y, Yoshihashi T, Takahashi K, Furuya K, Ohkohchi N, Oda T, Homma M. 2021. Drug-drug interaction between tacrolimus and vonoprazan in kidney transplant recipients. JCM. 10(17):3964.
- Tyzack JD, Kirchmair J. 2019. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem Biol Drug Des. 93(4):377–386.
- Wang J, Chen F, Jiang H, Xu J, Meng D, Geng P, Dai D, Hu J, Zhou Y, Zhou Q, et al. 2020. Inhibition and induction by poziotinib of different rat cytochrome P450 enzymes in vivo and in an in vitro cocktail method. Front Pharmacol. 11:593518.
- Wang Y, Jin Y, Yun X, Wang M, Dai Y, Xia Y. 2018. Co-administration with simvastatin or lovastatin alters the pharmacokinetic profile of sinomenine in rats through cytochrome P450-mediated pathways. Life Sci. 209:228–235.
- Wang Y, Wang C, Wang S, Zhou Q, Dai D, Shi J, Xu X, Luo Q. 2020. Cytochrome P450-based drug-drug interactions of vonoprazan in vitro and in vivo. Front Pharmacol. 11:53.
- Werk AN, Cascorbi I. 2014. Functional gene variants of CYP3A4. Clin Pharmacol Ther. 96(3):340–348.
- Yamasaki H, Kawaguchi N, Nonaka M, Takahashi J, Morohashi A, Hirabayashi H, Moriwaki T, Asahi S. 2017. In vitro metabolism of TAK-438, vonoprazan fumarate, a novel potassium-competitive acid blocker. Xenobiotica. 47(12):1027–1034.
- Yoneyama T, Teshima K, Jinno F, Kondo T, Asahi S. 2016. A validated simultaneous quantification method for vonoprazan (TAK-438F) and its 4 metabolites in human plasma by the liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 1015–1016:42–49.