
Abstract
Objectives. In normal conditions, sudden heart rate acceleration provokes a rapid reduction in ventricular action potential duration (APD). The protracted APD rate adaptation favors early afterdepolarizations and precipitates arrhythmia. Nevertheless, it is uncertain as to whether the rate-dependent changes of ventricular repolarization can be adversely modified by arrhythmogenic drugs (quinidine and procainamide) and hypokalemia, in comparison to the agents with safe therapeutic profile, such as lidocaine. Design. The rate adaptation of QT interval and monophasic APD obtained from the left ventricular (LV) and the right ventricular (RV) epicardium was examined during rapid cardiac pacing applied in isolated, perfused guinea-pig heart preparations. Results. At baseline, an abrupt increase in cardiac activation rate was associated with a substantial reduction of the QT interval and ventricular APD in the first two cardiac cycles, which was followed by a gradual shortening of repolarization over subsequent pacing intervals. The time constants of the fast (τfast) and slow (τslow) components of the APD dynamics determined from a double exponential fit were longer in RV compared to LV chamber. Quinidine, procainamide, and hypokalemia prolonged ventricular repolarization and delayed the rate adaptation of the QT interval and APD in LV and RV, as evidenced by increased τfast and τslow values. In contrast, lidocaine had no effect on the dynamic changes of ventricular repolarization upon heart rate acceleration. Conclusions. The rate adaptation of ventricular repolarization is delayed by arrhythmogenic interventions, such as quinidine, procainamide, and hypokalemia, but not changed by lidocaine, a clinically safe antiarrhythmic agent.
Introduction
In normal conditions, heart rate acceleration during exercise or emotional stress provokes a reduction in ventricular action potential duration [Citation1], an effect that ensures sufficiently long diastolic interval in order to allow an adequate ventricular filling and maintain coronary blood flow. The prolonged diastolic interval also eliminates the possibility for encroachment of the next ventricular depolarization on the T wave of a preceding beat (the “R-on-T phenomenon”), which would otherwise initiate arrhythmia. Importantly, attenuated shortening of ventricular repolarization in tachycardia can result in the “too long” action potential duration for a given level of increased heart rate, an effect that favors arrhythmogenic early afterdepolarizations [Citation1,Citation2]. The impaired rate adaptation of ventricular repolarization, therefore, is likely to be maladaptive and contributing to cardiac electrical instability.
There are several lines of clinical evidence in support of this notion. The QT interval shortening during tachycardia is significantly attenuated in patients with coronary artery disease which are prone to exercise-induced ventricular tachyarrhythmia (VT), compared to those without VT [Citation3,Citation4]. In survivors of acute myocardial infarction, the attenuated QT response to exercise-induced tachycardia has been shown to discriminate patients at high risk of sudden cardiac death from those at low risk [Citation5]. A depressed adaptation of QT interval to spontaneous variations in cardiac cycle length during 24-hour ambulatory ECG monitoring is observed in patients without structural heart disease who were resuscitated from ventricular fibrillation [Citation6]. Furthermore, a flat slope of the QT/RR relationship has been shown to independently predict arrhythmic death in post-infarcted patients treated with amiodarone [Citation7]. With arrhythmogenic disorder such as the long QT syndrome, the QT reduction in response to a brief tachycardia provoked by brisk standing is blunted [Citation8], and in the animal models of long QT, a spontaneous run of torsade de pointes is often initiated shortly after a sudden heart rate acceleration [Citation2]. Overall, these observations suggest that abnormal rate adaptation of ventricular repolarization is an important arrhythmic determinant [Citation1].
It remains incompletely understood as to whether the rate adaptation of ventricular repolarization can be adversely modified by drugs that facilitate arrhythmia, in comparison to the agents with safe therapeutic profiles. In this regard, one particular example are Na+ channel blockers, which include both clinically safe class Ib agents (such as lidocaine) and the drugs known to increase propensity for ventricular tachyarrhythmia (VT) in susceptible patients, such as class Ia (e.g. quinidine, procainamide) and class Ic agents (flecainide) [Citation9–11]. The striking differences in the safety profile among class I agents are likely attributed to their dissimilar effects at the ion channel level – whilst lidocaine is a selective INa blocker, quinidine, procainamide, and flecainide show more complex effects that include both INa blockade and inhibition of repolarizing K+ currents, such as the delayed rectifier (IK), in cardiac cells [Citation9,Citation10]. The latter can potentially contribute to repolarization abnormalities, including the slowed rate adaptation of ventricular action potential duration, which would partly account for the adverse drug effects. In support of this notion, flecainide, class Ic agent with clinically documented arrhythmogenic effects [Citation10,Citation11], has been shown to attenuate the rate adaptation of repolarization in perfused guinea-pig heart model [Citation12]. Furthermore, the clinical studies suggest that QT shortening in exercise-induced tachycardia is attenuated in patients with a history of VT induced by class Ia antiarrhythmics [Citation13].
The objective of the present study was to further extend these observations by comparing changes in the rate adaptation of cardiac repolarization induced by class Ia (quinidine and procainamide) vs. class Ib drugs (lidocaine). In addition, for systematic purposes, this study also examined effects produced by hypokalemia, an electrolyte abnormality associated with clinically important arrhythmogenic risks [Citation14], and commonly used as a reference proarrhythmic challenge in experimental studies [Citation15,Citation16].
Material and methods
This study complies with the European Community Guidelines for the Care and Use of Experimental Animals, and was approved by the local ethics committee.
Isolated, Langendorff-perfused heart preparations
The experiments on isolated, perfused hearts were performed as described previously [Citation12]. Female Dunkin-Hartley guinea-pigs weighing 400–500 g were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and anticoagulated with heparin (1000 IU/kg i.p.). The chest was opened, the hearts were immediately excised, mounted on a Langendorff perfusion set-up (Hugo Sachs Elektronik-Harvard Apparatus GmbH, March-Hugstetten, Germany) and perfused via the aorta at a constant flow (15 ml/min) with carefully filtered, warmed physiological saline solution saturated with 95%O2 and 5%CO2. The perfusion solution contained (in mM) 118.0 NaCl; 4.7 KCl; 2.5 CaCl2; 25 NaHCO3; 1.2 KH2PO4; 1.2 MgSO4; and 10.0 glucose, and had a pH of 7.4. The aortic perfusion pressure (65–70 mm Hg) was measured with a ISOTEC pressure transducer and the coronary flow rate was determined using an ultrasonic flowmeter probe (Transonic Systems Inc., USA) placed just above the aortic cannula. The electrical activity of the heart preparations was assessed from the volume-conducted ECG as well as monophasic action potential recordings. Throughout the experiments, the heart preparations were kept immersed in the temperature-controlled, perfusate-filled chamber to minimize thermal loss. Aortic pressure, coronary flow rate, ECG and ventricular action potentials were continuously monitored using the 16-channel PowerLab system (ADInstruments, Oxford, UK).
Electrical stimulations and electrophysiological recordings
Arrhythmic changes are more readily to develop when the rapid pacing is applied in conditions of the initially slowed spontaneous cardiac beating rate [Citation2]. To account for these relations, in the present study, the experiments were performed on atrioventricular (AV)-blocked heart preparations with slowed intrinsic activation rate. Both atria were removed and the AV node was crushed mechanically with forceps. Thereafter, a bipolar stimulating electrode was introduced into left ventricular chamber in order to apply endocardial stimulations with 2 ms rectangular pulses of twice diastolic threshold current generated by a programmable stimulator (Hugo Sachs Electronik-Harvard Apparatus GmbH, March-Hugstetten, Germany). Electrical stimulation thresholds were measured both at baseline and upon drug administration or hypokalemic perfusion, and the stimulating current strength was adjusted appropriately whenever necessary. Monophasic action potentials were obtained from the basal epicardium of the left ventricular (LV) and the right ventricular (RV) lateral wall using spring-loaded pressure contact electrodes (Hugo Sachs Electronik-Harvard Apparatus GmbH, March-Hugstetten, Germany). The action potential duration was measured at 90% repolarization (APD90). The volume-conducted ECG was recorded with four Ag-AgCl ECG electrodes fitted in a mounting ring placed in the perfusate-filled chamber just below the heart preparation. A 30 min stabilization period was allowed prior to starting data collection.
Rate adaptation of ventricular repolarization
The dynamic changes of ventricular repolarization in response to a sudden increase in cardiac activation rate were assessed upon application of a train of 50 regular (S1) pulses at S1–S1 = 250 ms. Assuming the running cycle length close to 600 ms in spontaneously beating, AV-blocked heart preparations, initiation of the rapid pacing was associated with more than double increase in cardiac activation rate (from ∼100 beats/min to 240 beats/min). QT interval, as well as LV and RV APD90 were measured in each cardiac beat during the tachypacing period, and then plotted as a function of time from the beginning of stimulation. The obtained rate adaptation plots were fitted using double-exponential function: y = y0 + A1exp(-DI/τ1) + A2exp(-DI/τ2), where y represents APD90, y0 is a free-fitting variable, A1 and A2 are the amplitudes, and τ1 and τ2 are the time constants of the fast (A1 and τ1) and slow (A2 and τ2) exponential components obtained by a least squares fit. The curve fitting was performed using Igor Pro 6.0 software (WaveMetrics, Inc., Portland, OR, USA).
Drug infusions and hypokalemia
In total, 35 heart preparations were used in this study, in order to examine effects produced by quinidine, procainamide, lidocaine, and hypokalemia (8–9 experiments in each study group). For precise dosing, drug infusions were performed at a rate of 0.3 ml/min using a calibrated infusion pump, while perfusing the hearts with protein-free saline solution at a constant coronary flow rate (see above). Quinidine (5 μM), procainamide (10 μM), and lidocaine (5 μM) (all from Sigma-Aldrich, Germany) were infused over 30 min, at concentrations close to the maximum free (i.e. protein-unbound) therapeutic plasma levels [Citation9,Citation10]. Hypokalemia was induced by decreasing KCl concentration in perfusion solution from 4.7 mM to 2.5 mM. This change was shown to markedly increase arrhythmic susceptibility in perfused guinea-pig heart preparations in previous studies [Citation15].
Data analysis
Data are expressed as mean ± standard error of the mean. One-way ANOVA was used for multiple comparisons, and paired t-tests were used to compare two data sets. p values less than 0.05 were considered to be significant.
Results
Basal parameters of the rate adaptation of ventricular repolarization
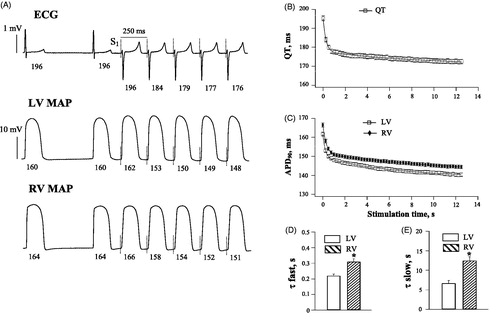
In , panel A shows representative ECG and ventricular monophasic action potential recordings obtained upon initiation of the rapid cardiac pacing in spontaneously beating, AV-blocked heart preparations, and panels B and C show the summary data that illustrate the rate adaptation of ventricular repolarization in basal conditions in 35 experiments. During spontaneous beating, the running cardiac cycle length was 610 ± 9 ms, the QT interval was 195 ± 1 ms, and action potential durations determined at LV and RV epicardium were 160 ± 1 ms and 165 ± 1 ms, respectively (LV vs. RV: p = 0.001). Cardiac pacing at accelerated rate was associated with progressive shortening of ventricular repolarization, wherein a total reduction of the QT interval and ventricular APD90 by the end of stimulation amounted about 20 ms. Overall, the rate adaptation of repolarization followed a bi-exponentially decaying time course, with 60–70% of the total reduction in QT interval and APD90 being attained during the first two cardiac cycles (, panels A, B and C); this was followed by a gradual monotonic shortening of repolarization over subsequent pacing intervals. With QT interval, the time constants of the fast and slow components (τfast and τslow, respectively) of the rate adaptation determined from a double exponential fit were 0.25 ± 0.02 s and 10 ± 1 s, respectively. With APD90, τfast (LV: 0.22 ± 0.01 s, RV: 0.31 ± 0.02 s; p < .001) and τslow (LV: 6.7 ± 0.7 s; RV: 12.5 ± 1 s; p < .001) were found to be longer at the RV compared to the LV epicardium (, panels D and E), indicating a delayed rate adaptation in RV chamber.
Figure 1. Basal electrophysiological recordings and the rate adaptation of QT interval and action potential duration in LV and RV chamber. Panel A shows representative ECG and monophasic action potentials (MAP) recorded at the left ventricular (LV) and the right ventricular (RV) epicardium upon an abrupt acceleration of the cardiac activation rate. In each recording, the numbers below traces indicate the duration (ms) of the QT interval and MAP measured during spontaneous beating (first cardiac cycle) and immediately upon initiation of the rapid S1–S1 pacing. The vertical dotted lines indicate the moments of the pacing stimulus (S1) application. In panels B and C, the mean QT values and ventricular action potential durations (APD90) from all experimental groups were determined in 50 S1 beats during pacing at a cycle length of 250 ms, and plotted vs. time from the beginning of stimulation. The QT and APD90 plots were fitted by a double exponential function, in order to determine the time constants (τ) of the fast and slow component of the rate adaptation. Panels D and E illustrate the LV vs. RV difference in τfast and τslow, respectively (*p < .05 vs. the LV value).
Quinidine
During spontaneous beating, quinidine had no effect on the running cycle length (Basal: 618 ± 32 ms, Quinidine: 646 ± 25 ms), but significantly prolonged the QT interval (Basal: 199 ± 4 ms, Quinidine: 221 ± 5 ms; p < .0001), and increased LV APD90 (Basal: 162 ± 2 ms, Quinidine: 177 ± 3 ms; p = .001), and RV APD90 (Basal: 167 ± 2 ms, Quinidine: 183 ± 3 ms; p = .002).
shows quinidine effects on the rate adaptation of ventricular repolarization during rapid pacing. Quinidine-induced prolongation of repolarization translated to the upward shift of the QT and APD90 rate adaptation plots, compared to baseline (, panels A, D and G). Importantly, quinidine slowed the dynamics of the QT and APD90 shortening upon an abrupt increase in cardiac activation rate. With QT interval (, panels B and C), τfast and τslow values were increased from 0.24 ± 0.01 s to 0.30 ± 0.02 s (p = .01) and from 8.3 ± 0.7 s to 18 ± 1 s (p = .005), respectively, upon drug infusion. With LV APD90 (, panels E and F), τfast and τslow were increased by quinidine from 0.16 ± 0.01 s to 0.22 ± 0.02 s (p = .03), and from 7.6 ± 0.7 s to 11.5 ± 0.9 s (p = .03), respectively. With RV APD90 (, panels H and I), τfast and τslow were increased by quinidine from 0.22 ± 0.01 s to 0.28 ± 0.02 s (p = .02), and from 13.9 ± 0.9 s to 19.0 ± 1.0 s (p = .001), respectively.
Figure 2. Effects of quinidine on the rate adaptation of QT interval and action potential duration in LV and RV chamber. The rate adaptation of QT interval (panels A, B and C), left ventricular (LV) action potential duration (APD90) (panels D, E and F), and right ventricular (RV) APD90 (panels G, H and I) was analyzed upon application of 50 pulses at S1–S1=250 ms at baseline and after quinidine infusion. In panels A, D and G, the first symbols staying apart from the main plots indicate the QT interval and ventricular APD90 determined in the last spontaneous cardiac beat prior to starting the LV pacing. *p < .05 vs. basal value. The same figure design is used in .
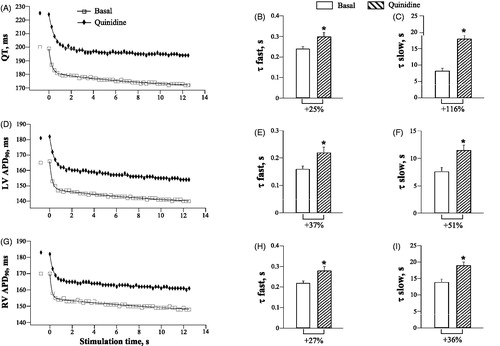
Even though quinidine delayed the APD90 rate adaptation in both ventricular chambers, it produced no change in RV vs. LV difference in the APD90 kinetics noted at baseline. The RV vs. LV difference in τfast (Basal: 0.06 ± 0.001 s, Quinidine: 0.06 ± 0.001 s) and τslow (Basal: 7.3 ± 0.7 s, Quinidine: 7.5 ± 0.7 s) for APD90 shortening during S1–S1 pacing was not modified by quinidine.
Procainamide
During spontaneous beating, procainamide had no effect on the running cycle length (Basal: 655 ± 22 ms, Procainamide: 656 ± 28 ms), but significantly prolonged the QT interval (Basal: 192 ± 3 ms, Procainamide: 202 ± 4 ms; p = .04), and increased LV APD90 (Basal: 160 ± 2 ms, Procainamide: 168 ± 2 ms; p = .01), and RV APD90 (Basal: 165 ± 2 ms, Procainamide: 171 ± 2 ms; p = .006).
shows procainamide effects on the dynamic changes of ventricular repolarization upon a sudden increase in cardiac activation rate. Consistent with prolonged repolarization, procainamide increased QT and ventricular APD90 values determined at comparable stimulation time points during rapid pacing (, panels A, D and G). The rate adaptation kinetics of the QT interval and APD90 were significantly delayed by procainamide. In particular, with QT interval (, panels B and C), τfast and τslow values were increased from 0.21 ± 0.01 s to 0.27 ± 0.01 s (p = .006), and from 9.0 ± 0.8 s to 13.1 ± 1.0 s (p = .005), respectively, upon drug infusion. With LV APD90 (, panels E and F), τfast and τslow were increased by procainamide from 0.17 ± 0.01 s to 0.35 ± 0.03s (p = .001), and from 8.3 ± 0.8 s to 11.7 ± 0.9 s (p = .02), respectively. With RV APD90 (, panels H and I), τfast and τslow were increased by procainamide from 0.26 ± 0.02 s to 0.36 ± 0.02 s (p = .01), and from 14.4 ± 1.0 s to 20.3 ± 1.0 s (p = .005), respectively.
Figure 3. Effects of procainamide on the rate adaptation of QT interval and action potential duration in LV and RV chamber.
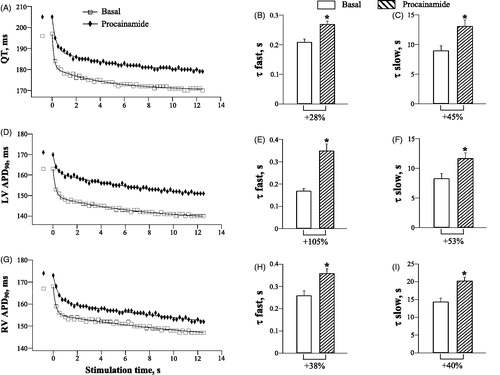
Owing to a greater relative increase in τfast for APD90 shortening in LV compared to RV chamber (, panels E and H), procainamide attenuated the RV vs. LV difference in τfast seen at baseline (Basal: 0.09 ± 0.001 s, Procainamide: 0.01 ± 0.001 s; p = .001). The RV vs. LV difference in the time constant for the slow component (τslow) of the APD90 shortening was not changed by procainamide (Basal: 6.1 ± 0.6 s, Procainamide: 8.6 ± 0.7 s).
Hypokalemia
Hypokalemia effects on the QT and APD90 dynamics were similar to those produced by quinidine and procainamide. During spontaneous beating, hypokalemic perfusion was associated with QT prolongation (Basal: 191 ± 3 ms, Hypokalemia: 204 ± 4 ms; p = .002), and an increase in LV APD90 (Basal: 159 ± 3 ms, Hypokalemia: 167 ± 3 ms; p = .03), and RV APD90 (Basal: 163 ± 2 ms, Hypokalemia: 168 ± 2 ms; p = .04). The QT and APD90 were also prolonged, compared to baseline, during rapid pacing (, panels A, D and G). Importantly, the kinetics of the QT and APD90 shortening upon an abrupt increase in cardiac activation rate was slowed by hypokalemia. For instance, with QT interval (, panels B and C), τfast and τslow values were increased from 0.26 ± 0.02 s to 0.37 ± 0.03 s (p = .005), and from 9.0 ± 0.8 s to 14.3 ± 1.0 s (p = .01), respectively, upon hypokalemic perfusion. With LV APD90 (, panels E and F), τfast and τslow were increased by hypokalemia from 0.27 ± 0.02 s to 0.36 ± 0.02 s (p = .03), and from 10.0 ± 0.9 s to 17.8 ± 1.0 s (p = .001), respectively. With RV APD90 (, panels H and I), τfast and τslow were increased by hypokalemia from 0.34 ± 0.03 s to 0.45 ± 0.03 s (p = .02), and from 13.0 ± 1.0 s to 17.0 ± 1.5 s (p = .04), respectively.
Figure 4. Effects of hypokalemia on the rate adaptation of QT interval and action potential duration in LV and RV chamber.
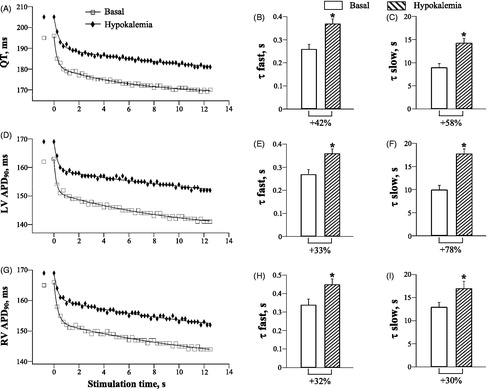
As hypokalemia produced a proportional increase in τfast for the pacing-induced APD90 shortening in LV and RV chamber (, panels E and H), the basal RV-to-LV difference in τfast was not changed upon hypokalemic perfusion (Basal: 0.07 ± 0.001 s, Hypokalemia: 0.09 ± 0.001 s). With regards to the slow component of APD90 rate adaptation, a relative increase in τslow in hypokalemic hearts tended to be larger in LV compared to RV epicardium (, panels F and I). Consequently, the RV-to-LV difference in τslow noted at baseline was eliminated by hypokalemia (Basal: 3.0 ± 0.2 s, Hypokalemia: −0.8 ± 0.1 s; p = .02).
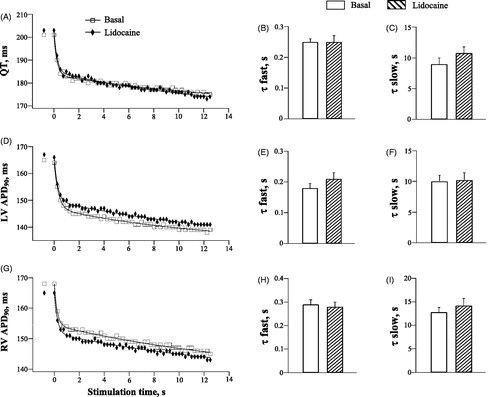
Lidocaine
Lidocaine had no effect on the QT interval and ventricular APD90 during spontaneous beating (data not shown), and did not modify the rate adaptation of ventricular repolarization (, panels A, D and G). The τfast and τslow values for the QT and APD90 shortening during rapid pacing were not changed by lidocaine (, panels B-C, E-F, and H-I).
Figure 5. Effects of lidocaine on the rate adaptation of QT interval and action potential duration in LV and RV chamber.
Discussion
Rate adaptation of ventricular repolarization and cardiac arrhythmia
Clinical studies have demonstrated the links between a protracted rate adaptation of ventricular repolarization and arrhythmogenic risks [Citation3–8]. A failure to rapidly attain appropriate APD90 shortening in transient tachycardia can be accounted for by multiple physiological factors. In normal conditions, an abrupt heart rate acceleration is associated with biphasic changes of repolarization, including a substantial APD90 reduction in the first few ventricular beats, followed by a slow phase of the gradual APD90 shortening to the new steady-state over subsequent cardiac cycles [Citation1,Citation17,Citation18]. The rapid APD90 reduction upon initiation of tachycardia is attributed to incomplete deactivation of IKs, the slow component of the delayed rectifier K+ current, and incomplete recovery of ICaL, the L-type Ca2+ current, in response to a sudden shortening of the diastolic interval [Citation1,Citation17]. Collectively, an increase in the outward current (IKs) along with a concomitant reduction in the inward current (ICaL) then contribute to APD90 drop immediately after the start of the rapid pacing. The subsequent slower phase of APD90 shortening is explained by alterations in ionic concentrations on both sides of the myocyte sarcolemma, which result from the fast excitation rate [Citation1,Citation17]. These changes include K+ accumulation in extracellular clefts, as well as increased intracellular Na+ and Ca2+ levels, which act to reduce APD90 via a complex mechanism that involves effects on the Na+–Ca2+ exchanger, Na+–K+ pump, and inactivation of the L-type Ca2+ current [Citation1,Citation17].
Clinical aspects
Class Ia Na+ channel blockers have been shown to induce postrepolarization refractoriness, an effect that prevents ventricular fibrillation [Citation19]. Nevertheless, in susceptible patients, this beneficial change can be outweighed by proarrhythmic modifications in ventricular repolarization. Quinidine therapy is recognized as one of the most common causes of the acquired long QT syndrome; this agent is estimated to prolong QT interval and provoke torsade de pointes in up to 5% of treated patients [Citation11]. Procainamide can prolong QT interval and occasionally precipitate VT [Citation11]. Apart from the drug therapies, another important risk factor for VT is hypokalemia, which represents a side effect of diuretic therapy in cardiac patients [Citation14].
Importantly, there is an indication that arrhythmogenic responses to class Ia agents are facilitated in the presence of impaired ventricular rate adaptation [Citation13]. In particular, the QT shortening during exercise-induced tachycardia is markedly attenuated in patients with a history of spontaneous VT induced by quinidine or procainamide therapies, as compared to those with no drug-induced arrhythmia [Citation13]. The attenuated QT shortening in the setting of preserved heart rate response to exercise translated to a paradoxical increase in the corrected QT value (QTc) in patients prone to arrhythmia, whereas in the control group the QTc was moderately reduced. Nevertheless, the time course of the QT rate adaptation was not explored in that study, and the potential impact of exercise-induced alterations in cardiac loads and coronary blood flow on the observed QT changes cannot be fully excluded. Likewise, owing to the clinical nature of the study, the local changes in repolarization of cardiac cells were not assessed. In this regard, the present animal study offers several advantages – the rate adaptation dynamics was accurately assessed by measuring appropriate metrics, such as τfast and τslow, from exponential plots, the QT changes on ECG were correlated to those in local APD90 determined both in LV and RV chambers, and all electrical recordings were taken in the controlled conditions of the whole heart perfusion.
Contributing mechanisms
Quinidine and procainamide have a complex electrophysiological profile, and can promote repolarization abnormalities [Citation20,Citation21] by reducing the outward K+ currents that govern phase 3 repolarization, such as IK, the delayed rectifier, and IK1, the inward rectifier [Citation22,Citation23]. Hypokalemia has been shown to suppress both IKr, the rapid component of the delayed rectifier, and IK1 [14]. A reduction in the delayed rectifier (IK), which is an important contributor to the initial phase of the rate adaptation [Citation1,Citation17], is likely to account for the attenuated APD90 shortening in the first few cardiac beats upon heart rate acceleration, by quinidine, procainamide, and hypokalemia, in the present study. As the initial phase of APD90 reduction is also partly determined by incomplete recovery of ICaL [Citation1,Citation17], it is noteworthy that both quinidine and procainamide exert an inhibitory effect on ICaL in ventricular myocytes [Citation22,Citation23]. The latter could have been an additional factor that accounts for the slowed kinetics of APD90 shortening upon administration of these drugs. Indeed, computer simulations and single myocyte studies suggest that ICaL blockade significantly prolongs τfast or even completely eliminates the rapid phase of APD90 reduction in tachycardia [Citation1,Citation17].
The slow phase of APD90 rate adaptation is likely modified by quinidine, procainamide, and hypokalemia through the effects on the Na+–K+ pump, Na+–Ca2+ exchanger, and the inward rectifier (IK1). Hypokalemia is known to inhibit the outward Na+–K+ pump current (INa–K) in cardiac cells [Citation16,Citation24], thus attenuating APD90 reduction during rapid pacing. This change is likely to be mediated through activation of the Ca2+-calmodulin kinase II (CaMKII) resulting from intracellular Na+ and Ca2+ overload upon inhibition of the Na+–K+ pump in cardiac myocyte sarcolemma [Citation16]. CaMKII then increases the late Na+ current and the L-type Ca2+ current, thus contributing to APD prolongation and facilitating early afterdepolarizations.
With quinidine and procainamide, the inhibition of the Na+–K+ pump can be produced either indirectly, as a consequence of reduced intracellular Na+ loading in the setting of INa blockade, or through the direct suppression of the ATP-hydrolytic activity of Na+–K+ ATPase [Citation25]. In addition, quinidine has been shown to partially block the outward current generated by the reverse-mode Na+–Ca2+ exchange [Citation26], which may have partly contributed to its effects in the present study. Interestingly, in isolated guinea-pig ventricular myocytes, the slow phase of APD90 rate adaptation during rapid pacing is almost completely abolished by a specific IK1 blocker, terikalant [Citation18]. As quinidine, procainamide, and hypokalemia are capable to reduce the inward rectifier current (IK1) in cardiac myocytes [Citation14,Citation22,Citation23], this mechanism should be considered to contribute to the effects of these interventions on the slow phase of APD90 dynamics.
Lidocaine is a clinically safe class Ib antiarrhythmic that acts as a selective INa blocker [Citation9,Citation10]. As such, it does not modify repolarizing K+ currents, which explains the lack of lidocaine effects on the QT and APD90 rate adaptation (). Furthermore, in contrast to quinidine and procainamide, which produce a sustained INa blockade, lidocaine rapidly dissociates from the Na+ channel. The time constant of INa recovery with quinidine and procainamide is in the range of 3–8 s, whereas with lidocaine it is as short as 0.09 s [Citation10]. This difference may imply much less capacity of lidocaine to interfere with intracellular Na+ accumulation and the associated ionic currents (such as INa-K and INaCa) during rapid pacing, which could have prevented its effects on the slow phase of APD90 rate adaptation.
Spatial heterogeneities in APD90 rate adaptation
Owing to the spatial variations in expression and function of the repolarizing K+ currents [Citation27], the APD90 rate adaptation kinetics is different at distinct recording sites within LV wall and across ventricular epicardium [Citation1,Citation28]. The present study adds to these findings by showing that significant interventricular differences in the rate adaptation exist in the guinea-pig heart. The basal action potential duration was found to be longer in RV vs. LV chamber, and the kinetics of APD90 shortening in response to heart rate acceleration was slower in RV (, panels C, D and E). Importantly, dissimilar APD90 dynamics at distinct ventricular sites can transiently amplify spatial repolarization heterogeneities and favor arrhythmia [Citation1,Citation28]. This raises a possibility that arrhythmogenic effects of quinidine, procainamide, and hypokalemia are partly related to the accentuated RV-to-LV non-uniformities in APD90 rate adaptation kinetics. The results from the present study, however, do not support this contention. Indeed, the basal RV-to-LV difference in τfast for APD90 reduction upon heart rate acceleration was not changed by quinidine and hypokalemia, and reduced by procainamide. Likewise, the RV-to-LV difference in τslow for APD90 reduction was not modified by quinidine and procainamide, whereas hypokalemia has abolished this difference. Overall, it appears that proarrhythmic effects of these interventions upon an abrupt heart rate acceleration are mainly attributed to the delayed APD90 rate adaptation at a given (either LV or RV) ventricular site, rather than to the accentuated spatial heterogeneities in repolarization dynamics.
Limitations
A delay in the rate adaptation of ventricular repolarization with quinidine, procainamide, or hypokalemia was not accompanied by initiation of the overt ventricular tachyarrhythmia in this study. This can be partly explained by the fact that VT initiation typically requires multiple hits on ventricular repolarization, for example, a combination of electrical remodeling related to cardiac disease, the APD-prolonging drug, and the electrolyte abnormality that reduces cardiac repolarization reserve, such as hypokalemia [Citation29]. Therefore, the lack of arrhythmogenic responses in this study is likely attributed to using normal heart preparations and testing effects of quinidine, procainamide, and hypokalemia separately, rather than in combination.
In cardiac patients, heart rate acceleration is usually caused by sympathetic activation which is an important arrhythmic trigger [Citation30]. As in the present study a sudden increase in cardiac activation rate was provoked by rapid pacing applied in denervated heart preparations, the adrenergic effects on the rate adaptation of ventricular repolarization were not addressed.
Conclusions
The rate adaptation of the QT interval and action potential duration in LV and RV chamber during rapid cardiac pacing is delayed by arrhythmogenic interventions, such as quinidine, procainamide, and hypokalemia, but not changed by lidocaine, a clinically safe antiarrhythmic agent.
Funding source
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Pueyo E, Husti Z, Hornyik T, et al. Mechanisms of ventricular rate adaptation as a predictor of arrhythmic risk. Am J Physiol Heart Circ Physiol. 2010;298:H1577–H1587.
- Vos MA, Verduyn SC, Gorgels APM, et al. Reproducible induction of early afterdepolarizations and torsade de pointes arrhythmias by d-sotalol and pacing in dogs with chronic atrioventricular block. Circulation. 1995;91:864–872.
- Cuomo S, De Caprio L, Acanfora D, et al. Relationship between QT interval duration and exercise-induced ventricular arrhythmias. Eur Heart J. 1989;10:622–627.
- Gill JS, Baszko A, Xia R, et al. Dynamics of the QT interval in patients with exercise-induced ventricular tachycardia in normal and abnormal hearts. Am Heart J. 1993;126:1357–1363.
- Yi G, Crook R, Guo XH, et al. Exercise-induced changes in the QT interval duration and dispersion in patients with sudden cardiac death after myocardial infarction. Int J Cardiol. 1998;63:271–279.
- Tavernier R, Jordaens L, Haerynck F, et al. Changes in the QT interval and its adaptation to rate, assessed with continuous electrocardiographic recordings in patients with ventricular fibrillation, as compared to normal individuals without arrhythmias. Eur Heart J. 1997;18:994–999.
- Smetana P, Pueyo E, Hnatkova K, et al. Individual patterns of dynamic QT/RR relationship in survivors of acute myocardial infarction and their relationship to antiarrhythmic efficacy of amiodarone. J Cardiovasc Electrophysiol. 2004;15:1147–1154.
- Viskin S, Postema PG, Bhuiyan ZA, et al. The response of the QT interval to the brief tachycardia provoked by standing: a bedside test for diagnosing long QT syndrome. J Am Coll Cardiol. 2010;55:1955–1961.
- Collinsworth KA, Kalman SM, Harrison DC. The clinical pharmacology of lidocaine as an antiarrhythymic drug. Circulation. 1974;50:1217–1230.
- Glaaser IW, Clancy CE. Cardiac Na+ channels as therapeutic targets for antiarrhythmic agents. Clin Exp Pharmacol Physiol. 2006;171:99–121.
- Frommeyer G, Eckardt L. Drug-induced proarrhythmia: risk factors and electrophysiological mechanisms. Nat Rev Cardiol. 2016;13:36–47.
- Osadchii OE. Flecainide attenuates rate adaptation of ventricular repolarization in guinea-pig heart. Scand Cardiovasc J. 2016;50:28–35.
- Kadish AH, Weisman HF, Veltri EP, et al. Paradoxical effects of exercise on the QT interval in patients with polymorphic ventricular tachycardia receiving type Ia antiarrhythmic agents. Circulation. 1990;81:14–19.
- Weiss JN, Qu Z, Shivkumar K. Electrophysiology of hypokalemia and hyperkalemia. Circ Arrhythm Electrophysiol. 2017;10:1–10.
- Osadchii OE. Impact of hypokalemia on electromechanical window, excitation wavelength and repolarization gradients in guinea-pig and rabbit hearts. PLoS One. 2014;9:e105599.
- Pezhouman A, Singh N, Song Z, et al. Molecular basis of hypokalemia-induced ventricular fibrillation. Circulation. 2015;132:1528–1537.
- Faber GM, Rudy Y. Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404.
- Williams BA, Dickenson DR, Beatch GN. Kinetics of rate-dependent shortening of action potential duration in guinea-pig ventricle; effects of IK1 and IKr blockade. Br J Pharmacol. 1999;126:1426–1436.
- Kirchhof PF, Fabritz CL, Franz MR. Postrepolarization refractoriness versus conduction slowing caused by class I antiarrhythmic drugs: antiarrhythmic and proarrhythmic effects. Circulation. 1998;97:2567–2574.
- Osadchii OE. Quinidine elicits proarrhythmic changes in ventricular repolarization and refractoriness in guinea-pig. Can J Physiol Pharmacol. 2013;91:306–315.
- Osadchii OE. Procainamide and lidocaine produce dissimilar changes in ventricular repolarization and arrhythmogenicity in guinea-pig. Fundam Clin Pharmacol. 2014;28:382–393.
- Salata JJ, Wasserstrom JA. Effects of quinidine on action potentials and ionic currents in isolated canine ventricular myocytes. Circ Res. 1988;62:324–337.
- Komeichi K, Tohse N, Nakaya H, et al. Effects of N-acetylprocainamide and sotalol on ion currents in isolated guinea-pig ventricular myocytes. Eur J Pharmacol. 1990;187:313–322.
- Aronsen JM, Skogestad J, Lewalle A, et al. Hypokalemia induces Ca2+ overload and Ca2+ waves in ventricular myocytes by reducing Na+–K+ ATPase α2 activity. J Physiol. 2015;593:1509–1521.
- Almotrefi AA, Basco C, Moorji A, et al. Class I antiarrhythmic drug effects on ouabain binding to guinea-pig cardiac Na+-K+ ATPase. Can J Physiol Pharmacol. 1999;77:866–870.
- Zhang YH, Hancox JC. Mode-dependent inhibition by quinidine of Na+-Ca2+ exchanger current from guinea-pig isolated ventricular myocytes. Clin Exp Pharmacol Physiol 2002;29:777–781.
- Chiamvimonvat N, Chen-Izu Y, Clancy CE, et al. Potassium currents in the heart: functional roles in repolarization, arrhythmia and therapeutics. J Physiol. 2017;595:2229–2252.
- Bueno-Orovio A, Hanson BM, Gill JS, et al. In vivo human left-to-right ventricular differences in rate adaptation transiently increase pro-arrhythmic risk following rate acceleration. PLoS One. 2012;7:e52234.
- Varro A, Baczko I. Cardiac ventricular repolarization reserve: a principle for understanding drug-related proarrhythmic risk. Br J Pharmacol. 2011;164:14–36.
- Fabritz L, Damke D, Emmerich M, et al. Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc Res. 2010;87:60–72.