
Abstract
Objectives. Although deuterium oxide (D2O) has preservative property on the extracted organ, whether D2O also protects the in situ myocardial injury remains unknown. Using cardiac microdialysis, local administration of D2O through dialysis probe was applied in situ rat heart. We examined the effect of the D2O on the myocardial injury induced ischemia, reperfusion, and chemical hypoxia. Methodology. We measured dialysate myoglobin levels during 30 min of coronary occlusion and reperfusion in the absence and presence of D2O. Furthermore, to confirm the effect of D2O on NaCN induced myocardial injury, we measured the dialysate myoglobin levels with local perfusion of NaCN in the absence and presence of D2O. Results. The dialysate myoglobin levels increased from 177 ± 45 ng/mL at baseline to 3030 ± 1523 ng/mL during 15–30 min of coronary occlusion and further increased to 8588 ± 1684ng/mL at 0–15 min of reperfusion. The dialysate myoglobin levels with 60 min local perfusion of NaCN increased to 1214 ± 279 ng/mL. D2O attenuated myocardial myoglobin release during 15–30 min of coronary occlusion and 0-30 min of reperfusion and 15–60 min of local perfusion of NaCN. Conclusions. D2O might have a beneficial effect of myocardium against ischemia, reperfusion and chemical hypoxia.
Introduction
Recently, in a rat heart transplant, an organ preservation solution containing D2O was found to exhibit more marked inhibitory effects against myocardial injury compared with the conventional University Wisconsin solution [Citation1]. Moreover, D2O increases glycolytic energy production, improves cellular energy maintenance [Citation2], and stabilizes plasma membrane [Citation3], membrane-bound proteins [Citation4], and cytoskeletal disruption [Citation5]. Relative to the rate of blood supply of D2O, the exchange of D2O between tissue and blood is extremely rapid [Citation6].
Although these suggest that D2O exhibits beneficial effects in organs in situ, there is little information regarding the protective effect of D2O in myocardial injury. The present work focused on the role of D2O on the induced impairment of in situ myocardial cells.
Until now, the cardiac microdialysis technique has been the method used for the local administration of pharmacological agents, including small and large molecular compounds, in the beating heart [Citation7,Citation8]. Furthermore, this method is suitable for examining the myocardial interstitial levels of various cardiac markers during ischemic reperfusion [Citation8]. The extent of myoglobin’s release into the interstitial space serves as an index of myocardial cell damage [Citation8,Citation9].
To clarify the in situ contribution of D2O to cardiomyocyte injury during ischemia and the following reperfusion, D2O was locally administered into anesthetized rat hearts in vivo, using a dialysis probe. The effects of D2O on myoglobin levels in the myocardial interstitial space were examined during coronary occlusion and following reperfusion.
Materials and methods
Animal preparation
The present study was conducted in accordance with the guidelines of the National Institutes of Health, “Guide for the Care and Use of Laboratory Animals” (Publication No. 85-23, Revised 2011). We have obtained necessary approvals for all animal procedures from the institutional Animal Care and Use Committee of Shiga University of Medical Science (2015-3-10). Male Sprague-Dawley rats at 14–16 weeks of age, weighing 370–470 g, were used. Pentobarbital sodium (30–40 mg i.p.) was used to anesthetize the rats. Based on previous experience [Citation10], pentobarbital sodium (15–20 mg/h) and butorphanol tartrate (0.025 mg/kg/h) were continuously infused intravenously into the right jugular vein, to achieve the required level of anesthesia. Heparin sodium (5 IU/kg/h) was administered to prevent coagulation. The rats were subjected to tracheotomy and ventilated with oxygen-mixed room air. The respiration rate was 70–80 min−1, and the tidal volume was 2.5 mL. Esophageal temperature was monitored as the body temperature. Maintenance of body temperature was accomplished using a lamp and heating pad. Mean arterial pressure (MAP), heart rate (HR), and continuous electrocardiogram (ECG) were monitored with a 3-lead ECG using PowerLab and LabChart (ADInstruments, Colorado, USA).
Dialysis methods
Full details of the procedure have been described previously [Citation8,Citation11]. In brief, handmade transverse dialysis probes were used. Polyethylene tubes were attached to both ends of the dialysis fiber (300 Å pore size, 0.215 mm OD, 0.175 mm ID, 5 mm long, sieving coefficient of human albumin during plasma perfusion: ∼0.9, Evaflux type 5 A, Kuraray Medical, Tokyo, Japan). With the help of a fine guiding needle, the dialysis probe was placed in the left ventricular wall. A snare occluder was placed around the LCA. The motion and color of the ventricular wall were examined to ascertain that the dialysis probe was in the ischemic region, during a brief coronary occlusion, and we have confirmed that the dialysis probe was located correctly.
We had dissolved the reagents (720 mg NaCl, 37 mg KCl, and 17 mg CaCl2) into 100 mL D2O (EURISO-TOP, Gif-sur-Yvette, France) to prepare a 100% D2O Ringer solution. A Ringer’s solution containing D2O was prepared by mixing 100% D2O Ringer’s solution and normal Ringer’s solution. The dialysis probes were perfused with Ringer’s solution or Ringer’s solution containing D2O at 5 µL/min using a microinjection pump (CMA/102, Carnegie Medicine, Sweden).
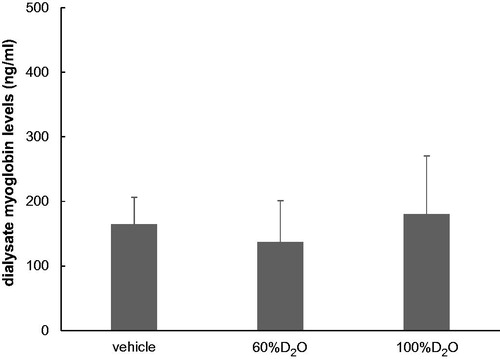
To confirm whether D2O exerted effects on the dialysate myoglobin levels at rest, the time course of the dialysate myoglobin levels in the presence or absence of D2O was measured. There were no significant differences between the levels at 90–120 min following probe implantation ().
Figure 1. Dialysate myoglobin levels at 90–120 min post-probe insertion (n = 3 each). The myoglobin levels in the 60%D2O and 100%D2O groups did not differ significantly in comparison to the vehicle group. Values are shown as mean ± standard error.
The sampling periods were 15 min, with a sampling volume of 75 μL, at baseline and also while coronary occlusion and reperfusion. D2O was administered locally, considering the dead space between the dialysis fiber and the sample tube, and the dialysate was sampled.
The myoglobin levels in the dialysate were measured and were considered to reflect the myocardial interstitial myoglobin levels. Immunochemistry (Cardiac Reader, Roche Diagnostics, Tokyo, Japan) was used for the measurement of myoglobin levels. The detection limit of myoglobin was 30 ng/mL.
Experimental protocols
Protocol 1: dialysate myoglobin levels induced by 30 min of coronary occlusion with varying concentrations of D2O perfusate
To examine whether D2O has effects on myocardial injury, the dialysates were sampled, and the myoglobin levels were measured in the ischemic region during 30 min of coronary occlusion in Ringer’s solution without D2O (vehicle; n = 7) and with D2O (20, 60, 80, and 100% D2O by volume; all n = 7). The dialysate was sampled from the ischemic region during the 30 min of coronary occlusion and the myoglobin levels were measured ().
Figure 2. Protocol of the experiments.
Protocol 2: effects of D2O on the dialysate myoglobin release induced by 30 min of coronary occlusion followed by 30 min reperfusion
To examine the role of D2O in myocardial damage during myocardial ischemia and reperfusion, myoglobin levels in the dialysate were measured in the presence of 60%D2O (60%D2O group; n = 8) and absence of D2O (vehicle group; n = 8) (). Following the collection of baseline samples for 15 min, the LCA was occluded for 30 min, followed by reperfusion through the release of the snare. The myoglobin levels were measured from dialysate samples collected every 15 min from the ischemic region during 30 min of coronary occlusion and 30 min of reperfusion.
Protocol 3: effects of D2O on dialysate myoglobin release induced by reperfusion
Protective effects of D2O on the myocardial injury during ischemia also affect reperfusion injury. To clarify these effects, D2O was administered just prior to reperfusion and dialysate myoglobin levels were measured during reperfusion.
The dialysate myoglobin levels were measured with the administration of 60%D2O immediately prior to coronary reperfusion. The collection was performed at 15–30 min of coronary occlusion as a baseline sample, and the tissues were then reperfused by releasing the snare. The dialysate myoglobin levels were measured during the coronary reperfusion in the absence (vehicle group; n = 8) and presence of 60%D2O (60%D2O group; n = 8).
Protocol 4: effects of D2O on dialysate myoglobin release induced by 30 mM sodium cyanide (NaCN)
To assess if D2O protects the myocardium against chemical hypoxia, local administration of D2O was performed. The dialysate myoglobin release induced by NaCN in the absence (vehicle group; n = 8) and presence of 60%D2O (60%D2O group; n = 8) was measured. In both groups, following the sampling of baseline dialysate, 30 mM NaCN was administered locally from the dialysis probe for 60 min. Myoglobin levels were measured in four consecutive dialysate samples.
At the end of the protocols, the rats were sacrificed by pentobarbital sodium overdosing, and the positioning of the dialysis probes in the myocardium was confirmed. A schematic of each experimental protocol is shown in .
Drugs
Freshly prepared drugs were used in all experiments. NaCN (Sigma, Tokyo, Japan) was dissolved in Ringer’s solution.
Statistical analysis
All the values are shown as the mean ± standard deviation unless otherwise specified. The dialysate myoglobin levels, HR, and MAP were statistically analyzed by two-way analysis of variance (ANOVA) with repeated measurements. Owing to the interaction, the analysis was performed by one-way ANOVA, followed by Tukey’s post hoc test. Differences between values were considered to be statistically significant when p < .05. The statistical software package GraphPad Prism (version 6.00; GraphPad Software, La Jolla, CA, USA) was used for the analysis.
Results
Protocol 1: dialysate myoglobin levels induced by 30 min of coronary occlusion at varying concentrations of D2O perfusate
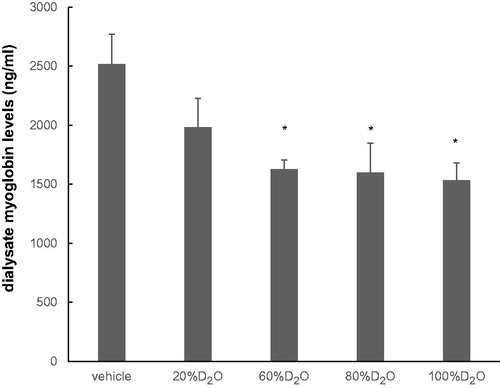
During the 30 min of coronary occlusion, myoglobin levels in the dialysate increased to 2516 ± 667 ng/mL in Ringer’s solution (vehicle group) (). Increasing the concentration of D2O increasingly suppressed the coronary-occlusion-induced release of myoglobin. The dialysate myoglobin levels were significantly suppressed between 60 and 100% D2O.
Figure 3. Dialysate myoglobin levels at 15–30 min of coronary occlusion at various concentrations of D2O (n = 7 each). The differences between the levels of dialysate myoglobin in the vehicle and D2O groups were significantly different (60, 80, and 100% D2O). Values are mean ± standard error. *p < .05 vs. vehicle.
Protocol 2: reduction of the myoglobin levels by D2O during 30 min coronary occlusion followed by 30 min reperfusion
Changes in HR, as well as MAP with time during the 30 min coronary occlusion and 30 min of reperfusion are shown in . In the vehicle control group, HR was significantly increased from 393 ± 15 beats min−1 at baseline to 422 ± 10 beats min−1 at 5 min of occlusion. Subsequently, it decreased to 395 ± 10 beats min−1 at 20 min of occlusion and was maintained at this level following reperfusion. HR was 399 ± 8 beats min−1 in the 60%D2O group, at baseline. These values increased significantly to 423 ± 13 beats min−1 after 5 min occlusion period and then decreased to 414 ± 4 beats min−1 at 20 min of occlusion, and these levels were maintained following reperfusion. The MAP values of 126 ± 7 mmHg in the vehicle group and 127 ± 6 mmHg in the 60%D2O group at baseline showed no significant change either during coronary occlusion or following reperfusion.
Table 1. Changes of HR and MAP in Protocol 2.
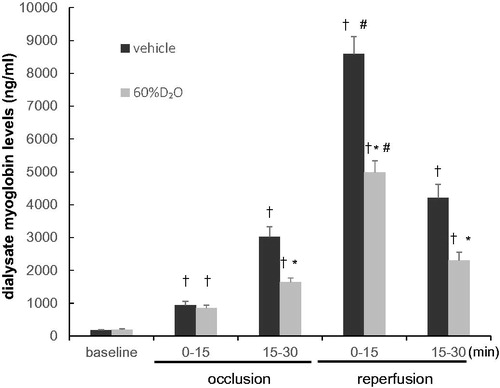
The changes in dialysate myoglobin concentration are shown in . In the control (vehicle) group, the dialysate myoglobin concentration at baseline was 177 ± 45 ng/mL. Following 15–30 min of coronary occlusion, this continued to increase, reaching 3030 ± 1523 ng/mL (p < 0.0001 compared to. baseline). Upon reperfusion, the dialysate myoglobin level increased even higher to 8588 ± 1684 ng/mL at 0–15 min reperfusion (p < .0001 vs. 15–30 min of occlusion) and then gradually decreased. The baseline dialysate myoglobin level in the 60%D2O group, was 197 ± 87 ng/mL. During occlusion, the level increased, reaching 1648 ± 345 ng/mL by 15–30 min of occlusion (p < .0001 compared to baseline). The dialysate myoglobin levels in the vehicle group were significantly different from the D2O group after coronary occlusion for 15–30 min, but not at baseline or at 0–15 min of occlusion. Following reperfusion, myoglobin in the dialysate increased further to 4989 ± 987 ng/mL at 0–15 min of reperfusion (p < .0001 compared to 15–30 min of occlusion) and then declined gradually. There was a significantly lower myoglobin level in the D2O group compared to the vehicle group during reperfusion.
Figure 4. Changes in dialysate myoglobin levels with time during the 30 min coronary occlusion and reperfusion in the vehicle group and 60%D2O group (n = 8 each). Exposure to 60%D2O suppressed the release of dialysate myoglobin induced by coronary occlusion and reperfusion. Values are mean ± standard error. *p < .05 vs. vehicle. †p < .05 vs. baseline. #p < .05 vs. 15–30 min of occlusion.
Protocol 3: D2O lowers myoglobin levels during the 30 min reperfusion after coronary occlusion for 30 min
The HR and MAP time courses were the same as those for protocol 2. HR was significantly increased from baseline to 5 min of occlusion. It subsequently decreased and was maintained following reperfusion. MAP did not alter significantly in either of the groups during coronary occlusion or the following reperfusion.
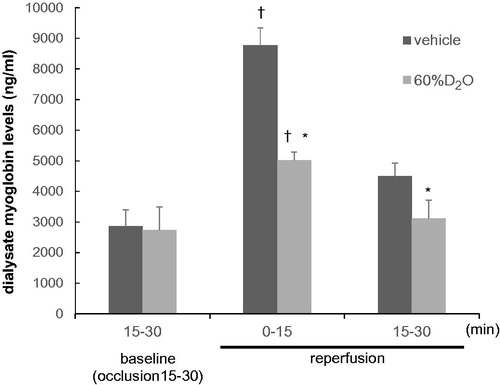
The changes in dialysate myoglobin concentration are shown in . The dialysate myoglobin level in the vehicle group was 2869 ± 1488 ng/mL prior to reperfusion (baseline: 15–30 min of occlusion). During reperfusion, it continued to increase, reaching 8768 ± 1628 ng/mL at 0–15 min of reperfusion (p < .0001 compared to pre-reperfusion) and then decreased slowly. In the 60%D2O group, myoglobin level in the dialysate was 1716 ± 354 ng/mL prior to reperfusion. After reperfusion for 0–15 min, this increased to 4948 ± 1037 ng/mL (p < .0001 compared to pre-reperfusion) and then declined gradually. At 0–15 and 15–30 min of reperfusion, the dialysate myoglobin levels were significantly lower in the D2O group than in the vehicle group.
Figure 5. Changes in dialysate myoglobin levels with time during the 30 min reperfusion in the vehicle and 60%D2O groups (n = 8 each). Exposure to 60%D2O suppressed the release of dialysate myoglobin induced by reperfusion. Values are shown as mean ± standard error. *p < .05 vs. vehicle. †p < .05 vs. baseline (15–30 min of occlusion).
Protocol 4: reduction of myoglobin levels by D2O during 60 min of local administration of NaCN
In the HR and MAP time courses, HR in the vehicle group was 413 ± 16 beats min−1, and MAP was 147 ± 19 mmHg at baseline. HR and MAP did not change significantly at any time point in protocol 4.
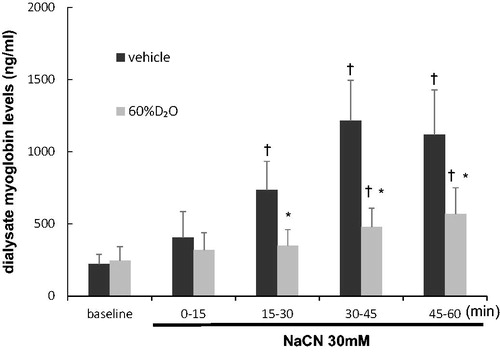
The changes in dialysate myoglobin levels are shown in . The dialysate myoglobin level in the vehicle group was 221 ± 68 ng/mL at baseline. Following the local administration of NaCN, the level of dialysate myoglobin continued to increase till a plateau is reached after 30–45 min of NaCN administration (1214 ± 279 ng/mL). In the 60%D2O group, the dialysate myoglobin concentration was 245 ± 96 ng/mL at baseline. Following the administration of NaCN, the levels of dialysate myoglobin were suppressed compared with those in the vehicle group. Significant differences were noticed in myoglobin levels in the dialysate between the vehicle and 60%D2O groups at 15–60 min following the administration of NaCN, but not at baseline or at 0–15 min.
Figure 6. Dialysate myoglobin levels as affected with time during the 60 min local administration of sodium cyanide (30 mM NaCN) in the vehicle group and 60%D2O group (n = 8 each). Exposure to 60%D2O suppressed the release of dialysate myoglobin induced by NaCN. Values are mean ± standard error. *p < .05 vs. vehicle. †p < .05 vs. baseline. NaCN: sodium cyanide.
Discussion
Employing a microdialysis method, the levels of myocardial interstitial myoglobin were measured in anesthetized rats. This study examined the effects of D2O on ischemia-reperfusion- and chemical-hypoxia-induced myocardial injury for the first time. In a dose-dependent manner, increasing concentrations of D2O depressed the ischemia-induced release of myoglobin with an approximate IC50 at 60% (). Similar effects have been shown, in which a D2O concentration of 35%–99.8% depressed the myocardial contraction and HR [Citation12]. Exposure to 60%D2O also suppressed the Ca2+ inflow into vascular smooth muscles [Citation13]. Therefore, this study examined the effects of 60%D2O during coronary occlusion on myoglobin efflux and also after reperfusion and local perfusion of NaCN.
Influence of D2O on the efflux of myoglobin during coronary occlusion
The levels of dialysate myoglobin increased during 0–30 min of coronary occlusion in a step-by-step manner. At 15–30 min of coronary occlusion, the levels of dialysate myoglobin had increased by ∼10-fold compared with that at baseline. The release of myoglobin into the interstitial space requires sarcolemmal disruption. A previous histochemical investigation using ionic lanthanum [Citation14] and NMR spectroscopy [Citation15] demonstrated sarcolemmal disruption at an early stage of ischemia. In addition, an immunochemical study observed that loss of myoglobin from the rat myocardium occurs after coronary occlusion for 30 min even without reperfusion, indicating that the release of myoglobin is induced by sarcolemmal disruption during ischemia [Citation16]. Therefore, the results suggested that D2O prevented substantial damage of the cardiomyocyte sarcolemma during 15–30 min of occlusion.
Lack of oxygen supply during regional ischemia blocks mitochondrial respiratory chain and oxidative phosphorylation. Synthesis of adenosine triphosphate (ATP) becomes restricted to the substrate-level phosphorylation in the glycolytic pathway, leading to lower activity of ATPases, and reduced active Ca2+ efflux and limited reuptake of Ca2+ by the endoplasmic reticulum, thereby producing Ca2+ overload in the cell [Citation17]. Hotta and Morales and Inoue et al. reported that the activity of myosin ATPase in D2O decreased to ∼60% of that in H2O, which led to the preservation of ATP [Citation18,Citation19]. Furthermore, 60%D2O suppressed the isometric contraction force of muscles from the left ventricle of rats [Citation13], which reduces cellular ATP demand. As a result, D2O improved the restoration of high-energy phosphates following reperfusion [Citation1]. The preservation of intracellular ATP levels by D2O might be major protective causes in myocardial injury during coronary occlusion.
Influence of D2O on the efflux of myoglobin during reperfusion injury
When the blood supply to a region is prevented, eventually all ischemic tissues present become necrotic [Citation20]. Restoration of the blood supply should minimize this damage; however, in practice, when coronary occlusion is relieved, the extent of reperfusion injury to reperfused tissues often increases with the restoration of the blood supply [Citation21]. Cardiac microdialysis showed that reperfusion markedly increased the release of myoglobin in the ischemic region. Although the precise mechanisms underlying this release of myoglobin were not verified in this study, the results suggest that D2O attenuated the release of substances during reperfusion. In previous studies, reperfusion augmented the release of myoglobin without any increase in the release of lactate or glycerol into the interstitium [Citation8]. Furthermore, reperfusion did not elevate the release of either catecholamine or acetylcholine at sympathetic and parasympathetic nerve endings in the ischemic myocardium [Citation22,Citation23]. During reperfusion, the surviving cardiomyocytes and also the nerve terminals rapidly recovered aerobic metabolism and eliminated these accumulated molecules, whereas the release of myoglobin continued via the disrupted membrane [Citation22,Citation23].
The reperfusion-induced release of myoglobin was suppressed by D2O administration. As reperfusion injury is influenced by ischemia preceding reperfusion, it was expected that the levels of myoglobin under D2O administration during ischemia and reperfusion may be lower than those during reperfusion. However, the levels of dialysate myoglobin during reperfusion did not differ between the two timings of D2O administration. Therefore, the protective effect of D2O on reperfusion injury may be independent of the extent of ischemic injury immediately prior to reperfusion.
D2O is a stable nonradioactive isotope of H2O, which contains ∼145 ppm of D2O. D2O has a higher density and more viscosity than H2O and has higher melting and boiling points [Citation24]. D2O has a variety of functions. The substitution of cellular H2O with D2O rapidly occurs and stabilizes the cytoskeleton [Citation25]. Furthermore, D2O restricts the influx of Ca2+ mainly through L-type Ca2+ channels [Citation26,Citation27] and Ca2+ efflux from the SR to the cytosol [Citation28]. The elevated cytosolic Ca2+ and subsequently activated Ca2+-dependent proteases serve a central role in cellular necrosis and apoptosis during periods of ischemia and reperfusion. Wakayama et al.[Citation1] reported that D2O prevented the elevation of cytosolic Ca2+ concentrations during cold preservation in vitro and inhibited the Ca2+-dependent activation of proteases in vivo [Citation1]. Owing to these characteristics, D2O has been investigated as an organ-preserving solution in organ transplantation [Citation29]. The efficacy of the preserving solution containing D2O for cold preservation of the liver, kidney, pancreas, and aorta has been reported [Citation2,Citation30–32]. Therefore, a protective effect of D2O on ischemia-reperfusion injury is expected in the in vivo rat myocardium.
In a clinical setting, the process of reperfusion can itself induce cardiomyocyte death [Citation21,Citation33]. Current therapeutic strategies for avoiding myocardial reperfusion injury have the ability to ameliorate the clinical outcomes in patients affected by acute myocardial infarction. Unfortunately, thus far, the results of the therapeutic focus on the isolated components of life-threatening myocardial ischemia-reperfusion injury, such as calcium overload [Citation34–37], oxidative stress [Citation34,Citation37], pH correction [Citation38,Citation39], and inflammation [Citation40,Citation41] have been discouraging. D2O attenuates the levels of dialysate myoglobin during reperfusion, which appears to be of importance to further investigate the therapeutic potential from the aspect of protection against reperfusion injury. Furthermore, this experiment showed that D2O protects against myocardial injury during ischemia and reperfusion injury separately and that D2O administration just prior to the release of coronary occlusion may provide effective protection against reperfusion injury. These results suggest that D2O application to coronary revascularization therapies via coronary arteries may exert beneficial effects of clinical practice.
The contribution of D2O to myoglobin efflux induced by NaCN
NaCN compromises mitochondrial respiration through cytochrome c oxidase inhibition, which led to a decrease in ATP production in a previous toxicological study [Citation42]. To investigate the protective role of D2O on energy failure in the efflux of large levels of myoglobin, the present study examined the effects of D2O on NaCN-induced myocardial myoglobin efflux. It has been reported that D2O is also considered to be effective in stabilizing the cytoskeleton structure [Citation5]. Our results showed that D2O suppresses the efflux of dialysate myoglobin induced by local perfusion of NaCN (). Therefore, it is likely that the potential preservation of ATP and cytoskeleton-stabilization by D2O attenuates myocardial injury induced by NaCN intoxication.
NaCN compromises mitochondrial respiration through the inhibition of cytochrome c oxidase, which led to a decrease in the production of ATP in a previous toxicological study [Citation42]. To investigate the protective role of D2O on energy failure in the efflux of large levels of myoglobin, the present study examined the effects of D2O on NaCN-induced myocardial myoglobin efflux. It was found that D2O suppressed the efflux of dialysate myoglobin induced by the local perfusion of NaCN (). Therefore, it is likely that the potential preservation of ATP by D2O attenuated the myocardial injury induced by NaCN intoxication.
The local pharmacological interventions used in the present study were selected in the context of possible myocardial injury during ischemia. The results of the local pharmacological interventions cannot be directly compared with the results of myocardial injury because of the complexity of the underlying mechanisms of ischemic injury. Furthermore, the coronary-occlusion-induced reduction in regional myocardial blood flow may affect the levels of dialysate myoglobin. It is difficult to isolate the change in the levels of dialysate myoglobin because of a decrease in the wash-out of extracellular fluid. In the present study, the coronary occlusion and local perfusion of NaCN-induced myoglobin efflux were suppressed to the same extent by D2O. Although the causality between coronary occlusion and local perfusion of NaCN remains to be elucidated, D2O was able to protect ischemic and anoxic myocardium.
Methodological considerations
We locally administered Ringer’s solution with D2O through a dialysis probe to avoid its systemic hemodynamic effects, and the solution constantly reached to the ischemic region during coronary occlusion. In the case of local administration, the active site of D2O is limited to a restricted region around the dialysis fiber and does not affect cardiac function. The area at risk was quantified to evaluate the efficacy of cardioprotective therapies. Therefore, the findings of the present study could not be directly compared with those of previous studies [Citation1,Citation43,Citation44]. However, the combination of local perfusion and sequential dialysate sampling provided information on the time course curves of D2O reactions in the ischemic region in vivo during coronary occlusion and following reperfusion.
In conclusion, D2O attenuated the release of myocardial myoglobin during coronary occlusion and reperfusion and local perfusion of NaCN. D2O is expected to offer promise as a protective approach to minimize the risk of myocardial ischemia-reperfusion injury, although additional studies are required.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Wakayama K, Fukai M, Yamashita K, et al. Successful transplantation of rat hearts subjected to extended cold preservation with a novel preservation solution. Trans Int. 2012;25:696–706.
- Fischer JH, Fuhs M, Miyata M, et al. Deuterium oxide (D2O) – a protective factor for cellular stabilization in hypothermic preservation of the liver? Chir Forum Exp Klin Forsch. 1980;129–133. German.
- Wenzel M, Hölscher B, Günther T, et al. Organ preservation by heavy water (D2O). Morphological and biochemical studies on heart and liver. J Clin Chem Clin Biochem. 1979;17:123–128.
- Ahlers J, Foret M, Lemm U. Does 2H2O also protect membrane-bound enzymes? Enzyme. 1983;30:70–73.
- Marsland D, Tilney LG, Hirshfield M. Stabilizing effects of D2O on the microtubular components and needle-like form of heliozoan axopods: a pressure-temperature analysis. J Cell Physiol. 1971;77:187–193.
- Thompson AM, Cavert HM, Lifson N. Kinetics of distribution of D2O and Antipyrine in isolated perfused rat liver. Am J Physiol. 1958;192:531–537.
- Delyani JA, Van Wylen DG. Endocardial and epicardial interstitial purines and lactate during graded ischemia. Am J Physiol Heart Circ Physiol. 1994;266:H1019–H1026.
- Kitagawa H, Yamazaki T, Akiyama T, et al. Microdialysis separately monitors myocardial interstitial myoglobin during ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2005;289:H924–30.
- Kawada T, Yamazaki T, Akiyama T, et al. Vagal stimulation suppresses ischemia-induced myocardial interstitial myoglobin release. Life Sci. 2008;83:490–495.
- Inagaki T, Akiyama T, Du CK, et al. Monoamine oxidase-induced hydroxyl radical production and cardiomyocyte injury during myocardial ischemia-reperfusion in rats. Free Radic Res. 2016;50:645–653.
- Sonobe T, Akiyama T, Du CK, et al. Contribution of calpain to myoglobin efflux from cardiomyocytes during ischaemia and after reperfusion in anaesthetized rats. Acta Physiol (Oxf). 2014;210:823–831.
- Kaiminer B. Effect of heavy water on different types of muscle and on glycerol-extracted psoas fibers. Nature. 1960;185:172–173.
- McWilliam TM, Liepins A, Rankin AJ. Deuterium oxide reduces agonist and depolarization-induced contraction of rat aortic rings. Can. Can J Physiol Pharmacol. 1990;68:1542–1547.
- Kupriyanov VV, Xiang B, Butler KW, et al. Energy metabolism, intracellular Na+ and contractile function in isolated pig and rat hearts during cardioplegic ischemia and reperfusion: 23Na- and 31P-NMR studies. Basic Res Cardiol. 1995;90:220–233.
- Askenasy N. Glycolysis protects sarcolemmal membrane integrity during total ischemia in the rat heart. Basic Res Cardiol. 2001;96:612–622.
- Nomoto K, Mori N, Shoji T, et al. Early loss of myocardial detected immunohistochemically following occlusion of the coronary artery in rats. Exp mol Pathol. 1987;47:390–402.
- Kalogeris T, Baines CP, Krenz M, et al. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317.
- Hotta K, Morales MF. Myosin B nucleoside triphosphatase in deuterium oxide. J Biol Chem. 1960;235:PC61–PC63.
- Inoue A, Fukushima Y, Tonomura Y. Effect of deuterium oxide on elementary steps in the ATPase reactions. Biochem. 1975;78:1113–1121.
- Altamirano F, Wang ZV, Hill JA. Cardioprotection in ischemia-reperfusion injury: novel mechanisms and clinical translation. J Physiol. 2015;593:3773–3788.
- Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135.
- Akiyama T, Yamazaki T, NInomiya I. Differential regional response of myocardial interstitial norepinephrine levels to coronary occlusion. Cardiovasc Res. 1993;27:817–822.
- Kawada T, Yamazaki T, Akiyama T, et al. Differential acetylcholine release mechanisms in the ischemic and non-ischemic myocardium. J Mol Cell Cardiol. 2000;32:405–414.
- Hirakura Y, Sugiyama T, Takeda M, et al. Deuteration as a tool in investigating the role of protons in cell signaling. Biochim Biophys Acta. 2011;1810:218–225.
- Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77:79–88.
- Prod’hom B, Pietrobon D, Hess P. Direct measurement of proton transfer rates to a group controlling the dihydropyridine-sensitive Ca2+ channel. Nature. 1987;329:243–246.
- Ikeda M, Hirono M, Kishio M, et al. Examination of microgravity effects using heavy water. J Gravit Physiol. 2000;7:63–64.
- Huxtable R, Bressler R. The effect of deuterium ion concentration on the properties of sarcoplasmic reticulum. J Membr Biol. 1974;17:189–197.
- Berwanger CS, Cleanthis TM, Hafez HM, et al. Deuterium oxide-based university of Wisconsin solution improves viability of hypothermically stored vascular tissue. Transplantation. 1998;65:735–737.
- Fischer JH, Knupfer P, Beyer M. Flush solution 2, a new concept for one-to-three-day htpothermic renal storage preservation. Functional recovery after preservation in Euro-Collins, Collins’ C2, hypertonic citrate, and F. 2 solution. Transplantation. 1985;39:122–126.
- Fischer JH, Jeschkeit S. Effectivity of freshly prepared or refreshed solutions for heart preservation versus commercial EuroCollins, Bretschneider’s HTK or University of Wisconsin solution. Transplantation. 1995;59:1259–1262.
- Hesse UJ, Gores PF, Florack G, et al. The use of D2O (heavy water)-based solution for hypothermic preservation of the pancreas. Transplant Proc. 1987;19:4167.
- Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J. Clin. Invest. 1985;76:1713.
- Kloner RA, Bolli R, Marban E, et al. Medical and cellular implications of stunning, hibernation, and preconditioning: an NHLBI workshop. Circulation. 1998;97:1848–1867.
- Avkiran M, Marber MS. Na(+)/H(+) exchange inhibitors for cardioprotective therapy: progress, problems and prospects. J Am Coll Cardiol. 2002;39:747–753.
- Miyamae M, Camacho SA, Weiner MW, et al. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am J Physiol. 1996;271:H2145–H2153.
- Bair FW, Tzivoni D, Dirksen MT, et al. Results of the first clinical study of adjunctive CAldaret (MCC-135) in patients undergoing primary percutaneous coronary intervention for ST-Elevation Myocardial Infarction: the randomized multicentre CASTEMI study. Eur Heart J. 2006;27:2516–2523.
- Lemaster JJ, Bond JM, Chacon E, et al. The pH paradox in ischemia-reperfusion injury to cardiac myocytes. EXS. 1996;76:99–114.
- Qian T, Nieminen AL, Herman B, et al. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol. 1997;273:C1783–C1792.
- Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–497.
- Atar D, Petzelbauer P, Schwitter J, et al. Effect of intravenous FX06 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction results of the F.I.R.E. (efficacy of FX06 in the prevention of myocardial reperfusion injury) trial. J Am Coll Cardiol. 2009;53:720–729.
- Thompson RW, Valentine HL, Valentine WM. Cytotoxic mechanisms of hydrosulfide anion and cyanide anion in primary rat hepatocyte cultures. Toxicology. 2003;188:149–159.
- Hirokata G, Fukai M, Wakayama K, et al. Dose heavy water attenuate cold preservation and reperfusion injury in canine kidney?. Low Temp Med. 2012;38:62–68.
- Grönros J, Kiss A, Palmer M, et al. Arginase inhibition improves coronary microvascular function and reduces infarct size following ischaemia-reperfusion in a rat model. Acta Physiol. 2013;208:172–179.