
Abstract
Objectives: This study aimed to evaluate the safety of CT-P13 in patients with rheumatoid arthritis (RA) during long-term treatment or after switching from innovator infliximab (IFX).
Methods: Patients who completed 54 weeks of treatment in a phase I/II study (PI/II) received CT-P13 at an initial dose of 3 mg/kg at Week 62, with dose increases permitted up to 10 mg/kg. The primary endpoint was adverse event (AE) incidence.
Results: Thirty-four of 38 patients in the maintenance group and 29 of 33 in the switch group reported at least one AE. Safety profiles in both groups were similar to those in PI/II. Eleven of 28 patients who were positive for anti-drug antibodies (ADA) at Week 62 discontinued the study before Week 110. Forty-one of 43 ADA-negative patients remained negative, and 10 of 28 ADA-positive patients became negative during the study. The mean DAS28 (ESR) at Week 134 was 3.166 in the maintenance group and 3.955 in the switch group.
Conclusions: CT-P13 was well tolerated in patients who maintained the treatment after 54 weeks and in patients who switched to CT-P13 after 54 weeks of IFX treatment. The study also demonstrated a stable clinical efficacy of CT-P13 in RA patients.
Introduction
CT-P13 is a biosimilar of innovator infliximab (IFX) and the first biosimilar antibody approved in Japan (in July 2014). In the phase I/II study in Japanese rheumatoid arthritis (RA) patients, pharmacokinetic (PK) equivalence between CT-P13 and IFX was confirmed as we reported previously [Citation1]. The data in the study showed that the safety and efficacy of CT-P13 throughout the 54-week dosing were similar to those in the PLANETRA study conducted in Europe, Latin America, Middle East, and Asia (not including Japan) [Citation2]. The results of the study also became the pivotal data basis for CT-P13 approval in Japan. As of December 1, 2015, CT-P13 is available in 66 countries, including Japan, for the treatment of immune-related inflammatory diseases such as RA.
The Position Statement of the ACR (American College of Rheumatology) [Citation3] requires that to use biosimilar product in the management of RA diseases safety and efficacy clinical data must be collected. Since infliximab is a chimeric antibody to TNF-α, it is crucial to evaluate changes in immunogenicity both during long-term treatment and after switching from IFX to CT-P13. However, only summary data have been reported from an extension study of CT-P13 phase III study, PLANETRA [Citation4], conducted only in non-Japanese patients.
Based on these circumstances, we planned this extension study to evaluate the safety of CT-P13 in RA patients undergoing a long-term treatment or switching from IFX after the phase I/II study. The study was conducted in patients who gave consent to participate in the extension study.
Patients and methods
Patient population
The study was conducted at 20 medical institutions throughout Japan, between December 2012 and February 2015, in RA patients who completed a 54-week treatment with CT-P13 or IFX in the phase I/II study. This extension study enrolled patients who signed informed consent and were judged by the investigators to be clinically stable and suitable for treatment with CT-P13 at the last visit in the phase I/II study. Details of patient eligibility criteria are provided online (see the Online Supplementary Appendix A).
Study design
This was an open-label, single-arm, multicenter, extension study. The primary endpoint was the safety of CT-P13 during long-term treatment and after switching from IFX to CT-P13 in RA patients who received CT-P13 or IFX concomitantly with methotrexate (MTX) in the Japanese phase I/II PK equivalence study. In addition, the long-term efficacy of CT-P13 was assessed as the secondary endpoint.
Patients who were confirmed to be eligible for extension of treatment based on the inclusion and exclusion criteria were enrolled to receive a 2-h intravenous infusion of 3 mg/kg CT-P13 at Week 62 (0×; the initiation week of extension study as well as 62 weeks from the initiation of phase I/II study) and eight weekly doses thereafter up to the study termination. Dose increases of CT-P13 were allowed up to 10 mg/kg after Week 70 (8×; 8 weeks from the initiation of the extension study), on the discretion of the investigator in charge, if insufficient effect or loss of response were observed. Throughout the study, MTX (4–16 mg/week; oral dose) and folic acid (≤10 mg/week; oral dose) were co-administered. Oral corticosteroid was permitted if the daily dose was no more than 10 mg equivalent of prednisolone.
This study was conducted in accordance with the ethical principles derived from the Declaration of Helsinki, and in compliance with the Good Clinical Practice guidelines. The study protocol and informed consent form were reviewed and approved by the Institutional Review Board at each site. Written informed consent was obtained from all patients. This study was registered with the JAPIC Clinical Trial Information Center (http://www.clinicaltrials.jp/user/cteSearch_e.jsp) (JapicCTI-142419).
Safety assessments
Safety parameters including adverse events (AEs), serious AEs (SAEs), vital signs (systolic and diastolic blood pressure, pulse rate, respiratory rate, and body temperature), physical findings, laboratory test results, infections (including tuberculosis), and infusion-related reactions were assessed at each visit, and anti-drug antibodies (ADA), proportion of patients whose interferon-gamma (INF-γ) release assay result became positive, and electrocardiogram findings were assessed at 24-week intervals. Serum samples for analysis were collected before each dose of CT-P13. Serum ADAs were analyzed by electroluminescence assay using the Meso Scale Discovery platform (MSD, Rockville, MD). This ADA assay system was able to detect both anti-IFX and anti-CT-P13 antibodies [Citation2].
Efficacy assessments
Efficacy parameters were assessed at 24-week intervals. These parameters included the American College of Rheumatology 20%, 50%, and 70% response rates (ACR20, ACR50, ACR70) [Citation5,Citation6], changes from baseline in disease activity score using a 28-joint count (DAS28) [Citation7], the proportions of patients achieving moderate or good response according to the European League Against Rheumatism (EULAR) response criteria [Citation8], simplified disease activity index (SDAI) [Citation9], clinical disease activity index (CDAI) [Citation10], and Health Assessment Questionnaire disability index (HAQ-DI) [Citation11].
Statistical analysis
All patients who received at least one dose of CT-P13 in the extension study were included in the safety analysis set. All AEs occurring between the start of study treatment and the time of assessment for the completion or discontinuation of study treatment were documented. The data were summarized in terms of the number of AEs, the number and percentage of patients experiencing each AE, as well as severity and relation to the study drug both in patients enrolled from the CT-P13 group of the phase I/II study (the maintenance group) and from the IFX group (the switch group). For laboratory parameters, the observed value and the changes from the value obtained immediately before the first study treatment at the phase I/II study (baseline) were summarized using descriptive statistics. Data on ADA are expressed in terms of the proportion of ADA-positive patients.
Efficacy analyses were performed on the full analysis set (FAS), which consisted of all patients who were eligible for the extension study and received at least one dose of the study drug. Patients who were ineligible for the phase I/II study were excluded from the FAS. For efficacy parameters, observed values and changes from baseline were summarized using descriptive statistics for each group. The non-responder imputation (NRI) procedure was applied for patients who were withdrawn due to insufficient response or adverse reactions, whereby missing continuous data were imputed by baseline data. Missing data generated due to other reasons were imputed using the last observation carried forward (LOCF) method.
Results
Patient disposition and disease characteristics
Of 82 patients who completed 54 weeks of treatment in the phase I/II study, 72 enrolled in the extension study, and 10 patients who did not provide informed consent were excluded (). One of the enrolled patients withdrew from the study before the initiation of study treatment because a surgical operation was performed for her sinusitis. Safety analysis was conducted for the remaining 71 patients who received CT-P13, consisting of 38 in the maintenance group and 33 in the switch group. Two patients were further excluded due to eligibility violations in the phase I/II study, giving a FAS comprising 69 patients (37 in the maintenance group and 32 in the switch group).
Figure 1. Patient disposition in the phase I/II study and the extension study.
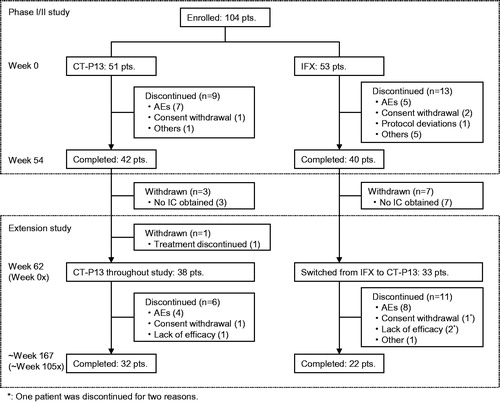
Of the 71 patients in the safety analysis set, seven patients discontinued the study treatment before Week 86 (24×); seven before Weeks 86 (24×)–110 (48×); and two before Weeks 110 (48×)–134 (72×) of the extension study. Fifty-five patients (32 in the maintenance group and 23 in the switch group) continued the study treatment through Week 134 (72×). Only one patient in the switch group discontinued study treatment after Week 134 (72×).
Demographic characteristics in the safety analysis set are shown in . All patients in both groups received combination treatment with MTX from the start of phase I/II study through extension study in accordance with the study protocol. Among demographic characteristics, only body weight at the start of extension study in the maintenance group was slightly different (higher) from that in the switch group. Patient’s assessments of pain (VAS) and health assessment questionnaire (HAQ) scores in the switch group were slightly higher than those in the maintenance group, but no significant difference was observed between the groups. The number of ADA-positive patients in the switch group was 16 (48.5%), which was higher than that in the maintenance group (12 patients; 31.6%).
Table 1. Baseline patient demographics and disease characteristics—safety analysis set.
Patients maintaining the CT-P13 treatment throughout the study
The duration [mean ± standard deviation (S.D.)] between the initial and final treatments in the extension study was 80.5 ± 19.7 weeks in the maintenance group. The reasons of study discontinuation were emergence of AEs (4 patients), lack of efficacy (1 patient), and consent withdrawal (1 patient). The mean age (mean ± S.D.) of patients was 54.5 ± 12.4 years. CT-P13 was administered to patients in the maintenance group every 8 weeks for 10.0 ± 2.4 (mean ± S.D.) times. The dose of CT-P13 was increased for 24 of 38 patients in the maintenance group, and the mean dose was 5.72 ± 3.00 mg/kg (mean ± S.D.) at Week 134 (72×). The dosage of MTX at Week 134 (72×) was 9.73 ± 2.94 mg/week (mean ± S.D.).
AEs reported in the phase I/II study (from Week 0 to Week 62) and the extension study (from Week 62 [0×] to Week 167 [105×]) are summarized in . There was no notable difference between the phase I/II study and the extension study in AEs, adverse drug reactions (ADRs), SAEs, infection, and infusion-related reactions. shows AEs leading to study discontinuation occurred in the extension study (from Week 62 [0×] to Week 167 [105×]). Infusion-related reactions and weight decreased were each reported as an SAE in one patient, the latter was considered unrelated to the study drug. Both events were resolved or improved. Four patients discontinued the study treatment due to AEs during the extension study. Infusion-related reaction, interferon-γ release assay positive, anaphylactic shock, and anti-neutrophil cytoplasmic antibody positive vasculitis were each reported in one patient, and all of these events, except interferon-γ release assay positive, were resolved. All of the AEs leading to study discontinuation were considered related to the study drug. No active tuberculosis or pneumocystis jiroveci pneumonia was reported.
Table 2. Summary of safety—safety analysis set.
Table 3. SAEs and AEs leading to study discontinuation occurred in extension phase—safety analysis set.
AEs reported in at least 5% of patients in the extension study (from Week 62 [0×] to Week 167 [105×]) are shown in . AEs frequently reported among the 38 patients in the maintenance group were nasopharyngitis (10 [26.3%] patients), upper respiratory tract inflammation (8 [21.1%] patients), herpes zoster (4 [10.5%] patients), and rash (4 [10.5%] patients). All of the moderate or severe AEs were reported only in one patient, except herpes zoster, which occurred in two patients (both events were moderate in severity). No grade 3 or grade 4 abnormality was observed in laboratory parameters. There was no clinically significant finding in vital sign, physical examination, and electrocardiogram.
Table 4. Adverse events reported in at least 5% of patients in either of the treatment groups—safety analysis set.
Positive rates of ADA at Week 110 (48×) and Week 134 (72×) in the maintenance group were 11.8% (4/34 patients) and 15.6% (5/32 patients). Twenty-five of 26 patients who were negative for ADA at the initiation of the extension study remained negative throughout the study, and 5 of 12 patients who were positive for ADA became negative during the study. All patients who became negative for ADA received an increased dose of CT-P13 (). Changes in ADA status at each study point are provided online in Supplementary Appendix B.
Figure 2. Changes in ADA status at baseline and Week 110 (48×) and the relationship between ADA status and dose of CT-P13 in patients who maintained the CT-P13 treatment throughout study (n = 38) and patients who switched from IFX to CT-P13 (n = 33) in the safety analysis set.
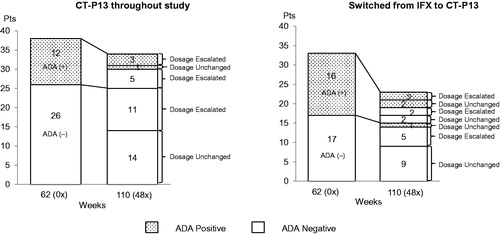
Three patients who were positive for ADA and one patient who was negative for ADA at Week 62 (0×) discontinued the study treatment before Week 110 (48×). All of these patients had received increased doses of CT-P13. Only one of the patients who were newly tested gave a positive result for ADA after Week 110 (48×). Five patients whose ADA test results changed from positive to negative continued to test negative through Week 134 (72×) to the end of study.
Although data varied among evaluation points, ACR response rates tended to show a slight improvement from the initiation of extension study through Week 134 (72×). The proportions of patients achieving ACR20, ACR50, and ACR70, respectively, were 73% (27/37 patients), 59.5% (22/37 patients), and 45.9% (17/37 patients) at Week 62 (0×); 81.1% (30/37 patients), 67.6% (25/37 patients), and 48.6% (18/37 patients) at Week 110 (48×); and 78.4% (29/37 patients), 70.3% (26/37 patients), and 54.1% (20/37 patients) at Week 134 (72×). The mean scores (mean ± S.D.) of DAS28 (ESR) at Week 62 (0×), Week 110 (48×), and Week 134 (72×) were 3.220 ± 1.328, 3.180 ± 1.693, and 3.166 ± 1.533, and the mean changes (mean ± S.D.) from baseline were −2.707 ± 1.589, −2.747 ± 1.727, and −2.761 ± 1.613, respectively. Although data varied among evaluation points, other parameters generally showed a stable clinical efficacy from the initiation of extension study through Week 134 (72×) (see the Online Supplementary Appendix C).
Patients switching from IFX to CT-P13
The duration (mean ± S.D.) between the initial and final treatments in the extension study was 69.0 ± 29.5 weeks in the switch group. The reasons of study discontinuation were emergence of AEs (8 patients), lack of efficacy (1 patient), lack of efficacy and consent withdrawal (1 patient), and other reason (1 patient). The mean age (mean ± S.D.) of patients was 57.2 ± 10.7 years. Patients in the switch group received CT-P13 every 8 weeks for 8.6 ± 3.6 (mean ± S.D.) times. The dose of CT-P13 was increased in 16 of 33 patients in the switch group, and the mean dose was 4.88 ± 2.87 mg/kg (mean ± S.D.) at Week 134 (72×). The dosage of MTX at Week 134 (72×) was 9.30 ± 3.28 mg/week (mean ± S.D.).
There was no notable difference between phase I/II study and the extension study in AEs, ADRs, SAEs, infection, and infusion-related reaction (). Enterocolitis, tendon rupture, interstitial lung disease, and colon cancer was each reported as an SAE in one patient, and all of these events except for colon cancer were resolved. Tendon rupture was considered attributable to complicated bone deformity with RA that had existed before the study and unrelated to the study drug. Eight patients discontinued the study treatment due to AEs during the extension study (). Infusion-related reaction (3 patients), pancytopenia (1 patient), RA (1 patient), interstitial lung disease (1 patient), colon cancer (1 patient), and pneumonia (1 patient) were reported. Except for colon cancer and RA, these AEs were resolved or improved. Pancytopenia and RA were considered unrelated to the study drug. No tuberculosis or pneumocystis jiroveci pneumonia was reported.
The AEs frequently reported among the 33 patients in the switch group were nasopharyngitis (9 [27.3%] patients), infusion-related reaction (4 [12.1%] patients), and osteoporosis (4 [12.1%] patients) (). All of the moderate or severe AEs were each reported only in one patient, except for tendon rupture in two patients (two moderate events), and RA in two patients (two moderate events). No grade 3 or grade 4 abnormality was observed in laboratory parameters. There was no clinically significant finding in vital sign, physical examination, or electrocardiogram.
Positive rates for ADA at Week 110 (48×) and 134 (72×) in the switch group were 21.7% (5/23 patients) and 17.4% (4/23 patients). Sixteen of 17 patients who were negative for ADA at the initiation of the extension study remained negative throughout the study, and 5 of 16 patients who were positive for ADA became negative during the study (). Changes in ADA status at each study point are provided online in Supplementary Appendix B.
Eight patients who were positive for ADA and two patients who were negative for ADA at Week 62 (0×) discontinued the study treatment before Week 110 (48×). Of these 10 patients, 5 ADA-positive patients and 1 ADA-negative patient received an increased dose of CT-P13, but no dosages were changed due to AEs for the other 4 patients. Only one of the patients who were negative at Week 62 (0×) became positive for ADA by Week 110 (48×). Four patients whose ADA test results changed from positive to negative continued to test negative through Week 134 (72×) to the end of study.
Although data varied among evaluation points, ACR response rates tended to show a slight improvement from the initiation of extension study through Week 134 (72×) (see the Online Supplementary Appendix C). The proportions of patients achieving ACR20, ACR50, and ACR70, respectively, were 62.5% (20/32 patients), 43.8% (14/32 patients), and 25.0% (8/32 patients) at Week 62 (0×); 59.4% (19/32 patients), 50.0% (16/32 patients), and 37.5% (12/32 patients) at Week 110 (48×); and 62.5% (20/32 patients), 53.1% (17/32 patients), and 40.6% (13/32 patients) at Week 134 (72×). The mean scores (mean ± S.D.) of DAS28 (ESR) at Weeks 62 (0×), 110 (48×), and 134 (72x) were 3.882 ± 1.496, 3.979 ± 1.721, and 3.955 ± 1.751, and the mean changes (mean ± S.D.) from baseline were −2.036 ± 1.305, −1.940 ± 1.664, and −1.964 ± 1.711, respectively. Although data varied among evaluation points, other parameters generally showed a stable clinical efficacy from the initiation of extension study through Week 134 (72×).
CT-P13 dose-increased patients
The AEs frequently reported (more than 10% of patients) in the 40 patients (24 in the maintenance group and 16 in the switch group) who received an increased dose of CT-P13 were nasopharyngitis (12 [30%] patients), herpes zoster (4 [10%] patients), and pneumonia (4 [10%] patients). Infusion-related reaction, enterocolitis, and interstitial lung disease were each reported in one patient as an SAE. Seven of 12 AEs (58.3%) leading to discontinuation of the study, infusion-related reactions (2 patients), anaphylactic shock (1 patient), anti-neutrophil cytoplasmic antibody positive vasculitis (1 patient), pneumonia (1 patient), RA (1 patient), and interstitial lung disease (1 patient) were reported in patients who received an increased dose of CT-P13. Although the frequency of infection in patients who received an increased dose of CT-P13 (24 [60.0%] patients) was slightly higher than those in patients who maintained the same dose of CT-P13 (14 [45.2%] patients), no severe AE was observed, and most of the AEs were mild in severity except for herpes zoster (2 moderate events), pneumonia (1 moderate event), and periodontitis (1 moderate event). There were no characteristic tendencies in AEs other than infection between dose-increased patients and dose-unchanged patients. In addition, no notable difference was observed in the profile of AEs between the maintenance group and the switch group.
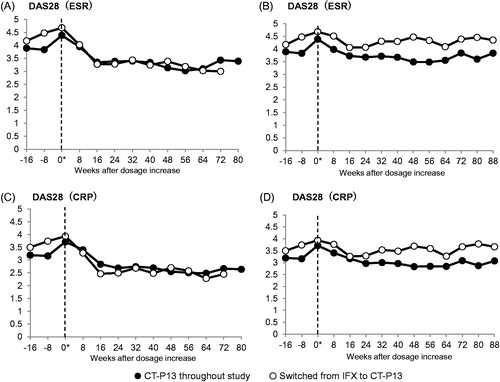
The mean scores (mean ± S.D.) of DAS28 (ESR) in dose-increased patients at the time of dose-increase and at 16 weeks after dose-increase were 4.39 ± 1.43 (n = 23) and 3.35 ± 1.60 (n = 19) in the maintenance group and 4.68 ± 1.20 (n = 16) and 3.27 ± 0.83 (n = 12) in the switch group, respectively, which demonstrated early improvements in both groups ().
Figure 3. Changes after dose-increase in DAS28 for patients who maintained the CT-P13 treatment throughout the study (solid circle; n = 23) and patients who switched from IFX to CT-P13 (open circle, n = 16). (A) DAS28 (ESR) in the FAS without LOCF and NRI. (B) DAS28 (ESR) in the FAS with the missing data imputed by the LOCF and NRI method. (C) DAS28 (CRP) in the FAS without LOCF and NRI. (D) DAS28 (CRP) in the FAS with the missing data imputed by the LOCF and NRI method. “0*” denotes the time of dose-increase.
ADA-positive patients
The AEs frequently reported (more than 10% of patients) in the 28 patients who were ADA-positive at Week 62 (0x) were nasopharyngitis (5 [17.9%] patients), infusion-related reaction (5 [17.9%] patients), and double-stranded DNA antibody positive (4 [14.3%] patients). The frequency of nasopharyngitis in ADA-positive patients was lower than those reported in ADA-negative patients (32.6%). Infusion-related reaction and double-stranded DNA antibody positive were reported only in ADA-positive patients. These events were observed in both the maintenance group and the switch group. Eleven of 28 patients (39.3%) who were ADA-positive at Week 62 (0×) discontinued study treatment before Week 110 (48×). The reasons for discontinuation were infusion-related reaction in four patients, lack of efficacy in two patients, and anaphylactic shock, anti-neutrophil cytoplasmic antibody positive vasculitis, pancytopenia, RA aggravated, and interstitial lung disease, respectively, in one patient .
The ACR response rates were lower in the ADA-positive patients than in the ADA-negative patients: the proportions of patients achieving ACR20 at Week 62 (0×) and 134 (72×) were 83.3% (35/42 patients) and 81.0% (34/42 patients) in the ADA-negative patients, and 44.4% (12/27 patients) and 55.6% (15/27 patients) in the ADA-positive patients.
Discussion
We evaluated the safety of CT-P13 in patients undergoing a long-term treatment over 54 weeks as well as in patients switching treatment from IFX to CT-P13 after 54 weeks, with types, frequencies, and severities of AEs as the primary endpoints.
The observed AEs in this extension study were all known, and the types and frequencies were similar to the results obtained in either the CT-P13 or IFX group in the phase I/II study [Citation1]. AEs leading to study discontinuation, anaphylactic shock, pneumonia, infusion-related reaction, RA, and interstitial lung disease in the phase I/II study were already reported. Overall, CT-P13 was well tolerated in this study. Since hypersensitivity reaction is one of the safety concerns related to IFX biosimilar, the frequency of infusion-related reaction and anaphylactic shock leading to study discontinuation are reported below. In the present study, the frequency of infusion-related reactions and anaphylactic shock leading to study discontinuation were 5.2% (2/38 patients) in the maintenance group and 9.1% (3/33 patients) in the switch group. However, in the phase I/II study, 3.9% (2/51 patients) in the CT-P13-treated group and 3.8% (2/53 patients) in the IFX-treated group were reported. The most frequent AEs in both groups were “Infections and infestations” (MedDRA SOC category). The frequency of infections in the dose-increased patients was higher than the dose-unchanged patients for both the maintenance group and the switch group. A study report indicated that patients who received IFX at an initial dose of 10 mg/kg are at a higher risk of contracting a severe infection compared with those who received IFX at 3 mg/kg [Citation12]. In this trial, the situation was different since all patients received 3 mg/kg for 54 weeks in the phase I/II study, and only after the 54 weeks did some of them received an increased dose. No severe infection and no worsening of infection were observed with the dose increase. No marked differences in the frequency and the severity of SAE, AEs leading to discontinuation of the study, or injection-related reactions were observed between the dose-increased patients and the dose-unchanged patients.
Clinically significant AEs observed in this trial were interstitial lung disease and colon cancer, occurring in one patient in the switch group. Since 25 cases of interstitial lung disease out of 5000 patients were reported in the IFX post-marketing survey [Citation13], careful attention should be paid to this AE during long-term treatment. Although it is not clear whether TNF-α inhibition has any effect on tumor immunity, there are numerous reports indicating that the long-term usage of immunosuppressant might increase the risk of cancer [Citation14–17]. Moreover, as previously stated in this paper, careful observation and appropriate treatment are required because the risk of infectious diseases increases with an increase in the dose of CT-P13.
ACR response rates improved during the course of this extension trial in both the maintenance group and the switch group. DAS28 (ESR) and DAS28 (CRP) improved shortly after the dose increase in patients given an increased dose. In general, other efficacy parameters also showed a stable clinical effect for 72 weeks from the study initiation. The ACR response rates were lower in the ADA-positive patients than in the ADA-negative patients, but still showed a slight improvement during the course of this extension trial.
As for patient characteristics, patient’s assessment of pain (VAS) and HAQ were worse in the switch group than in the maintenance group (). Other parameters on the disease status were also a little worse in the switch group than in the maintenance group. The average dose of CT-P13 at Week 134 (72x) in the maintenance group was 5.72 ± 3.00 mg, which was higher than that of 4.88 ± 2.87 mg in the switch group. The positive ratio of ADA was higher in the switch group (48.5%) than in the maintenance group (31.6%), but we believe this difference occurred by chance because the ratios in the previous studies were 25.5% in the CT-P13 group and 32.1% in the IFX group at Week 54 of the PI/II study [Citation1], and 49.1% in the CT-P13 group and 49.3% in the IFX group at Week 54 of the PLANETRA study [Citation4]. Given that (i) this extension study was not designed to compare the maintenance group and the switch group, (ii) the disease condition, one of the patient characteristics, was slightly different between the maintenance group and the switch group, and (iii) the dose of CT-P13 used differed, ranging from 3 to 10 mg/kg, among patients, it is difficult to compare the results between the two groups in the extension study. Thus, we decided to compare the results of both groups only between the extension study and the phase I/II study.
The phase I/II study used the same design as the PLANETRA study in order to demonstrate in Japanese patients the equivalence of PK between CT-P13 and IFX and to compare the efficacy and the safety of the products with those observed in the PLANETRA study [Citation2]. The dosage of CT-P13 was fixed at 3 mg/kg every 8 weeks until 54 weeks regardless of the disease conditions. According to the dose and administration of IFX approved in Japan, the dose can be increased up to 10 mg/kg after 6 weeks of treatment and the treatment interval can be shortened to 4 weeks when the response is insufficient or declined. Thus, in this extension trial, a dose increase was allowed up to 10 mg/kg after Week 8×. This dose setting might have affected the improvement trend in ACR during the course of this extension trial, resulting in the low percentages of patients achieving ACR20/50/70 (65.6/43.8/21.9%) in the switch group at Week 54 of the phase I/II study.
The dosage of MTX should be unchanged in each patient throughout the study, but the dosages varied among patients within the range of 6–16 mg/week. A dosage decrease was allowed when an AE occurred. Given the correlation between MTX dosages and suppression of RA disease activities, a possibility cannot be ruled out that these differences in MTX dosages among patients may have affected ADA production.
ADA status is important since patients testing positive for ADA may be required to change to another medication. The number of patients who were positive for ADA at the initiation of the extension study was 28/71 (39.4%), whereas the number of patients who were negative at the study initiation but became positive during the course of the study was 2/71 (2.8%). In this trial, infusion-related reactions were observed in two patients of the maintenance group and four patients in the switching group, with all of them testing positive for ADA. This was in line with the report showing that the risk of the infusion-related reaction increased in the patients positive for ADA [Citation18–20]. In the present study, all patients who experienced infusion-related reactions were positive for ADA before the initial dosing in the extension study. Since patients usually become positive for ADA at the early stage of IFX treatment, it is unlikely that patients who were ADA negative over one year would become positive for ADA, regardless of whether they switched to or continued with the CT-P13 treatment.
On the other hand, the percentages of patients testing positive for ADA at Week 110 (48x) in this extension study were 11.8% and 21.7%, respectively, in the maintenance group and the switch group, whereas those percentages at Week 54 in the phase I/II study were 28.9% and 45.5%, respectively, in the maintenance group and the switch group, indicating lower percentages of ADA-positive in this extension study. Five of the 12 positive patients in the maintenance group and 4 of the 16 positive patients in the switch group became negative at Week 86 (24×) of the extension study. The antibody titers of ADA were all low in those patients who became negative. In the PLANETRA extension study, which had a similar design to this trial and in which the measurement method of ADA was also the same, the percentages of patients testing positive for ADA were 50.4% and 46.4% at Week 86 (24×) and 49.6% and 49.6% at Week 110 (48×), respectively, in the maintenance group and the switch group, showing no marked decrease in the percentage of patients testing positive [Citation4]. However, we should take note of the fact that dose increases of CT-P13 were allowed in this extension study but not in the extension study of PLANETRA. Although the factors which affect the production of ADA were not yet clearly identified, a low-level trough value of IFX serum concentration at the study initiation or at the drug withdrawal has been reported to increase the risk of becoming positive for ADA [Citation21]. Another report stated that the risk of an early production of ADA became lower with a dose increase at an early stage or with a shortened dosing interval [Citation18]. No trough level was measured in this trial; however, we believe that the increased dose of CT-P13 might have decreased the level of disease activity and partly contributed to becoming ADA-negative, considering the fact that five of the eight dose-increased patients in the maintenance group and two of the four dose-increased patients in the switch group became negative at Week 110 (48×).
Since biosimilars are generally approximately 30% less expensive than reference products at launch in Japan, CT-P13 may be a cost-effective alternative to IFX. According to a recent budget impact analysis, the potential effects of introducing CT-P13 for the treatment of RA in European countries estimated significant savings [Citation22].
The major limitation of the present analysis is the study sample size, which was set to analyze the primary endpoint for phase I/II study. The extension study findings were not considered when calculating the sample size.
In summary, CT-P13 was generally well tolerated with persistent efficacy for RA during the long-term treatment over 54 weeks as well as after the switch to CT-P13 from IFX treatment after 54 weeks. AEs observed in the long-term treatment included malignant tumor, infusion-related reactions in ADA-positive patients and infectious diseases in dose-increased patients.
Conflict of interest
Y.T. has received grants, personal fees, and non-financial support from Nippon Kayaku Co., Ltd. during this study, personal fees from Nippon Kayaku Co., Ltd., and grants from Mitsubishi Tanabe Pharma Corp., Takeda Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Astellas Pharma Inc., Eisai Co., Ltd., Taisho Toyama Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., AbbVie GK, and Bristol-Myers K.K. outside the submitted work.
H.Y. has received grants and personal fees from Nippon Kayaku Co., Ltd. during this study, and personal fees from Nippon Kayaku Co., Ltd. outside the submitted work.
T.T. has received grants, personal fees, and non-financial support from Nippon Kayaku Co., Ltd., AbbVie GK, Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd. and Mitsubishi Tanabe Pharma Corp., grants and non-financial support from Daiichi Sankyo Co., Ltd., personal fees from Pfizer Japan Inc., grants from Takeda Pharmaceutical Co., Ltd. during this study; grants, personal fees, and non-financial support from Astellas Pharma Inc., and grants from Santen Pharmaceutical Co., Ltd., Teijin Pharma Ltd., Asahi Kasei Pharma Corp., Taisho Toyama Pharmaceutical Co., Ltd. and SymBio Pharmaceuticals Ltd. outside the submitted work.
M.I. has received grants and non-financial support from Nippon Kayaku Co., Ltd. during the conduct of the study; and personal fees from Nippon Kayaku Co., Ltd. outside the submitted work.
K.S. has received grants and non-financial support from Nippon Kayaku Co., Ltd. during this study.
Y.S. has received grants and non-financial support from Nippon Kayaku Co., Ltd. during this study.
S.J.L. is a full-time employee of Celltrion, Inc. and has received personal fees and non-financial support from Celltrion, Inc. during this study, as well as personal fees and non-financial support from Celltrion, Inc. outside the submitted work.
Y.N. is a board member with stock ownership of Nippon Kayaku Co., Ltd., and has received personal fees and non-financial support from Nippon Kayaku Co., Ltd. during this study, as well as personal fees and non-financial support from Nippon Kayaku Co., Ltd. outside the submitted work.
Supplementary material available online
Supplementary Appendix
Download MS Word (27.9 KB)Acknowledgments
The studies described herein were funded by Celltrion, Inc. and Nippon Kayaku Co., Ltd. The authors acknowledge all investigators at the following hospitals for their contribution to the study: Sapporo City General Hospital; Hokkaido Medical Center for Rheumatic Diseases; Inoue Hospital; Saitama Medical Centre, Saitama Medical University; School of Medicine, Keio University; Tokyo Medical and Dental University; Institute of Rheumatology, Tokyo Women’s Medical University; Niigata Rheumatic Center; Shizuoka Kousei Hospital; Aichi Medical University School of Medicine; Kyoto University Graduate School of Medicine; National Hospital Organization Osaka-Minami Medical Center; Matsubara Mayflower Hospital; Higashihiroshima Memorial Hospital; University of Occupational and Environmental Health; Kyushu University Graduate School of Medical Sciences; Nagasaki University Graduate School of Biomedical Sciences; Sasebo Chuo Hospital; Shimin-No-Mori Hospital; Kagoshima Red Cross Hospital. All costs associated with development of this manuscript were funded by Nippon Kayaku Co., Ltd.
References
- Takeuchi T, Yamanaka H, Tanaka Y, Sakurai T, Saito K, Ohtsubo H, et al. Evaluation of the pharmacokinetic equivalence and 54-week efficacy and safety of CT-P13 and innovator infliximab in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2015;25(6):817–24.
- Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613–20.
- Committee on Rheumatologic Care. American College of Rheumatology Position Statement. February 2015.
- Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre EB, Lanzon A, et al. Efficacy and safety of CT-P13 (infliximab biosimilar) over two years in patients with rheumatoid arthritis: comparison between continued CT-P13 and switching from infliximab to CT-P13 [abstract]. Arthritis Rheumatol. 2013;65(12):3319.
- Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38:727–35.
- Felson DT, Anderson JJ, Lange MLM, Wells G, and Lavalley MP. Should improvement in rheumatoid arthritis clinical trials be defined as fifty percent or seventy percent improvement in core set measures, rather than twenty percent? Arthritis Rheum. 1998;41:1564–70.
- Prevoo MLL, van’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, and van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:44–8.
- van Riel PLCM, van Gestel AM, and van de Putte LBA. Development and validation of response criteria in rheumatoid arthritis: steps towards an international consensus on prognostic markers. Br J Rheumatol. 1996;35(Suppl 2):4–7.
- Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, et al. A simplified disease activity index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford). 2003;42:244–57.
- Aletaha D, Nell VPK, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther. 2005;7:R796–806.
- Fries JF, Spitz P, Kraines RG, and Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23:137–45.
- Westhovens R, Yocum D, Han J, Berman A, Strusberg I, Geusens P, et al. The safety of infliximab, combined with background treatments, among patients with rheumatoid arthritis and various comorbidities: a large, randomized, placebo-controlled trial. Arthritis Rheum. 2006;54(4):1075–86.
- Takeuchi T, Tatsuki Y, Nogami Y, Ishiguro N, Tanaka Y, Yamanaka H, et al. Postmarketing surveillance of the safety profile of infliximab in 5000 Japanese patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67:189–94.
- Penn I. Malignancies associated with immunosuppressive or cytotoxic therapy. Surgery. 1978;83(5):492–502.
- Penn I. Cancer following cyclosporine therapy. Transplant Proc. 1987;19(1):2211–13.
- Kinlen L. Immunosuppressive therapy and acquired immunological disorders. Cancer Res. 1992;52(19 Suppl):5474s–6s.
- Jones M, Symmons D, Finn J, and Wolfe F. Does exposure to immunosuppressive therapy increase the 10 year malignancy and mortality risks in rheumatoid arthritis? A matched cohort study. Br J Rheumatol. 1996;35(8):738–45.
- Bendtzen K, Geborek P, Svenson M, Larsson L, Kapetanovic MC, and Saxne T. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 2006;54:3782–9.
- Wolbink GJ, Vis M, Lems W, Voskuyl AE, de Groot E, Nurmohamed MT, et al. Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:711–15.
- Krintel SB, Grunert VP, Hetland ML, Johansen JS, Rothfuss M, Palermo G, et al. The frequency of anti-infliximab antibodies in patients with rheumatoid arthritis treated in routine care and the associations with adverse drug reactions and treatment failure. Rheumatology. 2013;53:299–307.
- Ducourau E, Mulleman D, Paintaud G, Miow Lin DC, Lauféron F, Ternant D, et al. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. 2011;13:R105.
- Brodszky V, Baji P, Balogh O, and Péntek M. Budget impact analysis of biosimilar infliximab (CT-P13) for the treatment of rheumatoid arthritis in six Central and Eastern European countries. Eur J Health Econ. 2014;15(Suppl 1):S65–71.