
Abstract
Objectives: The objective of this study is to evaluate the efficacy and safety of long-term (64 weeks; 52-week extension of a 12-week study) baricitinib treatment in Japanese patients with active rheumatoid arthritis (RA) despite methotrexate therapy.
Methods: Patients (N = 145) with active RA were randomized to placebo, 1mg, 2mg, 4mg, or 8mg baricitinib for the first 12 weeks. During the 52-week extension period, patients on 4mg or 8mg baricitinib remained on the same dose and all other patients were re-randomized to 4mg or 8mg baricitinib. Most patients on 8mg baricitinib were switched to 4mg by week 64 (protocol amendment); data analysis was based on the treatment group at the beginning of the extension period.
Results: Increases in the American College of Rheumatology (ACR) response rates (ACR20, ACR50, and ACR70) observed during the first 12 weeks were maintained during the extension period, accompanied by improvements in ACR core components. At week 64, a large proportion of patients (>40%) had low disease activity. Most treatment-related adverse events were mild or moderate; herpes zoster was the most common reason (11/27 patients) for discontinuation.
Conclusions: The efficacy and safety profile of baricitinib was maintained during long-term treatment of Japanese patients with RA and background methotrexate therapy.
Clinicaltrials.gov NCT01469013; Funding: Eli Lilly and Incyte
Introduction
Inhibition of the intracellular Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway is an effective strategy for the treatment of rheumatoid arthritis (RA) [Citation1–3]. Baricitinib is an oral, reversible inhibitor of the JAK family of protein tyrosine kinases [Citation4–6] that is a promising treatment for RA.
In recent studies, baricitinib was effective at reducing the signs and symptoms of RA and had an acceptable safety profile [Citation7–10]. In a multinational Phase 2b study with patients who had active RA despite methotrexate (MTX) therapy, a greater proportion of patients receiving baricitinib achieved an American College of Rheumatology 20% (ACR20) response after 12 weeks, compared with placebo [Citation9]. Importantly, the clinical efficacy observed at weeks 12 and 24 of this study, including ACR responses and disease activity, was maintained or improved in patients during the extension period of the study for up to 128 weeks, with acceptable safety and tolerability [Citation11,Citation12].
Given the prevalence of RA in Asian countries [Citation13], we believe that it is important to evaluate the efficacy and safety of baricitinib specifically in Asian populations. Recently, Tanaka et al. [Citation5] reported the results of a 12-week, double-blind, placebo-controlled study investigating the efficacy of baricitinib in Japanese patients with active RA who had an inadequate response to MTX therapy. In this study, significantly more patients treated with baricitinib (4 mg and 8 mg groups combined) were able to achieve an ACR20 response compared with placebo (77% versus 31%, p < .001). In addition, baricitinib was generally well tolerated and the safety profile was consistent with studies in non-Japanese patients. Here, we report the results of the extension study of Tanaka et al. For the extension study, patients receiving placebo, 1 mg, or 2 mg baricitinib in the first 12 weeks were re-randomized to receive 4 mg or 8 mg baricitinib, and patients who received 4 mg or 8 mg baricitinib remained on the same dose. During the course of the extension study, a protocol amendment was implemented and patients receiving 8 mg were switched to 4 mg until study completion. The objective of this extension study was to evaluate the efficacy and safety of long-term (64 weeks) baricitinib treatment in Japanese patients with active RA despite MTX therapy.
Materials and methods
Study design
This was a Phase 2b, randomized, placebo-controlled study (clinicaltrials.gov NCT01469013), with a 12-week, double-blind treatment period followed by a 52-week, single-blind extension period (). The results from the 12-week treatment period have been reported by Tanaka et al. [Citation5] and the results of the extension period are presented here. The study was conducted at 24 sites in Japan from November 2011 to December 2013, and was approved by the institutional review board or ethics committee at each site. The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and all applicable laws and regulations in Japan. All participants provided written, informed consent before any study-related procedures.
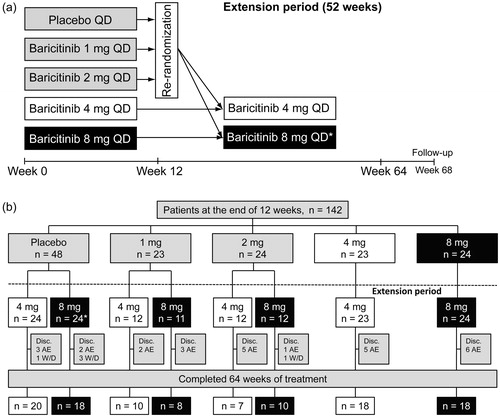
Figure 1. (a) Study design for the 64-week study of baricitinib in Japanese patients with active rheumatoid arthritis. Patients who received placebo, or 1 mg or 2 mg of baricitinib during the first 12 weeks of the study were re-randomized to receive either 4 mg or 8 mg of baricitinib during the extension period. * Patients assigned to 8 mg baricitinib at the beginning of the extension period were switched to 4 mg baricitinib by the end of the study, as per protocol amendment. QD: once daily. (b) Patient flow diagram for the 52-week extension period of the study. * One patient from the placebo group was re-randomized to the 8 mg baricitinib group, but due to an adverse event, this patient was withdrawn from the extension study before receiving study medication. AE: adverse event; Disc.: discontinuations; W/D: withdrawal.
Study population
The main inclusion criteria were as follows: diagnosis of adult-onset RA, according to the 2010 ACR/European League Against Rheumatism (EULAR) classification criteria [Citation14]; presence of active RA, defined as at least six swollen and at least six tender joints based on the 66/68 joint count, and a C-reactive protein (CRP) measurement >0.5 mg/dL or an erythrocyte sedimentation rate (ESR) > 28 mm/hr; and use of methotrexate for ≥12 weeks with a stable dose (6–16 mg/week) established for ≥8 weeks prior to randomization. The main exclusion criteria were prior use of conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs) other than MTX and/or sulfasalazine in the 8 weeks before randomization and significant hematological or chemical abnormalities identified during screening (see Tanaka et al. for details [Citation5]).
Treatment protocol
Patients were randomized 2:1:1:1:1 to receive placebo or 1 mg, 2 mg, 4 mg, or 8 mg once daily (QD) oral baricitinib during the first 12 weeks of the study (see Tanaka et al. for details [Citation5]). After completion of the double-blind treatment period at week 12, patients were eligible to participate in the 52-week, investigator- and patient-blinded extension period. In this extension period, patients receiving placebo, 1 mg, or 2 mg baricitinib during the first 12 weeks of the study were re-randomized to receive either 4 mg or 8 mg oral baricitinib QD and patients already receiving 4 mg or 8 mg baricitinib remained on the same dose (). Based on the evaluation of data across baricitinib Phase 2 RA studies, which indicated that maximal efficacy could be achieved with 4 mg QD [Citation9], a protocol amendment was implemented during the extension period and the 8 mg dose of baricitinib was removed from evaluation. Following this amendment, patients on 8 mg baricitinib were switched to 4 mg baricitinib for the remainder of the study, to week 64 or early discontinuation. Investigators and patients remained blinded to the dose at week 12 and the date on which the dose was switched to 4 mg. Although patients who received 8 mg at the beginning of the extension period were eventually switched to 4 mg, the data were analyzed and reported based on the patients’ treatment groups at re-randomization for the extension period. In the 8 mg group, adverse events that were reported after patients were switched to 4 mg were still included as part of the 8 mg group during safety analyses. In the extension period, efficacy and safety assessments were conducted at weeks 14, 16, 28, 40, 52, and 64. Safety assessments were also conducted at a follow-up visit, 4 weeks after completion of the 64-week treatment period (week 68), or 4 weeks after early discontinuation. The follow-up safety data comprised data from weeks 64 to 68 as well as data following early discontinuation.
Efficacy measures
Relief of signs and symptoms of RA was assessed using the ACR20 responder index, where the ACR20 response is defined as an improvement of ≥20% from baseline in the number of tender and swollen joints as well as an improvement in at least three of the following five measures: Physician’s Global Assessment of Disease Activity; Patient’s Global Assessment of Disease Activity; patient’s assessment of arthritis pain; patient’s assessment of physical function, reported using the Health Assessment Questionnaire – Disability Index (HAQ-DI); and high-sensitivity CRP (hsCRP). Other efficacy outcomes included the following: ACR50 and ACR70 responses; individual ACR core components (listed above); 28-joint Disease Activity Score (DAS28), based on the level of hsCRP (DAS28-CRP) or on the ESR (DAS28-ESR); and simplified disease activity index (SDAI). Low disease activity was defined as DAS28-CRP or DAS28-ESR <3.2 and SDAI ≤11.0. Remission was defined as DAS28-CRP or DAS28-ESR <2.6 and SDAI ≤3.3. Remission in terms of disease-associated disability was defined as HAQ-DI ≤0.5. Details of the efficacy measurements are provided in Tanaka et al. [Citation5]. Efficacy measures were assessed based on the proportion of patients who met the pre-defined criteria for response or score, or by the change over time.
Safety measures
Treatment-emergent adverse events (TEAEs) were coded and summarized using the Medical Dictionary for Regulatory Activities (Version 16.1) [Citation15]. Laboratory measures assessed included levels of hemoglobin, neutrophils, lymphocytes, creatinine, aspartate aminotransferase (AST), alanine transaminase (ALT), high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, and creatine phosphokinase (CPK). Post-baseline changes in laboratory measures were described using the National Institute of Health Common Terminology Criteria for Adverse Events (CTCAE; V4.0) [Citation16].
Statistical analysis
Calculation of sample size for the study is detailed in Tanaka et al. [Citation5]. Efficacy and safety analyses were conducted using the full analysis set, defined as all patients who received any amount of baricitinib during the extension period. Analyses were performed using SAS® Version 9.2 (SAS Institute Inc, Cary, NC).
Results
Patient disposition and characteristics
A total of 142 patients completed the first 12 weeks of the study and were re-randomized to receive either 4 mg or 8 mg of baricitinib in the extension period (). Of these, 109 patients completed both the initial study and the extension period. At the beginning of the extension period, there were 71 patients in the 4 mg group and 71 patients in the 8 mg group. However, one patient from the placebo group who was re-randomized to the 8 mg baricitinib group withdrew from the extension period due to an adverse event before receiving study medication. Most patients were switched to 4 mg by the end of the extension period (Supplementary Table 1). Demographic and clinical characteristics for the study population at baseline are presented in Tanaka et al. [Citation5], according to treatment received during the initial 12-week treatment period, and in , according to treatment received at the beginning of the extension period. Demographic and clinical characteristics were generally well matched between the two treatment groups. Most of the patients who were enrolled in the study were women with a mean age greater than 50 years and a mean duration of RA greater than 5 years ().
Table 1. Baseline (week 0) demographics and patient characteristics.
Efficacy
ACR response rate
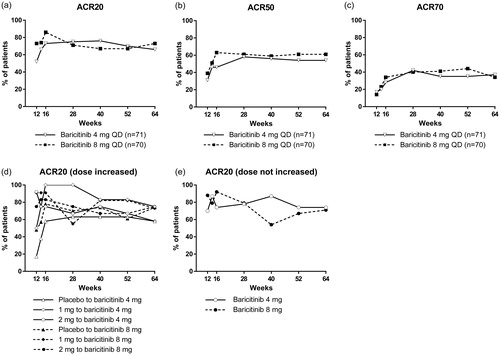
The percentage of patients with an ACR20 response increased between baseline and week 12 for all baricitinib treatment groups [Citation5]; overall, there was a further increase in both the 4 mg and 8 mg groups during the extension period (). This increase can be attributed mainly to patients who were prescribed placebo during the first 12 weeks; following the increase in baricitinib dose (to 4 or 8 mg) during the extension period, there was an increase in the percentage of patients in these groups who achieved an ACR20 response (). For the groups treated with 2 mg, 4 mg or 8 mg of baricitinib during the first 12 weeks, the percentage of patients achieving an ACR20 response during the extension period did not increase by the same magnitude as for the groups initially prescribed placebo (). From week 14 to week 64, each treatment group had a similar percentage of patients with an ACR20 response (at least 65%, ). The percentage of patients with an ACR50 or ACR70 response also increased between baseline and week 12 for all baricitinib treatment groups [Citation5]; this increase was maintained, and improved, in both the 4 mg and 8 mg groups during the extension period (). From week 28, both treatment groups had a similar percentage of patients with ACR50 (approximately 60%) and ACR70 (approximately 40%) responses.
Figure 2. Change in ACR20 (a), ACR50 (b), and ACR70 (c) response rates during the 52-week extension period of the study. ACR20 response rate in groups stratified according to dosage received during the first 12 weeks; shown are patients who received a higher dose of baricitinib during the extension period (d) and patients whose dosage of baricitinib was not increased upon entering the extension period (e). For patients who received at least one dose of study drug during the extension period and discontinued before week 64, the non-responder imputation method was used for analysis. ACR: American College of Rheumatology; QD: once daily.
ACR core components
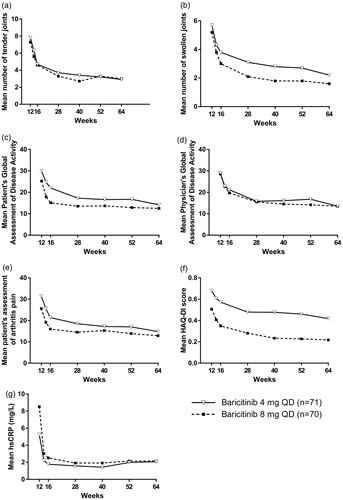
All ACR core components were improved by baricitinib between weeks 12 and 64 of the study (). All core components, including Physician’s Global Assessment of Disease Activity, Patient’s Global Assessment of Disease Activity, patient’s assessment of arthritis pain, HAQ-DI, and hsCRP, improved between weeks 12 and 64, and generally the lowest values during this period were observed at week 64 ().
Figure 3. Change in ACR core components during the 52-week extension period of the study. Changes in number of tender joints (a), number of swollen joints (b), Patient’s Global Assessment of Disease Activity (c), Physician’s Global Assessment of Disease Activity (d), patient’s assessment of arthritis pain (e), HAQ-DI score (f), and hsCRP (g). ACR: American College of Rheumatology; HAQ-DI: Health Assessment Questionnaire-Disability Index; hsCRP: high-sensitivity C-reactive protein.
Disease activity and disease-associated disability
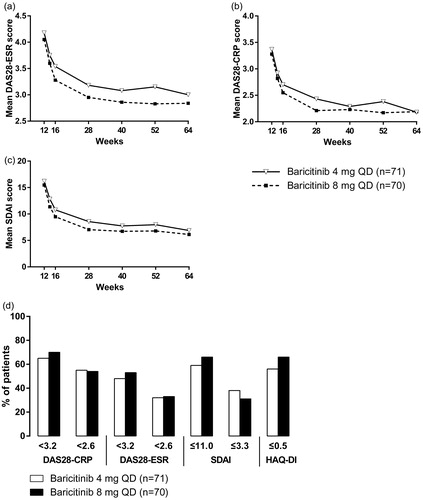
During the extension period, reductions were observed in several measures of disease activity and disease-associated disability (). A large proportion of patients (48–70%) treated with baricitinib were classified as being in a state of low disease activity, as defined by numerous composite measures (). The percentages of patients classified as being in a state of low disease activity or remission were similar in both treatment groups at week 64 for DAS, reported as DAS28-CRP and DAS28-ESR, and for SDAI (). The percentage of patients classified as having remission in terms of disease-associated disability, as measured by HAQ-DI, was also relatively high and similar in both treatment groups ().
Figure 4. Measures of disease activity and disease-associated disability. Changes in DAS28-ESR (a), DAS28-CRP (b), and SDAI (c) during the 52-week extension period of the study. Percentage of patients with disease activity and disease-associated disability scores associated with low disease activity or remission at week 64 of the study (d). For patients who received at least one dose of study drug during the extension period and discontinued before week 64, the non-responder imputation method was used for analysis at week 64. CRP: C-reactive protein; DAS28: disease activity score at 28 joints; ESR: erythrocyte sedimentation rate; HAQ-DI: Health Assessment Questionnaire – Disability Index; QD: once daily; SDAI: simplified disease activity index.
Safety and tolerability
Adverse events
Baricitinib had an acceptable safety profile in the extension period of the study. A total of 65 (92%) patients from the 4 mg group and 69 (99%) patients from the 8 mg group reported at least one TEAE, with the majority of TEAEs being mild or moderate in severity (). The most commonly reported TEAEs were nasopharyngitis, increases in blood creatine phosphokinase, hyperlipidemia, herpes zoster, and hypercholesterolemia. Most incidences of herpes zoster were mild or moderate in severity. Malignancies were reported in two patients; these were one case of rectal cancer and one case of chondrosarcoma (diagnosed after completion of the study). There were no deaths reported throughout the study.
Table 2. Summary of adverse events from week 12 to week 64.
Serious adverse events (SAEs) were reported by 11% of patients in the 4 mg group and 17% of patients in the 8 mg group (), with some patients experiencing more than one event (Supplementary Table 2). Similar numbers of patients in both the 4 mg and 8 mg groups were discontinued from the extension period due to an adverse event (AE; ). Patients who developed herpes zoster were discontinued from the study (as specified by the protocol) and this was the most common reason for discontinuation from the extension period (11 of 27 patients; Supplementary Table 3). One patient in the 8 mg group who was discontinued due to herpes zoster was found to have a Grade 1 chondrosarcoma, which was surgically removed and showed no evidence of metastatic disease. Three herpes zoster events were reported as SAEs, including one with a rash distributed beyond the primary and adjacent dermatomes (8 mg group), and one complicated by facial nerve palsy (8 mg group). The two patients who were discontinued due to Pneumocystis pneumonia had modest elevations in serum β-d-glucan and abnormalities on chest computed tomography imaging. Both were diagnosed at the same study site, had lymphopenia at baseline, and recovered following treatment. One patient reported no symptoms during the event and neither patient had bronchoscopy to determine the presence of Pneumocystis organisms; one had sputum PCR positive for Pneumocystis pneumonia at the study site that was not confirmed by hospital testing. A total of 48 patients experienced at least one AE that led to study drug interruption, comprising 18 patients from the 4 mg group and 30 patients from the 8 mg group (). The most common AEs (based on System Organ Class and Preferred Term) resulting in study drug interruptions were nasopharyngitis and lymphopenia. A total of seven patients (three from the 4 mg group and four from the 8 mg group) had lymphopenia that resulted in study drug interruption.
Laboratory measures
The changes in laboratory measures in the extension period of the study were generally consistent with those at week 12 reported previously by Tanaka et al. [Citation5]. Neutrophil and lymphocyte counts had plateaued below baseline by week 64, whereas platelet count, AST, ALT, cholesterol, creatinine, and CPK levels had plateaued above baseline (). Hemoglobin levels were stable in both treatment groups.
Table 3. Mean change in laboratory measures from baseline (week 0) to weeks 24, 44, and 64, and follow-up.
Most mean changes in laboratory values observed at week 64 were lessening at follow-up (), which for most patients, was at week 68; for patients who were discontinued from the study early, a follow-up visit was completed 4 weeks after the last dose of baricitinib. Platelet count and HDL cholesterol levels plateaued above baseline by week 64, but by follow-up, these measures had dropped below baseline levels ().
Changes in laboratory measures during the extension period were also assessed by shifts in CTCAE grade (). The grades of anemia were mostly stable, except one patient in the 4 mg group experienced Grade 3 anemia during the extension period of the study (no anemia at baseline). The same patient was discontinued from the study 103 days after the first dose due to two episodes of Grade 3 lymphopenia.
Table 4. Shift table for laboratory values for maximum CTCAE (V4.0) grade at any time during weeks 12–64.
Of 22 patients who experienced Grade 3 lymphopenia, most (17 of 22) had lymphopenia at baseline. Most cases were transient (only five of 22 episodes were observed at the final study visit, including follow-up). Nine of the 22 patients (four in the 4 mg group and five in the 8 mg group; eight of nine with lymphopenia at baseline) discontinued early from the study. Four patients discontinued due to a second episode of Grade 3 lymphopenia (as per the protocol), after restarting baricitinib following an interruption of study drug. The remaining five patients discontinued due to AEs; one had interstitial lung disease (8 mg group), two had Pneumocystis pneumonia (one in the 4 mg group and one in the 8 mg group), and two had herpes zoster (both in the 8 mg group; discontinuation required as per protocol; lymphopenia occurred after the onset of zoster).
Increases in AST or ALT observed during the extension period were predominantly from no grade to Grade 1, and none reached Grade 3 or higher ().
Discussion
For Japanese patients with active RA despite MTX therapy, long-term (at least 52 weeks) baricitinib treatment effectively reduced the signs and symptoms of RA. At the beginning of the extension period (Week 12), the percentage of patients with ACR20/50/70 responses in each treatment group was variable; by week 12, a substantial percentage of patients treated with baricitinib had already achieved ACR20/50/70 responses, whereas the corresponding percentage was much lower for patients treated with placebo [Citation5]. At the end of the extension period (week 64), more than half of the patients had achieved an ACR20 response, irrespective of their treatment group in the first 12 weeks. These results suggest that during the extension period, for patients previously treated with placebo, baricitinib was able to reduce the signs and symptoms of RA; in patients previously treated with baricitinib, improvements achieved during the first 12 weeks were maintained throughout the extension period. Further, baricitinib was well tolerated during the extension period and there were no major differences in safety between doses. These results highlight the potential of baricitinib as a long-term treatment for Japanese patients with RA.
The efficacy findings of this extension study were similar to the initial short-term treatment [Citation5] and to long-term treatment in a multinational population [Citation9]. In our extension study, all improvements in RA observed following the initial 12 weeks of active treatment were maintained (or further improved) during the next 52-week period (). This pattern is similar to the results at weeks 24, 76, and 128 from a trial of baricitinib in a multinational population with active RA despite MTX therapy [Citation11]. In addition, maintenance of efficacy with baricitinib up to 24 weeks has also been observed in patients with RA and an inadequate response to biologic DMARDs [Citation8] or conventional synthetic DMARDs [Citation7], and up to 52 weeks in DMARD-naïve patients [Citation10]. Consistent with the findings of the present study, sustained efficacy has been described in Japanese patients with active RA and background MTX therapy after a median of 1185 d of treatment with the pan-JAK inhibitor tofacitinib [Citation17].
The safety findings of this extension period were generally consistent with the initial 12-week treatment period [Citation5], and with other international baricitinib studies with short-term and long-term treatment [Citation8,Citation9,Citation11]. Overall, baricitinib was well tolerated, and the nature of TEAEs and the incidence and nature of SAEs were similar between the 4 mg and 8 mg groups. Although there were no incidences of herpes zoster during the initial 12-week treatment period [Citation5], herpes zoster was reported for 11 (7.8%) patients in the extension period and was the most common reason for discontinuation from the study (as required by protocol for herpes zoster events). In the longer study of Japanese patients with RA treated with tofacitinib described above, the incidence of herpes zoster was 19.3% [Citation17]. Both tofacitinib and baricitinib have been associated with an increased risk of herpes zoster, with the highest risk being observed in Japanese patients [Citation18,Citation19]; the incidence rate (per 100 patient-years) of herpes zoster in patients from Japan and South Korea was 9.2 for tofacitinib and 6.5 for baricitinib (Japan only). Further studies are required to identify the factors responsible for the increased risk of herpes zoster in Japanese patients. The American College of Rheumatology guidelines for the treatment of RA recommend the use of herpes zoster vaccines prior to initiation of DMARD therapy [Citation20]. Non-live herpes zoster vaccines are currently in development and if approved, may constitute an advance in the treatment of RA. In our study, there was no difference in the incidence of herpes zoster infection between the 4 mg group (five patients) and the 8 mg group (six patients). None of these patients had a documented history of herpes zoster and none had ever been vaccinated against herpes zoster. Two patients were discontinued from the extension study due to diagnoses of Pneumocystis jirovecii pneumonia; one case was asymptomatic with no microbiological evidence for the organism’s presence, and neither case was confirmed by invasive methods such as bronchoscopy.
Shifts of CTCAE grade were noted for a number of laboratory parameters, including hemoglobin, lymphocyte counts, AST, and ALT. During the first 12 weeks of the study, a higher proportion of patients receiving 8 mg of baricitinib had post-baseline decreases in hemoglobin (based on CTCAE grade) compared with patients receiving placebo, 1 mg, 2 mg, or 4 mg of baricitinib [Citation5]. Hemoglobin remained stable during the extension period in both treatment groups. The reduction in neutrophils observed in the extension period was also observed in the first 12 weeks of the study [Citation5] as well as in other studies of baricitinib [Citation8,Citation9], with similar shifts in CTCAE grade observed, and prior data indicate that transient margination of neutrophils contributed to this observation [Citation21]. Declines in lymphocyte count were observed in some patients during the extension period. These were predominantly seen in patients with lymphopenia at baseline.
The strengths of this study include the long-term administration of baricitinib for 64 weeks, the high completion rate, and that both investigators and patients were blinded even during the extension period. The study was limited by the relatively small sample size and the lack of controls during the extension period. In addition, because of a protocol amendment, most patients in the 8 mg baricitinib group were switched to 4 mg baricitinib during the extension period, which limits interpretation of dose responses.
In conclusion, long-term, 64-week baricitinib treatment was effective and well tolerated in Japanese patients with RA and background MTX therapy. The efficacy and safety profile of baricitinib was maintained throughout the 52-week extension period and was consistent with the short-term results of this study and with other studies of baricitinib in non-Asian populations. Given the prevalence of RA in Asia, the results from this study highlight the potential of baricitinib as a long-term treatment for RA in this population.
Conflicts of interest
T. I., Z. C., D. S., T. R., and W. M. are employees of and/or own stock in Eli Lilly and Company or Eli Lilly Japan K.K. Y. T. has received consulting fees, speaking fees, and/or honoraria from AbbVie, Daiichi-Sankyo, Chugai, Takeda, Mitsubishi-Tanabe, Bristol-Myers Squibb, Astellas, Eisai, Janssen, Pfizer, Asahi-kasei, Eli Lilly, GlaxoSmithKline, UCB, Teijin, MSD and Santen, and has received research grants from Mitsubishi-Tanabe, Takeda, Chugai, Astellas, Eisai, Taisho-Toyama, Kyowa-Kirin, AbbVie, and Bristol-Myers Squibb.
JADN_baricitinib_extension_study_Supplemental_Material.docx
Download MS Word (39.3 KB)Additional information
Funding
Related Research Data
References
- Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–60.
- Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377–86.
- Tanaka Y. Recent progress and perspective in JAK inhibitors for rheumatoid arthritis: from bench to bedside. J Biochem. 2015;158:173–9.
- Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–307.
- Tanaka Y, Emoto K, Cai Z, Aoki T, Schlichting D, Rooney T, et al. Efficacy and safety of baricitinib in Japanese patients with active rheumatoid arthritis receiving background methotrexate therapy: a 12-week, double-blind, randomized placebo-controlled study. J Rheumatol. 2016;43:504–11.
- Kubo S, Nakayamada S, Tanaka Y. Baricitinib for the treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2016;12:911–19.
- Dougados M, van der Heijde D, Chen Y-C, Greenwald M, Drescher E, Liu J, et al. Baricitinib, an oral Janus kinase (JAK)1/JAK2 inhibitor, in patients with active rheumatoid arthritis (RA) and an inadequate response to CDMARD therapy: results of the phase 3 RA-Build study. Ann Rheum Dis. 2015;74:79.
- Genovese MC, Kremer J, Zamani O, Ludivico C, Krogulec M, Xie L, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243–52.
- Keystone EC, Taylor PC, Drescher E, Schlichting DE, Beattie SD, Berclaz PY, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rheum Dis. 2015;74:333–40.
- Fleischmann R, Schiff M, van der Heijde D, Ramos-Remus C, Spindler A, Stanislav M, et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Rheumatol. 2017;69:506–17.
- Keystone EC, Taylor PC, Genovese MC, Schlichting D, De La Torre I, Beattie SD, et al. Safety and efficacy of baricitinib through 128 weeks in an open-label, long-term extension study in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;64:S1049–S50.
- Taylor P, Genovese M, Keystone E, Schlichting D, Beattie S, Macias W. Baricitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: safety and efficacy in an open-label, long-term extension study. Ann Rheum Dis. 2014;73:A31.
- Alamanos Y, Drosos AA. Epidemiology of adult rheumatoid arthritis. Autoimmun Rev. 2005;4:130–6.
- Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81.
- Medical Dictionary for Regulatory Activities. Available from: http://www.meddra.org/.
- US Department of Health and Human Services NIoH, National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. 2009.
- Yamanaka H, Tanaka Y, Takeuchi T, Sugiyama N, Yuasa H, Toyoizumi S, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open-label, long-term extension study. Arthritis Res Ther. 2016;18:34.
- Winthrop KL, Yamanaka H, Valdez H, Mortensen E, Chew R, Krishnaswami S, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66:2675–84.
- Winthrop KL, Lindsey S, Weinblatt M, Takeuchi T, Hyslop D, Issa M, et al. Herpes Zoster in patients with moderate to severe rheumatoid arthritis treated with baricitinib. Arthritis Rheumatol. 2016;68(Suppl10).
- Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2016;68:1–25.
- Shi JG, Chen X, Lee F, Emm T, Scherle PA, Lo Y, et al. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol. 2014;54:1354–61.