
1. Introduction
The clinical complications of sickle cell anemia (HbSS or HbSβ⁰thalassemia, SCA) result from a cascade of events that starts with sickle hemoglobin (HbS) polymerization (), used with permission from original article[Citation1]). Supportive therapies like antibiotic prophylaxis improve survival in children through prevention of death from overwhelming infection but have not increased overall life expectancy [Citation2,Citation3]. Available SCA-modifying therapies prior to 2017 decreased HbS concentration by either increasing fetal hemoglobin (HbF) levels (hydroxyurea, HU) or increasing hemoglobin A (HbA) levels (transfusion). Since 2017, however, the FDA has approved three additional SCA treatments. L-glutamine, voxelotor, and crizanlizumab interrupt SCA pathophysiology at different stages along the biochemical cascade ()). Curative options like hematopoietic stem cell transplant (HSCT) and gene therapy strive to eliminate the production of HbS, but neither are available to the majority of patients with SCA because of limited donors and clinical trial enrollment restrictions, respectively.
Figure 1. (a) Sickle Cell Pathophysiology. The pathophysiology of sickle cell anemia is complex and stems from the polymerization of HbS that occurs during periods of hypoxemia, dehydration, acidosis, and pyrexia. Polymers of sickle hemoglobin cause the characteristic shape change of the erythrocyte and lead to hemolysis, abnormal rheology, cellular adhesion and decreased nitric oxide availability. These changes result in anemia, vaso-occlusion and vasoconstriction that are the cause of SCD-associated end organ damage. (b) Sickle Cell Therapies Block Sickling Cascade. Voxelotor binds to the alpha subunit of the hemoglobin molecule and prevents circulatory desaturation which ultimately inhibits HbS polymerization. Hydroxyurea increases fetal hemoglobin levels which prevent HbS polyermization. Crizanlizumab block P-selectin and interrupts cellular adhesion. Mechanism for L-glutamine has not been fully elucidated to date. Adapted in part from [Citation1] with permission of Springer Nature
![Figure 1. (a) Sickle Cell Pathophysiology. The pathophysiology of sickle cell anemia is complex and stems from the polymerization of HbS that occurs during periods of hypoxemia, dehydration, acidosis, and pyrexia. Polymers of sickle hemoglobin cause the characteristic shape change of the erythrocyte and lead to hemolysis, abnormal rheology, cellular adhesion and decreased nitric oxide availability. These changes result in anemia, vaso-occlusion and vasoconstriction that are the cause of SCD-associated end organ damage. (b) Sickle Cell Therapies Block Sickling Cascade. Voxelotor binds to the alpha subunit of the hemoglobin molecule and prevents circulatory desaturation which ultimately inhibits HbS polymerization. Hydroxyurea increases fetal hemoglobin levels which prevent HbS polyermization. Crizanlizumab block P-selectin and interrupts cellular adhesion. Mechanism for L-glutamine has not been fully elucidated to date. Adapted in part from [Citation1] with permission of Springer Nature](/cms/asset/793d31cb-d1ea-4b06-b818-96dce1c791d7/ieop_a_1819238_f0001_b.gif)
Because of its favorable safety profile and efficacy, HU is recommended for people of all ages with SCA. The number of people treated with HU has increased slowly since the 1995 publication of the Multi-Center Study of Hydroxyurea which showed that vaso-occlusive episodes (VOE) in adults treated with HU were reduced by 44% compared to placebo[Citation4]. Subsequent trials in infants, children, and adolescents showed similar benefits which led the National Heart, Lung and Blood Institute (NHLBI), in 2014, to recommend that all infants 9 months of age and older with SCA be offered HU, regardless of SCA-related complications[Citation5].
In July 2017, L-glutamine became the first new medication approved by the FDA to treat people with SCA in almost 20 years. While the exact mechanism of action for L-glutamine has not been fully elucidated, glutamine is a known precursor for nicotinamide adenine dinucleotide (NAD+) synthesis[Citation6]. Increased levels of NAD+ within sickled erythrocytes help prevent oxidative damage, thereby decreasing hemolysis and vaso-occlusion. A Phase 3 double blind, placebo controlled randomized clinical trial (RCT) of L-glutamine revealed a 25% reduction in VOE in the L-glutamine group compared to placebo (mean 3 and 4 VOEs per year, respectively). Two thirds of the people enrolled in the Phase 3 study were being treated with HU[Citation7]. L-glutamine is FDA approved for people 5 years of age and older.
The FDA approved two additional therapies in 2019, crizanlizumab and voxelotor. Crizanlizumab blocks P-selectin, an adhesion molecule that plays a role in painful VOEs in people with SCA. In a Phase 2 RCT, monthly infusions of crizanlizumab decreased the rates of painful VOE by 45% compared to placebo [Citation8,Citation9]. Crizanlizumab is currently FDA approved for those 16 years and older; studies in younger children are ongoing (NCT03474965). Voxelotor, currently approved for use in individuals 12 years of age and older, is an oral compound that binds to the alpha globin subunit of hemoglobin, causing an allosteric change in the hemoglobin molecule that increases its oxygen affinity, thereby preventing HbS polymerization[Citation10]. Voxelotor received approval after a Phase 3 double-blind RCT revealed that 51% of study participants in the 1500 mg voxelotor group attained the primary study outcome of an increased hemoglobin level of at least 1 g/dL above baseline, compared to 7% in the placebo group[Citation11]. VOEs decreased, though not statistically significantly, in the voxelotor group compared to placebo. Ongoing trials are investigating the clinical effects of voxelotor (NCT04218084, NCT02850406, NCT04188509, NCT03573882, NCT04335721, NCT04400487). Over two-thirds of the participants in the voxelotor and crizanlizumab trials were treated with HU [Citation8,Citation11].
The only currently available cure for SCA is HSCT. Matched sibling donor (MSD) HSCT for SCA has the highest survival rates and lowest rates of complications such as graft versus host disease (GVHD) [Citation12,Citation13]. Increasing age is associated with worsened survival; every year of age raised the risk of graft failure or death by 10%[Citation12]. Similarly, rates of GVHD are significantly lower in children less than 16 years of age compared to older candidates[Citation12]. Given the scarcity of MSD, studies evaluating alternative donor HSCT from matched unrelated donors (MUD) or haploidentical donors are ongoing and have shown mixed results. Haploidentical HSCT has low rates of GVHD, but high rates (40–50%) of graft rejection. In contrast, MUD HSCT has relatively low rates of rejection, but higher rates of GHVD. Thus, MUD or haploidentical HSCT should only be performed in the context of a research study[Citation13]. Gene therapy is an attractive curative option for SCA because less than 20% of affected individuals have a MSD for HSCT. The first report of gene therapy used to treat SCA was published in early 2017 [Citation14] and there are several open gene therapy trials in the US (NCT03282656, NCT03964792, NCT04293185, NCT02247843, NCT04091737, NCT02186418, NCT03745287, and NCT02140554).
2. Expert opinion
With increasing therapeutic options, the ideal scenario for children with SCA would be one similar to childhood acute lymphoblastic leukemia (ALL) risk stratification: treatment intensity varies with risk level. Thus, children with low-risk SCA would receive less intense therapy (), while the highest risk patients would proceed directly to curative therapy, especially if a MSD was available for HSCT. Risk-based therapy for ALL has improved survival and decreased late effects of therapy in the low risk and standard risk groups. As the number of therapies for SCA increases, SCA healthcare providers should strive for similarly personalized treatment for children with SCA. Currently, there are a number of barriers to making this a reality. First, crizanlizumab and voxelotor are approved for ages 16 and 12 years and older, respectively. While studies in younger children are ongoing, FDA authorization or clinician comfort with utilizing these new forms of therapy in an off-label fashion are likely 3–5 years away provided that the clinical pediatric studies show similar results as the studies in adolescents and adults. An additional challenge is the lack of a validated predictor of overall SCA severity that is available prior to the onset of SCA-related complications. The definition of ‘severe SCA’ is often debated, though most hematologists agree that the inclusion criteria for HSCT clinical trials () are a reasonable place to start. Balancing the risks of potentially lethal curative therapy with benefits of curing a disease that with the known morbidity and early mortality in adulthood is challenging without a validated global risk stratifier for SCA complications[Citation3]. One of the most fascinating and frustrating aspects of SCA care is how people with the same single amino acid substitution can have different clinical complications of varied severity that occur at different stages of life. The only currently available validated predictor of a severe SCA outcome is an abnormal velocity on transcranial Doppler ultrasonography, which identifies children at highest risk for stroke; predictors for recurrent VOE or ACS are not currently available. HbF levels and alpha-globin gene number have frequently been studied as possible early predictors of severe SCA, but results of these studies have varied depending on outcome variables and study sample sizes[Citation15]. Absolute reticulocyte count (ARC) above 200 K/µL between the ages of 2 and 6 months is associated with triple the risk of hospitalization before age 3 years and higher rates of stroke and death in a historical SCA newborn cohort. ARC was the only predictor studied before infants developed SCA-related symptomatology, allowing it to function as a true predictor of SCA severity[Citation15].
Table 1. Indications for MSD Hematopoietic stem cell transplant: reproduced from [Citation16] with permission of Elsevier.
Figure 2. Proposed Risk Based Therapy for Sickle Cell Anemia. SCA treatment intensity should vary with risk level, once a validated severity risk marker is available. Hydroxyurea (HU) should be considered standard of care for all individuals of SCA, regardless of risk category, as well as supportive care measures like fever education and antibiotic prophylaxis. High risk individuals should proceed to MSD HSCT early in life to minimize SCA complications as well as HSCT-related complications like graft versus host disease. If MSD HSCT is not available, then experimental treatment options like alternative donor HSCT or gene therapy should be discussed. Once appropriate age is reached, then other therapies could be considered. Medium risk SCA could be treated with MSD HSCT, but the risk-benefit ratio is likely unfavorable for alternative donor HSCT. Again, other therapies could be considered once the approved age is attained. Children at low risk for SCA should continue HU
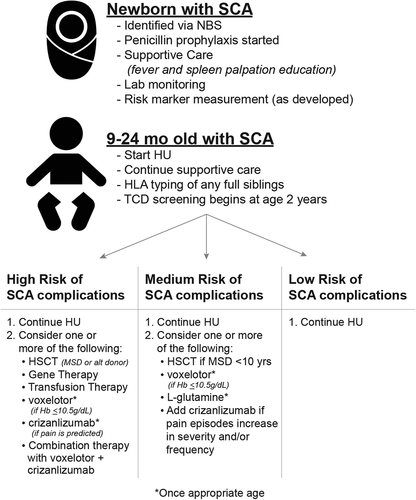
Risk-based therapy for adults with SCA is more challenging given the end organ injury that is present due to decades of erythrocyte sickling, hypoxia, and vaso-occlusion. Given the benefits of HU that have been elucidated over the past 25 years of its use, all adults with SCA should receive HU as disease modifying-therapy, regardless of SCA severity or complications. The addition of the remaining 3 FDA-approved treatments should be based on clinical and laboratory complications that are still present after HU dosing has been maximized [Citation17,Citation18]. Since nearly two-thirds of patients enrolled in each of the seminal trials for the new agents were being treated with HU, support for utilization of combination therapy for SCA can be found in these studies. L-glutamine and crizanlizumab decreased VOE and should be considered as additional therapy for adults who continue to have VOEs despite maximum tolerated dosing of HU. Voxelotor should be added to HU for those adults who continue to have significant anemia.
Despite over 25 years of experience in utilizing HU as a treatment for SCA and its proven benefits in preventing SCA-related complications, HU continues to be underutilized due to a combination of provider- and patient-related factors. Some providers are concerned about the side effects of HU or being able to obtain laboratory results at recommended intervals to monitor for myelosuppression [Citation19,Citation20]. Patient-reported adherence barriers include: concern about side effects, lack of perceived benefit, and lack of knowledge about HU[Citation21]. Because HU was initially used as a chemotherapeutic agent, many of these side effects (nausea, hair loss) are rare in people with SCA since the dosing strategies are different for SCA and malignancies. Even so, when many parents and patients search the web for HU, they find these and other concerning chemotherapy-related side effects (like decreased spermatogenesis and malignancy risk) that make them hesitant to take HU on a consistent basis.
In contrast, L-glutamine, crizanlizumab, and voxelotor have not been used as chemotherapy and were developed as SCA treatments, so patients and providers may be more likely to utilize these new SCA-specific treatments. A validated SCA severity risk marker would allow SCA treatment to vary in intensity, based on risk category. High risk individuals could be offered curative therapy (if available) early in life or multi-drug therapy once the appropriate age for each agent is attained. Medium risk SCA could be treated with MSD HSCT, but the risk-benefit ratio is likely unfavorable for alternative donor HSCT. Other therapies could be considered once the approved age is attained. HU should be considered standard of care for all individuals of SCA, regardless of risk category, as should supportive care measures like fever education and antibiotic prophylaxis. Children at low risk for SCA should continue HU. Hopefully, as the number of SCA modifying and curative therapies increase, more innovative treatment strategies will be tested and lead to improved quality of life and increased life expectancy for individuals with SCA.
Declaration of interest
E Meier has received funding from the Centers for Disease Control and Prevention (CDC), USA as well as consultancy fees from CVS Caremark. Further, she has served on the Speakers Bureau and Advisory Board of Global Blood Therapeutics and has served on the Data Monitoring Committees of Novartis and the NHLBI. She has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Meier ER, Rampersad A. Pediatric sickle cell disease: past successes and future challenges. Pediatric Res. 2017;81(1–2):249–258.
- Quinn CT, Rogers ZR, McCavit TL, et al. Improved survival of children and adolescents with sickle cell disease. Blood. 2010 Apr 29;115(17):3447–3452.
- Lanzkron S, Carroll CP, Haywood C Jr. Mortality rates and age at death from sickle cell disease: U.S., 1979-2005. Public Health Rep (Washington, DC: 1974). 2013 Mar-Apr;128(2):110–116.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. 1995 May 18;332(20):1317–1322.
- National Heart Lung and Blood Institute (NHLBI). Evidence-based management of sickle cell disease: expert panel report, 2014. 2014. http://www.nhlbi.nih.gov/health-pro/guidelines/sickle-cell-disease-guidelines.
- Niihara Y, Zerez CR, Akiyama DS, et al. Oral L-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am J Hematol. 1998 Jun;58(2):117–121.
- Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018 Jul 19;379(3):226–235.
- Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017 Feb 2;376(5):429–439.
- Han J, Saraf SL, Gordeuk VR. Systematic review of crizanlizumab: a new parenteral option to reduce vaso-occlusive pain crises in patients with sickle cell disease. Pharmacotherapy. 2020 Jun;40(6):535–543.
- Han J, Saraf SL, Gordeuk VR. Systematic review of voxelotor: a first-in-class sickle hemoglobin polymerization inhibitor for management of sickle cell disease. Pharmacotherapy. 2020 Jun;40(6):525–534.
- Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019 Aug 8;381(6):509–519.
- Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017 Mar 16;129(11):1548–1556.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood. 2014 May 15;123(20):3089–3094. quiz 210.
- Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene Therapy in a Patient with Sickle Cell Disease. N Engl J Med. 2017 Mar 02;376(9):848–855.
- Meier ER, Fasano RM, Levett PR. A systematic review of the literature for severity predictors in children with sickle cell anemia. Blood Cells Mol Dis. 2017 Jun;65:86–94.
- Meier ER. Treatment options for sickle cell disease. Pediatr Clin North Am. 2018;65(3):427–443.
- Rai P, Ataga K. Drug therapies for the management of sickle cell disease F1000. Res. 2020;11(9):F1000Faculty Rev–592.
- Ballas S. The evolving pharmacotherapeutic landscape for the treatment of sickle cell disease. Mediterr J Hematol Infect Dis. 2020;12(1):e2020010.
- Lanzkron S, Haywood C Jr., Hassell KL, et al. Provider barriers to hydroxyurea use in adults with sickle cell disease: a survey of the Sickle Cell Disease Adult Provider Network. J Natl Med Assoc. 2008 Aug;100(8):968–973.
- Brandow AM, Jirovec DL, Panepinto JA. Hydroxyurea in children with sickle cell disease: practice patterns and barriers to utilization. Am J Hematol. 2010 Aug;85(8):611–613.
- Oyeku SO, Driscoll MC, Cohen HW, et al. Parental and other factors associated with hydroxyurea use for pediatric sickle cell disease. Pediatr Blood Cancer. 2013 Apr;60(4):653–658.