
ABSTRACT
Objective: We report the characterization of MCLA-117, a novel T cell-redirecting antibody for acute myeloid leukaemia (AML) treatment targeting CD3 on T cells and CLEC12A on leukaemic cells. In AML, CLEC12A is expressed on blasts and leukaemic stem cells.
Methods: The functional capacity of MCLA-117 to redirect resting T cells to eradicate CLEC12APOS tumor cells was studied using human samples, including primary AML samples.
Results: Within the normal hematopoietic compartment, MCLA-117 binds to cells expressing CD3 and CLEC12A but not to early myeloid progenitors or hematopoietic stem cells. MCLA-117 induces T cell activation (EC50 = 44 ng/mL), T cell proliferation, mild pro-inflammatory cytokine release, and redirects T cells to lyse CLEC12APOS target cells (EC50 = 68 ng/mL). MCLA-117-induced targeting of normal CD34POS cells co-cultured with T cells spares erythrocyte and megakaryocyte differentiation as well as preserves mono-myelocytic lineage development. In primary AML patient samples with autologous T cells, MCLA-117 robustly induced AML blast killing (23–98%) at low effector-to-target ratios (1:3–1:97).
Conclusion: These findings demonstrate that MCLA-117 efficiently redirects T cells to kill tumour cells while sparing the potential of the bone marrow to develop the full hematological compartment and support further clinical evaluation as a potentially potent treatment option for AML.
1. Introduction
Acute myeloid leukaemia (AML) is the third most common leukaemia worldwide, but over the past four decades has seen little progress in disease outcomes [Citation1–Citation3]. The yearly incidence of AML in European adults is five to eight per 100,000 people. In the United States four to five per 100,000 people are newly diagnosed with AML each year, with 10,000 people dying each year from AML. The five-year survival in young adults is approximately 40%, but this drops significantly with age to less than 25% in older patients. The majority of patients achieving morphologic complete remission after induction chemotherapy subsequently relapse within three years [Citation4,Citation5]. These patients therefore urgently require the development of new agents that have a more effective mode of action.
Blinatumomab is a CD19xCD3 bispecific T cell engager (BiTE) which is reported to benefit certain patient populations with non-Hodgkin lymphoma (NHL) and acute lymphoblastic leukaemia (ALL) [Citation6,Citation7]. A T cell engager is a type of bispecific antibody that specifically binds to a tumour-associated antigen on the tumour on the one hand, and to CD3 on T cells on the other. Such T cell-redirecting antibody redirects polyclonal T cells – irrespective of MHC restriction – via the tumour-associated target to induce efficient tumour cell killing. Preclinical studies have reported that a T cell engager mode of action may be effective for different tumour-associated antigens expressed on various tumour types [Citation8]. Moreover, preclinical studies suggest that T cell-redirecting bispecific antibodies may have the potential to kill heterogeneous tumours, quiescent cancer stem cells as well as chemotherapy-resistant tumours [Citation9–Citation12]. Today, blinatumomab is the only regulatory approved T cell engager.
There are several T cell engagers under clinical development for the treatment of AML [Citation13], which include a CD33xCD3 BiTE and CD123xCD3 dual-affinity retargeting (DART) molecules [Citation14,Citation15]. CD33 and CD123 are broadly expressed on myeloid leukaemia blasts and leukaemic progenitors [Citation16]. While CD33 targeting has been explored extensively in AML [Citation17,Citation18], these approaches have faced major setbacks due to the agents’ ineffectiveness or associated toxicities [Citation19–Citation22]. An antigen focused upon by our group is a target specifically expressed on AML blasts and leukaemic stem cells (LSCs) and is the C-type lectin domain family 12 member A or CLEC12A (also referred to as human myeloid inhibitory C-type lectin, hMICL, C-type lectin-like molecule-1, CLL-1, or CD371) [Citation23,Citation24]. CLEC12A is a myeloid differentiation antigen that is expressed on leukemic cells in 90–95% of cases of either de novo or relapsed AML, irrespective of subtype [Citation23,Citation25,Citation26]. CLEC12A is selectively expressed on CD34POSCD38NEG LSCs but it is not expressed on normal haematopoietic stem cells (HSCs), erythroid precursors or megakaryocytes [Citation24,Citation27]. This is in contrast to CD33 and CD123, which are also expressed on HSCs [Citation28]. Recent studies have shown the potential of the CLEC12A antigen to target and eradicate CLEC12APOS leukaemic cells, either via an anti-CLEC12A antibody drug conjugate concept depleting CLEC12A expressing cells or a CLEC12AxCD3 T cell engager concept inducing T cell-mediated lysis of CLEC12APOS tumour cell lines [Citation29–Citation31].
Because of its restricted expression profile, T cell-mediated CLEC12A targeting has the potential to selectively eradicate leukaemic cells (including stem cells) without affecting normal HSCs, thereby permitting normal haematopoiesis to be re-established after treatment and potentially limiting haematological toxicity.
Many of the T cell engager formats face potential issues because they are heavily engineered, including a short serum half-life, immunogenicity and difficulties in terms of manufacture [Citation32]. To overcome these potential issues, we have developed a CLEC12AxCD3 bispecific antibody that has a native human IgG format: MCLA-117. MCLA-117 is a full-length human IgG1 bispecific antibody, lacking Fc effector function and containing a common light chain (cLC). Here we report the functional characterization of MCLA-117.
2. Materials and methods
2.1. Bispecific antibodies
MCLA-117 is a full-length human IgG1 bispecific antibody specific for CLEC12A and CD3 containing a common light chain. The negative control antibodies were a cLC monospecific antibody against tetanus toxoid (TTxTT IgG, isotype control [Citation33]) and a cLC bispecific antibody against tetanus toxoid and CD3 (MockxCD3 or TTxCD3 IgG, using the same CD3 Fab as in MCLA-117); the positive control antibody was a bivalent cLC monospecific antibody against CD3 (CD3xCD3, also using the same CD3 Fab as in MCLA-117). In initial batches of the bispecific antibodies, electrostatically engineered heavy chains were used for transient production, resulting in bispecific IgG1 with >95% purity [Citation34]. Later batches were produced using heavy chain Fc engineering comprising substitutions L351D and L368E in one heavy chain combined with L351K and T366K in the other heavy chain [Citation35]. Unless otherwise specified, the IgG batches of MCLA-117, MockxCD3 and TTxTT IgG lacked Fc effector function upon introduction of amino acid substitutions in the CH2 regions.
2.2. Affinity measurement
MCLA-117 was immobilized on a CM5 chip (GE Healthcare) through a mouse anti-human IgG monoclonal antibody (BD Biosciences). Recombinant purified CLEC12A-His (Sino Biologicals) or recombinant CD3δε-Fc protein was subsequently run over the surface using a Biacore T100 instrument (GE Healthcare).
2.3. In vivo pharmacokinetic study
C57BL/6J mice received a single intravenous dose of 1 mg/kg MCLA-117. MCLA-117 serum levels were quantified at various time points using an anti-human IgG ELISA (ZeptoMetrix).
2.4. Cell lines
HL60 cells (DSMZ) were cultured in IMDM (GIBCO Invitrogen, 21980–065) containing 2mM L-glutamine and 10% FBS (Integro). CHO-K1 (DSMZ), Jurkat E6.1 (LGC) and J.RT-T3.5 (LGC) cell lines were maintained in DMEM/F12 (GIBCO Invitrogen, 11320–033) supplemented with 2mM l-glutamine and 10% FBS (ThermoFisher Scientific). The CLEC12A-CHO-K1 cell line was generated by stable integration of pcDNA3.1+vector (ThermoFisher Scientific) encoding human CLEC12A.
2.5. Clinical samples
AML patient samples (Supplemental Table 1) and healthy donor bone marrow were obtained in accordance with the Declaration of Helsinki and RUMC institutional guidelines and regulations. For storage in liquid nitrogen, cells were resuspended in IMDM medium containing 7% dimethyl sulfoxide (DMSO; Sigma-Aldrich, W387509) and 10% foetal bovine serum (FBS, Integro). Upon thawing, cells were incubated in 10% human serum (HS, PAA laboratories) containing 250 µg/mL DNAse (Roche, 11284932001) and 1.25 mM MgCl2 for 10 min. Subsequently, cells were washed with excess volumes of medium.
2.6. Cytotoxicity assay with purified T cells
Human peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood (PB) collected from healthy donors or AML patients by density gradient centrifugation (see Supplemental Table 1 for patient characteristics). CD3POS T cells were purified from freshly isolated or cryopreserved PBMCs by negative selection by using magnetic cell sorting (MACS, Miltenyi, 130–096-535). Cytotoxicity assays were performed in IMDM supplemented with 10% AB+ HS (Sanquin) in 96-well plates (NuncTM, ThermoFisher Scientific). Unless otherwise specified, 105 T cells were used. CFSE-labelled HL60 or primary AML blast target cells were used (ThermoFisher Scientific, C11-57). Prior to CFSE labelling, AML blasts were pre-cultured overnight in assay medium supplemented with 100 ng/mL GM-CSF (Immunotools, 11343125), 100 ng/mL G-CSF (Amgen Inc, Neupogen®), 50 ng/mL IL-3 (Cellgenix), 25 ng/mL SCF (Immunotools, 11343325) and 20 ng/mL Flt3L (Immunotools, 11343305). Subsequently, T cells and target cells were co-cultured in the same cytokine-supplemented assay medium in the presence of MCLA-117 or control IgGs. After 24–72 hours, T cell activation and the number of remaining CFSEPOS target cells were quantified by flow cytometry (for gating strategy see Supplemental Figure 1). The percentage of specific lysis was calculated as follows: Specific lysis (%) = 100 – [(100 x target cell number in condition with IgG)/(average target cell number in condition without IgG)].
2.7. PBMC-based monocyte cytotoxicity assay
PBMCs (2 x 105) were incubated in IMDM with 10% AB+ HS for 48 hours in the presence of MCLA-117 or control IgGs. Prior to harvesting, the assay plates were centrifuged (500 g, 5 minutes) to collect supernatant for cytokine quantification. After a subsequent incubation at 2-8°C for 1 hour, cells were stained in the assay medium without washing. The specific lysis of monocytes, B cells and natural killer (NK) cells was quantified by flow cytometry in a fixed volume collected from each assay well (Supplemental Figure 2).
2.8. T cell proliferation assay
CFSE-labelled CD3POS T cells were co-cultured with CD14POS MACS-purified (Miltenyi, 130–050-201) autologous monocytes in IMDM with 10%AB+ HS with or without test IgG at an effector-to-target (E:T) ratio of 5:1. After 5 days, the fraction of proliferating T cells was visualized as CFSELOW T cells by flow cytometry.
2.9. Cytokine analysis
The levels of IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, GM-CSF, TNF-α and IFN-γ in culture supernatants were measured using the Luminex® platform (Life Technologies, LHC0001).
2.10. Cytotoxicity assay with healthy donor bone marrow cells
CD3POS T cells and CD34POS cells were isolated from healthy donor bone marrow mononuclear cells (obtained from orthopaedic patients or purchased from Lonza, 2M-125C) by positive selection using MACS (Miltenyi, 130–046-702 and 130–050-101, respectively). Pre-activated T cells were generated by activating purified T cells for 6 days in plates pre-coated with anti-CD3 (1 µg/mL, BD Bioscience, 555329) and anti-CD28 (2 µg/mL, BD Bioscience, 555725) in IMDM supplemented with 10% A+ HS (Sanquin blood bank) and 30 IU/mL IL-2 (Chiron, Proleukin®) followed by overnight rest in plates with 30 IU/mL IL-2. On the next day, pre-activated T cells were co-cultured with autologous CD34POS cells at a 10:1 E:T ratio in IMDM supplemented with 10% A+ HS in 96-well plates (Corning Costar, 3799) in the presence of MCLA-117 or control condition. After 16 hours, impact of MCLA-117 was quantified by flow cytometry for various CD34POS progenitor populations and colony forming unit (CFU) assays as described below. For quantification by flow cytometry, flow count fluorospheres (Beckman Coulter, 7547053) were used. For gating strategy see Supplemental Figures 4 and 5.
2.11. Colony forming unit (CFU) assays
Upon cytotoxicity assay with CD34POS cells (described above) all remaining cells/well were harvested. Co-cultures of T cells and CD34POS cells were seeded upon washing in MethoCult GF H84435 (Stemcell Technologies, 84435) for analysis of erythroid and mono-myelocytic colony outgrowth and MegaCultTM-C (complete kit with cytokines (04971) with added human low-density lipoproteins (40 µg/mL, 02698), both from Stemcell Technologies) for analysis of megakaryocyte colony formation according to the manufacturer’s instructions. To this end, upon washing of the cells the number of cells in the no-IgG condition were counted and for both the MethoCult and MegaCultTM-C assays, co-cultures were seeded at two different concentrations. For the MCLA-117 condition, an equal volume of cells were seeded as were seeded for the control IgG-treated co-cultures. For the MethoCult assay, 1 × 103 and 5 × 102 CD34POS cells of the non-IgG condition were seeded in triplicate in 35 mm culture dishes (Corning, 430165), and for the MegaCult-C assay, 7.5 × 103 and 2.5 × 103 CD34POS cells were seeded per chamber of a double-chamber culture slide and two slides prepared per condition. The number of erythroid, mono-myelocytic and megakaryocyte colonies were scored after 13–15 day culture, and subsequently the MethoCult cultures were harvested and analysed by flow cytometry for monocytic and myelocytic markers.
2.12. Cytotoxicity assay with primary AML samples
Primary AML patient samples (5 x 105 cells) were cultured in the presence of MCLA-117 or control bispecific IgG in IMDM containing 10% AB+ HS, 2.5 ng/mL GM-CSF, 12.5 ng/mL G-CSF (Neupogen, Amgen), 6.25 ng/mL IL-3 (Immunotools, 11340035), 3.0 ng/mL SCF and 2.5 ng/mL Flt3L. After 24 hours, 20 ng/mL IL-15 (Miltenyi, 130–095-766) was added to the culture. After 10 days, AML blast lysis and T cell expansion was quantified by flow cytometry.
The research was conducted and the human samples were collected according to the Declaration of Helsinki and RUMC institutional guidelines and regulations. Additional materials and methods are described in the Supplemental section.
3. Results
3.1. MCLA-117 binds specifically to CLEC12APOS and CD3POS cells within the normal haematopoietic compartment
MCLA-117 is a T cell-redirecting bispecific antibody that has been developed to treat AML that binds to CD3 on T cells and CLEC12A on the AML blasts and LSCs. MCLA-117 is a human full-length IgG1 bispecific antibody based on the common light chain bispecific IgG format ()). Its CLEC12A-specific cLC Fab was identified using phage display technology and its CD3-binding cLC Fab is derived from a human anti-CD3 antibody for which the original light chain was replaced by the cLC. To force heavy chain heterodimerization, oppositely charged amino acid residues were introduced into the CH3 domain of the two different heavy chains [Citation35].
Figure 1. MCLA-117, a CLEC12AxCD3 full length IgGs bispecific specifically binds to CLEC12APOS and CD3POS target cells.
(a) Schematic representation of MCLA-117 and the contribution of each domain to the functionality of MCLA-117. cLC: common light chain; 'DE' 'KK': amino acid substitutions L351D, L368E and L351K, T366K introduced to promote heavy chain heterodimerization. (b) Binding of MCLA-117 to CLEC12A and CD3 was determined on a panel of tumour cell lines by flow cytometry. HL60: CLEC12APOS promyelogenous cell line; CLEC12A-CHO-K1: CHO-K1 cell line stably expressing human CLEC12A (validated with reference anti-CLEC12A antibody, data not shown); CHO-K1: CLEC12ANEG parental CHO-K1 cell line; Jurkat E6.1: CD3POS T cell line; J.RT-T3.5: CD3NEG T cell line. (c) MCLA-117 binding profile in normal peripheral blood by flow cytometry. Gating strategy for analysed subsets were CD4 T cells: CD4POS; CD8 T cells: CD8POS; NK cells: CD3NEGCD56POS; NKT cells: CD3POSCD56POS; B cells: CD19POS; myeloid dendritic cells (DC): BDCA1POSCD19NEG; granulocytes: based on SSC-FSC; monocytes: CD14POSCD33POS. (d) MCLA-117 binding within CD34POS progenitor compartment in normal bone marrow by flow cytometry, gating strategy is indicated in figure. HSC: haematopoietic stem cell; MPP: multipotent progenitor; CMP: common myeloid progenitor; GMP: granulocyte-macrophage progenitor; MEP: megakaryocyte-erythroid progenitor; CLP: common lymphoid progenitor. Data shown for MCLA-117 and isotype control IgG formatted with the final Fc format (silenced Fc effector function).
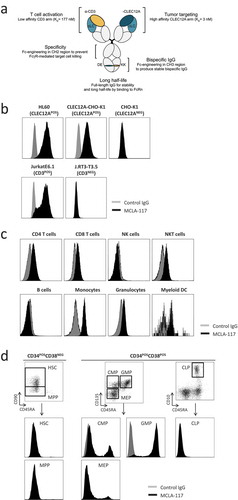
Flow cytometry using tumour cell lines showed that MCLA-117 bound specifically to CLEC12A and CD3 ()). Via its CLEC12A arm, it bound the CLEC12APOS HL60 cells as well as to CHO-K1 cells transfected with human CLEC12A, but not to parental CHO-K1 cells. Via its CD3 arm, it bound to CD3POS Jurkat E6.1 cells, but not to CD3NEG Jurkat J.RT-T3.5 cells.
The affinity of MCLA-117 for its target proteins was 3 nM for CLEC12A and 177 nM for CD3, as determined by surface plasmon resonance technology using recombinant proteins. To investigate whether the engineered Fc region of MCLA-117 had a potential impact on pharmacokinetics (PK), the in vivo half-life of MCLA-117 was determined in C57BL/6J mice and found to be 226 hours (9.4 days). This half-life is within the range of in vivo half-lives typically observed for human IgG1 antibodies in rodents, indicating that the specific design of the human cLC antibody had no effect on the non-specific elimination pathways for IgG.
Analysis of the binding profile of MCLA-117 in normal peripheral blood showed that MCLA-117 bound to CD3-expressing CD4POS and CD8POS T cells as well as to CD3POSCD56POS natural killerT (NKT) cells ()). In accordance with the previously reported expression profile for CLEC12A [Citation23,Citation36,Citation37], MCLA-117 also bound to monocytes, myeloid dendritic cells (mDC), plasmacytoid dendritic cells (pDC) and granulocytes, but not to natural killer (NK) cells or B cells (()) and data not shown). In normal bone marrow, the binding profile of MCLA-117 within the CD34POS progenitor compartment ()) also resembled the reported CLEC12A expression profile [Citation24]. MCLA-117 bound uniformly to granulocyte-macrophage progenitor (GMP) cells, whereas it bound only a minor fraction of the common myeloid progenitor (CMP) and megakaryocyte-erythroid progenitor (MEP) subsets. Notably, MCLA-117 binding was not observed within the CD34POSCD38NEG compartment, which includes the multipotent progenitors (MPP) and pluripotent haematopoietic stem cells (HSC).
3.2. MCLA-117 induces CLEC12A-dependent T cell activation and lysis of CLEC12APOS HL60 cells
Next, we determined the capacity of the CLEC12AxCD3 bispecific IgG format to induce CLEC12A antigen-dependent T cell activation and lysis of CLEC12APOS target cells in a cytotoxicity assay. The effector cells were healthy donor-derived resting T cells and the target cells were CLEC12APOS HL60 cells. Firstly, a MCLA-117 prototype with a wild-type (WT) Fc region (intact Fc effector function) dubbed as MCLA-117* was analyzed and compared with MockxCD3 control IgG formatted with a WT Fc region (labelled as MockxCD3*). MCLA-117* induced upregulation of CD69 () and CD25 (data not shown) on CD4 and CD8 T cells after 24 and 48 hours. T cell-mediated lysis of HL60 cells was already evident after 24 hours and >95% HL60 cell cytotoxicity was measured after 48 hours ()). Target cell lysis was dependent on the effector-to-target ratio with a maximum lysis at E:T ratios of 5:1 or higher (Supplemental Figure 3). Notably, the MCLA-117*-induced lytic activity was CLEC12A antigen-mediated (,)); only at later time points some T cell activation and HL60 target cell lysis was observed for the MockxCD3* control. Nevertheless, this revealed that the bispecific IgG format with WT Fc could induce Fcγ receptor-mediated T cell activation without the need for CLEC12A binding (, 72 hours).
Figure 2. MCLA-117 mediates CLEC12A-dependent activation of T cells and redirects these T cells to induce lysis of a CLEC12APOS HL60 cells.
Resting T cells derived from healthy donors were purified by negative selection and co-cultured with CFSE-labelled HL60 cells in the presence of antibodies or PBS. Firstly, the activity of a prototype MCLA-117 with wild-type (WT) Fc effector function, dubbed as MCLA-117*, was analyzed and compared to MockxCD3* control IgG (formatted with WT Fc function) (a-b). Subsequently MCLA-117, Fc effector function silenced, was functionally evaluated (c-e). (a-b) Cells were co-incubated at an E:T ratio of 5:1 together with MCLA-117* and MockxCD3* (both WT Fc function) at an IgG concentration of 1,000 ng/mL. The capacity of MCLA-117* function to induce T cell activation (a) and target cell lysis (b) was quantified after 24/48/72 hours by flow cytometry relative to the PBS condition. The data shown are from two representative healthy donors out of >10 tested. (c-e) Cells were co-incubated at an E:T ratio of 5:1 with indicated IgGs – all with silenced Fc effector function – at a range of IgG concentrations. The capacity of MCLA-117 (with silenced Fc effector function) to induce activation of CD4 and CD8 T cells (c, d) and target cell lysis (e) was quantified after 48 hours by flow cytometry relative to the PBS condition. The data shown are from a representative donor, whereby a total of six donors were tested in three independent assays.
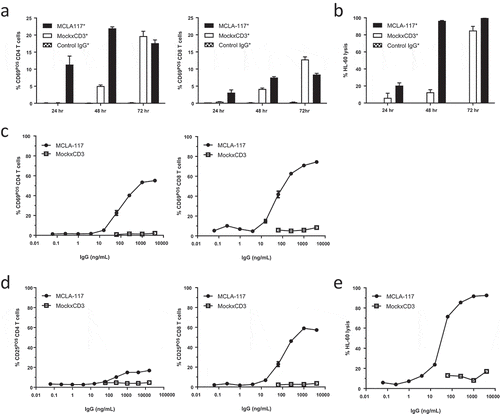
3.3. MCLA-117 with silenced Fc effector function induces specific lysis of CLEC12APOS AML cell line
To circumvent that MCLA-117 induced non-specific Fcγ receptor-mediated cell lysis, a bispecific IgG format lacking Fc effector function was generated by introducing the amino acid substitutions into the CH2 region of both IgG1 heavy chains, referred to as MCLA-117 (no asterisk). The introduction of these amino acid substitutions into MCLA-117 abrogated binding to CD16, CD32 and C1q, and diminished binding to CD64 > 200-fold. In cytotoxicity assays using normal resting T cells and CLEC12APOS HL60 cells, MCLA-117 induced concentration-dependent upregulation of CD69 ()) and CD25 ()) on CD4 T cells and CD8 T cells. The most sensitive read-out for MCLA-117-induced activity was upregulation of CD69 on CD8 T cells with a mean half-maximal effective concentration (EC50) of 44 ng/mL (). More importantly, MCLA-117 induced a concentration-dependent T cell-mediated lysis of HL60 cells with >80% lysis at concentrations of 100 ng/mL and above, and a mean EC50 of 68 ng/mL (), ). While activation of CD4 and CD8 T cells by the initial version of the MockxCD3 antibody with wild-type Fc effector function was already seen at day two (), 1,000 ng/mL), no T cell activation was observed with the Fcγ receptor-silenced MockxCD3 antibody after 48 hours, not even at 8,000 ng/mL (-)). All together this shows that the T cell cytotoxicity induced by Fcγ receptor-silenced MCLA-117 (-)) was CLEC12A antigen-specific.
Table 1. Mean EC50 values of MCLA-117-induced T cell activation and target cell lysis.
Collectively, these data demonstrated the capacity of MCLA-117 lacking Fc effector function to mediate CLEC12A-specific T cell activation and to effectively redirect these T cells to induce lysis of CLEC12APOS HL60 cells.
3.4. MCLA-117 induces specific lysis of CLEC12APOS monocytes by autologous T cells
The capacity of MCLA-117 to induce redirected lysis of CLEC12APOS monocytes by resting autologous T cells was determined in PBMC cultures. MCLA-117 induced dose-dependent activation of CD4 T cells and CD8 T cells ()), and lysis of monocytes (from 100 ng/mL, )). Importantly, the MCLA-117-induced activity was CLEC12A-specific as the MockxCD3 control did not induce observable T cell activation or monocyte lysis at concentrations up to 10,000 ng/mL (-)). Furthermore, the lytic activity of MCLA-117-activated cytotoxic T cells was selective for CLEC12A-antigen expressing cells, as B cell and NK cell fractions were not affected, even at the highest tested MCLA-117 concentrations () and data not shown).
Figure 3. MCLA-117 induces CLEC12A-mediated lysis of monocytes, and inflammatory cytokine release, and has the capacity to induce CLEC12A-antigen specific T cell proliferation.
(a-c) Healthy donor-derived PBMC samples were co-cultured with MCLA-117 or MockxCD3 antibody at the indicated concentration range and compared to the PBS condition. T cell activation (a), monocyte lysis by autologous T cells (b) and B cell lysis (c) were assessed after 48 hours using flow cytometry. (d) Monocytes derived from healthy donors were purified by negative selection and co-cultured with CFSE-labelled resting autologous T cells at an E:T ratio of 5:1 with MCLA-117 and MockxCD3 antibody at 1,000 ng/mL (anti-CD3 IgG was formatted with WT Fc effector function) for 5 days. T cell proliferation was demonstrated by the reduction of CFSE-positive T cells. (e) Healthy donor-derived PBMCs were cultured with 1,000 ng/mL MCLA-117, MockxCD3 or anti-CD3 (CD3) antibody for 48 hours. Subsequently, supernatant was harvested to measure the human cytokine levels in these supernatants using a human 10-plex Luminex setup. Cytokine levels were detectable for the six cytokines shown. Data shown in (a-d) are from a single donor and are representative of four independent donors from multiple independent experiments. Data shown in (e) are from two representative healthy PBMC donors, with duplicates for each donor, whereby PBMCs from eight healthy donors were tested.

These data demonstrated that MCLA-117 could efficiently induce CLEC12A-restricted redirected lysis of CLEC12APOS monocytes via redirection and activation of autologous T cells.
3.5. CLEC12A-antigen specific induction of cytokine release and T cell proliferation
The capacity of MCLA-117 to induce T cell proliferation was studied upon co-incubation of monocytes with autologous resting T cells. MCLA-117 efficiently induced CLEC12A-antigen specific proliferation of both CD4 and CD8 T cells at 100 and 1,000 ng/mL ()). After five days of co-culture, the MockxCD3 control did not induce any T cell proliferation, proving once more that MCLA-117 function is CLEC12A-antigen specific.
Next, levels of cytokines were measured in these PBMC cultures after 48 hours of MCLA-117 exposure at 1,000 ng/mL. MCLA-117 induced the release of IL-1β, TNF-α, IFN-γ, IL-6 and IL-10 in the range of 10–200 pg/mL, and IL-8 release in the range of 10,000–30,000 pg/mL ()). In general, MCLA-117-induced cytokine levels were lower than those obtained after stimulation with the anti-CD3 bivalent antibody. The IL-8 release is understood to be – at least partially – derived from the MCLA-117-lysed monocytes as monocytes have been demonstrated to produce IL-8 when exposed to IL-1 and/or TNFα [Citation38,Citation39]. The MCLA-117-induced cytokine release was CLEC12A-dependent as cytokine release in the presence of the MockxCD3 control was absent or limited.
These data demonstrated that MCLA-117 could induce autologous T cells to proliferate and release cytokines upon co-incubation with CLEC12APOS monocytes.
3.6. MCLA-117-induced targeting of normal CD34+ cells spares erythrocyte and megakaryocyte differentiation as well as retains potential to develop the mono-myelocytic lineage
As CLEC12A is expressed in the mono-myelocytic progenitor compartment within the normal CD34POS compartment we aimed to evaluate the impact of MCLA-117 on normal haematopoiesis. To this end, cytotoxicity assays with healthy donor bone marrow-derived CD34POS cells and pre-activated autologous T cells were performed. Pre-activated T cells were used to have a short 16 hour cytotoxicity assay window which minimizes the impact of in vitro differentiation of the CD34POS cells on the outcome of the assay. We observed that MCLA-117 specifically lysed > 85% of the CLEC12APOSCD34POS cells, while leaving the CLEC12ANEG CD10POS or CD10NEG cell fractions unaffected ()). Detailed analysis of the various CD34POS progenitor fractions revealed that MCLA-117 targeted the CLEC12A-expressing GMPs (3x-fold reduction, )). MCLA-117 did neither significantly impact the cell fractions partially expressing CLEC12A (CMP and MEP) nor the CLEC12A-negative HSC, MPP and common lymphoid progenitors (CLP). Interestingly, MCLA-117 only lysed a fraction of CD123POS cells, showing that a large subset of CD123POSCLEC12ANEG cells is not affected by MCLA-117, a subset that comprises CMPs ()).
Figure 4. MCLA-117 spares the potential of normal bone marrow progenitor cells to develop the complete myeloid and erythroid lineage.
CD34POS hematopoietic progenitor cells from healthy donor bone marrow were co-cultured with autologous pre-activated T cells in the presence of 200 ng/mL MCLA-117 or MockxCD3 at an E:T ratio of 10:1 for 16 hours. (a) Using a quantified flow cytometry setup, MCLA-117-induced lysis was determined relative to non-IgG condition for the indicated cell fractions. Each data point represents the mean value for an independent bone marrow donor tested in triplicate (n = 4). Gating strategy is described in Supplementary Figure 4. (b) Analyses of the impact of MCLA-117 on different CD34POS bone marrow populations after 16 hour cytotoxicity assay, showing absolute number for three donors. Gating of CD34POS progenitor cells is depicted in Supplementary Figure 5. Quantification of absolute number in the different test conditions was calculated by integration of the frequency and absolute count of total CD34POS cells determined with flow count fluorospheres. Data show mean of duplicate measurement. (c) Scatter plots showing impact of MCLA-117 on CLEC12A- and/or CD123-expressing subsets after 16 hours cytotoxicity assay for indicated conditions, gated on CD34POSLINNEG cells. One representative donor out of two shown. (d) Capacity of CD34POS cells upon 16 hours cytotoxicity assay with MCLA-117 to give rise to granulocyte-macrophage colonies (CFU-GM), burst-forming units-erythrocyte (BFU-E) and megakaryocyte colonies (CFU-Mk) was quantified after two weeks culture in semi-solid medium, from four different donors. Control cultures are either non-IgG treated co-cultures or CD34POS cells seeded alone. Each data point represents the mean value of triplicate cultures (CFU-GM and BFU-E) or quadruplicates (CFU-Mk) per donor. (e) Flow cytometric analysis of CD15POSCD33POS and CD14POSCD36POSCD33POS cell content of the two week MethoCult cultures described in (d). Data show the frequency of monocytic and myelocytic cells out of total CD33POS cells for three different donors. HSC: haematopoietic stem cell; MPP: multipotent progenitor; CMP: common myeloid progenitor; GMP: granulocyte-macrophage progenitor; MEP: megakaryocyte-erythroid progenitor; CLP: common lymphoid progenitor. Statistical analysis was performed using paired student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
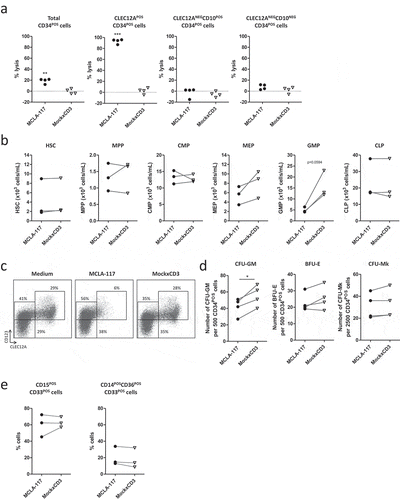
Next, the ability of the normal CD34POS cell fraction to give rise to monocytes, granulocytes, erythrocytes and megakaryocytes upon MCLA-117-induced redirected lysis was evaluated at a functional level using CFU assays. The outgrowth of the erythroid (BFU-E; burst-forming units-erythroid) or megakaryocyte (CFU-Mk; colony forming unit-megakaryocyte) colonies from the bone marrow CD34POS hematopoietic progenitor cells were not affected by MCLA-117 ()). Notably, despite that MCLA-117-induced redirected lysis caused a strong reduction in GMPs only a modest reduction of 24% in the outgrowth of granulocyte-macrophage colonies (CFU-GM) was observed at functional level. Interestingly, the GM-progenitor cells, unaffected by MCLA-117, retained their capacity to develop both monocytic (CD14POSCD36POSCD33POS) and myelocytic (CD15POSCD33POS) cells ()), as determined by flow cytometric analysis of the CFU-GM colonies.
Taken together, these data show that MCLA-117 does not affect erythroid megakaryocytic development. Furthermore, while it efficiently eradicates CLEC12APOSCD34POS progenitors, granulocyte and monocyte development potential is retained upon MCLA-117 treatment.
3.7. MCLA-117 induces redirected lysis of primary AML blasts by autologous T cells
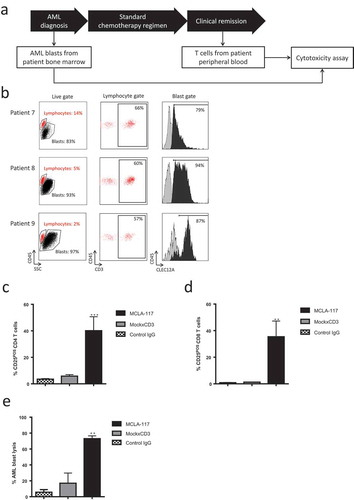
Next, we determined the capacity of MCLA-117 to induce AML patient-derived T cells to lyse CLEC12APOS target cells. The T cell compartment from AML patients in morphologic clinical remission showed normal composition of naïve, central memory (CM), effector memory (EM) and effector memory cells with reacquired CD45RA (EMRA+) (Supplemental Figure 6a-b, and Supplemental Table 1, patients 1–6). Analysing the capacity of AML patient T cells to mediate MCLA-117 cytotoxicity of CLEC12APOS target cells revealed no differences between the ability of AML patient-derived T cells and that of healthy donor-derived T cells to induce MCLA-117-mediated T cell activation and lysis of CLEC12APOS cells (Supplemental Figure 6c-e).
The capacity of MCLA-117 to induce CLEC12A-antigen specific lysis of primary AML blasts by resting autologous T cells collected from the same AML patient at a later time point was determined ()), patients listed in Supplemental Table 1, patients 7–9). The primary AML blast samples taken at diagnosis contained a high fraction (>80%) of blasts expressing CLEC12A ()). The AML blasts were co-cultured with autologous resting T cells taken during clinical remission at an E:T ratio of 5:1 for 48 hours. MCLA-117 induced potent CLEC12A-specific activation of CD4 and CD8 T cells as well as >70% AML blast lysis (-)). These data revealed that MCLA-117 could efficiently induce redirected CLEC12A-dependent lysis of CLEC12APOS primary AML blasts by autologous T cells.
Figure 5. MCLA-117 efficiently induces redirected lysis of CLEC12APOS primary AML blasts by autologous T cells.
Resting CD3POS T cells isolated from the peripheral blood of AML patients in clinical remission were co-cultured for 48 hours with CFSE-labelled CLEC12APOS autologous primary AML blasts collected at AML diagnosis. The E:T ratio was 5:1 and IgGs were added at a concentration of 1,000 ng/mL: MCLA-117, MockxCD3 or control IgG. The extent of AML blast lysis induced by each IgG relative to the PBS condition was quantified using flow cytometry. (a) Diagram showing stage of AML at which the AML blast samples and autologous T cells were obtained. (b) Expression of CLEC12A antigen on primary AML blast samples (AML blast defined as CD45±SSC±). The histograms show anti-CLEC12A IgG binding for each patient sample, with lymphocytes indicated in grey and AML blasts in black. The gate for CLEC12A positivity was set based on the CLEC12A-negative lymphocyte fraction; percentage CLEC12A positive blasts is given. (c-d) Activation status of AML patient CD4POS (c) and CD8POS T cells (d). (e) Cytotoxicity of CLEC12APOS primary AML blasts mediated by autologous primary T cells when incubated with indicated IgG (relative to PBS). The data shown are mean ± SE of three AML patient samples (patients 7–9, see Supplemental Table 1) tested in triplicate. Data were analysed by ANOVA relative to PBS: ** p < 0.01, *** p < 0.001.
3.8. MCLA-117 redirects lysis of primary AML blasts in diagnostic samples with low E:T ratios
We then determined the ability of MCLA-117 to induce lysis of AML blasts by autologous T cells in primary AML samples collected at diagnosis. In the primary AML samples tested (listed in Supplemental Table 1, patients 10–19), the numbers of T cells were relatively scarce compared with the numbers of AML blasts, resulting in low effective E:T ratios (1:3–1:97, ). FACS analysis revealed patient heterogeneity in terms of CLEC12A expression (). The primary AML samples were cultured for 10 days in the presence of a cytokine cocktail to support AML blast and T cell survival during the assay. After 10 days of culture, the median AML blast recovery in the absence of MCLA-117 was 41–1591%. MCLA-117 and MockxCD3 control IgG were added once at the onset of culture. T cell expansion and AML blast lysis was quantified by flow cytometry at day 7 and day 10. Not only did MCLA-117 efficiently induce CLEC12A-mediated T cell expansion (range of fold expansion 6–157), it also induced AML blast lysis at day 7 and day 10 (). At day 10, MCLA-117 had efficiently induced AML blast lysis (23–98%) in 10/11 patient samples. Although the MockxCD3 control antibody also reduced the numbers of tumour cells in some samples, in those samples the levels of T cell activation were much lower than those observed for MCLA-117 (median of 48- vs 2- fold T cell expansion by MCLA-117 vs MockxCD3). Notably, MCLA-117 was active even in AML samples with very low effector-to-target ratios ( and ). For the one AML sample in which no blast lysis was observed (patient 16), also the level of T cell expansion was only 15-fold compared to the median 48-fold T cell expansion as observed for the 10 MCLA-117 responsive samples.
Table 2. MCLA-117 induces AML blast lysis and T cell expansion in primary AML samples.
Figure 6. MCLA-117 induces AML blast killing in primary AML samples.
Primary AML patient samples taken at AML diagnosis were cultured for 10 days in medium supplemented with a cytokine cocktail to support AML blast survival. MCLA-117 or MockxCD3 antibodies were added at 200 ng/mL. For each patient, the CD45xCD33 plots show the live gate for day 0 and for each IgG at day 10. The T cells are shown in red, the AML blasts in blue (defined as CD45±SSC±), with the percentages of T cells and AML blasts indicated. Histograms at day 0 show for each patient the CLEC12A expression levels of AML blast cells. Staining using an anti-CLEC12A antibody is shown in black and staining using an isotype control antibody is shown in grey.
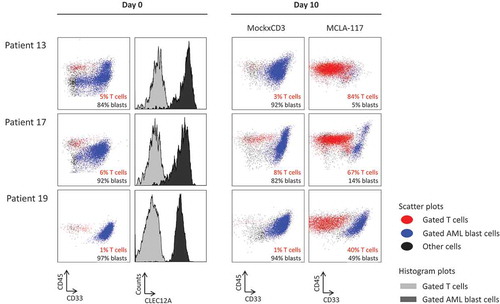
In conclusion, MCLA-117 efficiently induced CLEC12A-mediated lysis of AML blasts by T cells present in AML patient bone marrow samples, even at very low E:T ratios, and also provoked robust T cell proliferation.
4. Discussion
Here we report the characterization of MCLA-117, a full-length Fc effector silenced CLEC12AxCD3 bispecific human IgG1 antibody that has potential as a new therapeutic agent to treat all subtypes of AML by targeting CLEC12A expressed on AML blasts and leukemic stem cells. Our findings show that MCLA-117 is a potent bispecific antibody that efficiently activates and redirects T cells to lyse CLEC12A-expressing AML cells. In 10/11 primary AML patient samples, this CLEC12AxCD3 T cell engager efficiently induced CLEC12A-mediated lysis of AML blasts by redirecting resident autologous T cells. In this ex vivo cytokine-supported system, AML samples were cultured for 7–10 days in the presence of MCLA-117. This assay enabled us to establish MCLA-117 efficacy in two ways: it revealed that MCLA-117 had the capacity to induce expansion of AML patient T cells in these samples, and it showed that those MCLA-117-redirected T cells were able to redirect cytolytic activity towards AML blasts through cytolysis (presented in ). No correlation was found between MCLA-117 efficacy and CLEC12A expression on the AML blasts, or on the E:T ratios in the AML sample. We emphasize, however, that MCLA-117 was able to induce significant T cell expansion and blast lysis even in those AML samples that had low initial E:T ratios (1:45–1:97) or relatively low levels of CLEC12A expression. Comparison of the MCLA-117 versus MockxCD3 conditions in the ex vivo assay revealed that the MCLA-117-induced activity is CLEC12A antigen-mediated; robust T cell proliferation and AML blast cell lysis for MCLA-117 was observed compared to minimal activity for MockxCD3 in 9/10 primary AML samples. For the one non-responding sample that lacked both AML blast lysis and robust MCLA-117-induced T cell expansion (patient 16), we suspect that this AML patient may have had severe T cell dysfunction.
Although we used healthy donor T cells in many of our initial experiments, we also performed cytotoxicity assays using AML patient-derived T cells to test the capability of MCLA-117 to redirect T cells in a disease setting. Specific defects have been reported for AML T cells, including impaired immunological synapse formation and reduced T cell costimulatory capacity [Citation40,Citation41]. Here, we have shown that MCLA-117 can redirect AML patient-derived T cells obtained at morphologic clinical remission (n = 6) as efficiently as it redirects healthy donor-derived T cells to induce CLEC12A-specific lysis of CLEC12APOS tumour cells. Moreover, we found that MCLA-117 could effectively redirect the autologous T cells in primary de novo AML samples to lyse AML blasts. Our results advance the field of T cell engagement against the landscape of observations reported, for example the CD33xCD3 BiTE molecule, i.e. showing that T cells from de novo AML patients can induce T cell engager-mediated AML blast cytotoxicity [Citation14,Citation42]. Although our ex vivo experimental setting is different from that of these previous studies [Citation14,Citation42], we have demonstrated here that AML patient-derived T cells can be functionally activated by a full-length CD3-engaging bispecific IgG, resulting in T cell activation and proliferation, in a HSC-sparing manner and at low E:T ratios. Such functional activation of autologous T cells via MCLA-117 effectively enlarges the effector T cell compartment, thus facilitating more favourable E:T ratios for efficient eradication of AML blasts and LSCs in patients.
In normal bone marrow we showed that MCLA-117 binds in the bone marrow uniformly to GMP cells, while it only binds a minor fraction of the CMP and MEP subsets. More importantly, MCLA-117 does not bind to the CD34POSCD38NEG compartment which includes pluripotent HSCs. In line with the minor CLEC12A expression on the CMP and MEP fractions we report that MCLA-117 did not affect the development of erythroid nor megakaryocytic lineage in our in vitro CFU assays. Furthermore, although MCLA-117 strongly reduced the number of CLEC12APOS GMP cells in the ex vivo cytotoxicity assay, we observed in the follow-up CFU assays that upon MCLA-117 treatment the remaining CD34POS bone marrow cells retained their ability to give rise to both the monocytic and myelocytic lineages, permitting mono-myelocytic outgrowth arising from CD34POSCLEC12ANEG progenitor cells, including HSCs and MPPs, which are not observably impacted by the treatment of MCLA-117 [Citation24,Citation43,Citation44].
Although it is anticipated that haematological toxicity following administration of MCLA-117 to patients might be limited to neutropenia and reduction of monocyte counts [Citation30], our results demonstrate that upon MCLA-117 redirected lysis neutrophils and monocytes can re-establish from the remaining CD34POS cells. We hypothesize that the re-establishment of the normal haematopoietic compartment can arise from MEP and CMP cells which are not affected by MCLA-117, as well as from the HSC cells negative for CLEC12A expression. This potential repopulation of haematopoietic cells after treatment contrasts with the likely situation for treatments that target CD33 or CD123 [Citation14,Citation15]. Compared with CLEC12A, CD33 and CD123 are more widely expressed on normal CD34POS progenitors, including CMPs, GMPs and pluripotent HSCs [Citation28]. Our data show that CLEC12A targeting spares the normal CD123POS CMPs which are a target for CD123 directing therapies. In vivo studies in humanized mice have shown that CD33 and CD123 targeting chimeric antigen receptor (CAR)-transduced T cells induce lysis in the CD34POSCD38NEG stem cell fraction [Citation45,Citation46]. As a consequence, haematological toxicity concerns have been raised for therapies targeting CD33 and CD123. There is the possibility that these therapies could induce thrombocytopaenia, neutropaenia and anaemia, in addition to the risk of HSC eradication, which would prohibit efficient re-establishment of the normal haematopoietic compartment.
To increase the specificity of MCLA-117, the Fc region of MCLA-117 has been modified to prevent binding to FcγR and C1q, without affecting binding to the FcRn receptor. The results of our monocyte assays indeed showed that MCLA-117-induced T cell activation and associated cytokine release is restricted to CLEC12A expressing cells. The Fc silencing dampens antibody dependent cellular cytotoxicity (ADCC) and C1q-mediated complement-dependent cytotoxicity (CDC) of CD3POS and/or CLEC12APOS cells. Our data also show that MCLA-117-activated T cells lyse CLEC12APOS cells selectively (e.g. B cells and NK cells were unaffected in PBMC cultures). The fact that MCLA-117 affinity for CLEC12A (3 nM) appears to be 60-fold higher than that for CD3 (177 nM) is predicted to facilitate the preferential opsonization of the AML blasts with MCLA-117. Moreover, the low affinity for CD3 is expected to be critical in terms of minimizing the risk of cytokine release syndrome. The CD19xCD3 bispecific T cell engager blinatumomab is also known to have a much higher affinity for the tumor target than for CD3 (1.5 nM for CD19 and 267 nM for CD3) [Citation47]. A disadvantage of blinatumomab is that as a single chain Fv antibody it has a short serum half-life, and therefore requires administration by continuous infusion to achieve effective systemic levels [Citation48]. In contrast, our PK study in mice confirmed that MCLA-117, a full-length IgG1, has a normal FcRn-mediated in vivo half-life of 9–10 days. Therefore, we expect that – unlike other T cell engager concepts – effective systemic levels of MCLA-117 in patients can be achieved by weekly or biweekly intravenous administration, thereby reducing patient burden of continuous infusion.
5. Conclusion
In conclusion, these results warrant further investigation of MCLA-117 as a promising new T cell-mediated immunotherapy for all subtypes of AML. MCLA-117 is expected to selectively target myeloid blasts while sparing normal HSCs. Based on its potential to eradicate residual LSCs, MCLA-117 could also be considered for the management of minimal residual disease in AML. MCLA-117 is currently being investigated in a phase I clinical study (MCLA-117-CL01, NCT03038230) to evaluate the preliminary safety and efficacy of MCLA-117 in adult AML patients.
Author contributions
Contributions: P.F.L. designed the research, analysed the data and wrote the manuscript; B.N.H., S.T. and J.D.G. designed and performed the in vitro and ex vivo experiments and analysed the data; H.V. contributed to the ex vivo data analysis and designed and analysed the normal bone marrow in vitro data; M.L. performed the in vitro and ex vivo experiments and analysed the data; L.J.A.H., A.K. and R.C.R. contributed to CD3 discovery; R.P.D. contributed to the PK study; J.K., A.S., M.T. and T.L. contributed to the conceptual design and reviewed the manuscript; H.D. and A.B.H.B designed the research and reviewed the manuscript. All authors drafted and/or critically revised the manuscript and approved the final draft for submission.
Declaration of interest
PF van Loo, H Veninga, LJA Kaldenberg, A Kramer, RC Roovers, J de Kruif, RP Doornbos, A Sirulnik, M Throsby, T Logtenberg and ABH Bakker are employed by Merus N.V. (Utrecht, The Netherlands). Research performed by BN Hangalapura, S Thordardottir, JD Gibbins, M Leenders and H Dolstra was sponsored by Merus N.V. (Utrecht, The Netherlands). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
MCLA_117_manuscript_-_Suppl_Figures.pdf
Download PDF (728.9 KB)Acknowledgments
The authors thank Dr N. Schaap from the Department of Hematology of the Radboud University Medical Center for providing clinical samples for this study.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Arrighi JF, Soulas C, Hauser C, et al. TNF-alpha induces the generation of Langerin/(CD207)+ immature Langerhans-type dendritic cells from both CD14-CD1a and CD14+CD1a- precursors derived from CD34+ cord blood cells. Eur J Immunol. 2003;33(7):2053–2063.
- Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med. 2009;361(13):1235–1248.
- Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29(5):487–494.
- Juliusson G, Lazarevic V, Hörstedt A-S, et al. Acute myeloid leukemia in the real world: why population-based registries are needed. Blood. 2012;119(17):3890–3899.
- Oran B, Weisdorf DJ. Survival for older patients with acute myeloid leukemia: a population-based study. Haematologica. 2012;97(12):1916–1924.
- Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321(5891):974–977.
- Topp MS, Gökbuget N, Zugmaier G, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120(26):5185–5187.
- Zhang X, Yang Y, Fan D, et al. The development of bispecific antibodies and their applications in tumor immune escape. Exp Hematol Oncol. 2017;6:12.
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69(12):4941–4944.
- Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36(6):458–467.
- Riethmuller G. Symmetry breaking: bispecific antibodies, the beginnings, and 50 years on. Cancer Immun. 2012;12:12.
- Kim CH, Axup JY, Lawson BR, et al. Bispecific small molecule-antibody conjugate targeting prostate cancer. Proc Natl Acad Sci U S A. 2013;110(44):17796–17801.
- Hoseini SS, Cheung NK. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer J. 2017;7(4):e552.
- Aigner M, Feulner J, Schaffer S, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27(5):1107–1115.
- Al-Hussaini M, Rettig MP, Ritchey JK, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood. 2016;127(1):122–131.
- Testa U, Riccioni R, Militi S, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980–2988.
- Hauswirth AW, Florian S, Printz D, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest. 2007;37(1):73–82.
- Jurcic JG. What happened to anti-CD33 therapy for acute myeloid leukemia? Curr Hematol Malig Rep. 2012;7(1):65–73.
- Giles F, Estey E, O’Brien S. Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer. 2003;98(10):2095–2104.
- Feldman EJ, Brandwein J, Stone R, et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J Clin Oncol. 2005;23(18):4110–4116.
- Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res. 2011;17(20):6417–6427.
- Lapusan S, Vidriales MB, Thomas X, et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Invest New Drugs. 2012;30(3):1121–1131.
- Bakker AB, van Den Oudenrijn S, Bakker AQ, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004;64(22):8443–8450.
- van Rhenen A, van Dongen GA, Kelder A, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110(7):2659–2666.
- Zhao X, Singh S, Pardoux C, et al. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica. 2010;95(1):71–78.
- Larsen HO, Roug AS, Just T, et al. Expression of the hMICL in acute myeloid leukemia-a highly reliable disease marker at diagnosis and during follow-up. Cytometry B Clin Cytom. 2012;82(1):3–8.
- Kikushige Y, Miyamoto T. TIM-3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. Int J Hematol. 2013;98(6):627–633.
- Taussig DC, Pearce DJ, Simpson C, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092.
- Lu H, Zhou Q, Deshmukh V, et al. Targeting human C-type lectin-like molecule-1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew Chem Int Ed Engl. 2014;53(37):9841–9845.
- Leong SR, Sukumaran S, Hristopoulos M, et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. 2017;129(5):609–618.
- Zheng B, Yu SF, Del Rosario G, et al. An anti-CLL-1 antibody-drug conjugate for the treatment of acute myeloid leukemia. Clin Cancer Res. 2019;25(4):1358–1368.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212.
- de Kruif J, Kramer A, Visser T, et al. Human immunoglobulin repertoires against tetanus toxoid contain a large and diverse fraction of high-affinity promiscuous V(H) genes. J Mol Biol. 2009;387(3):548–558.
- Gunasekaran K, Pentony M, Shen M, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–19646.
- De Nardis C, Hendriks LJA, Poirier E, et al. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J Biol Chem. 2017;292(35):14706–14717.
- Marshall AS, Willment JA, LinHH, et al. Identification and characterization of a novel human myeloid inhibitory C-type lectin-like receptor (MICL) that is predominantly expressed on granulocytes and monocytes. J Biol Chem. 2004;279(15):14792–14802.
- Lahoud MH, Proietto AI, Ahmet F, et al. The C-type lectin Clec12A present on mouse and human dendritic cells can serve as a target for antigen delivery and enhancement of antibody responses. J Immunol. 2009;182(12):7587–7594.
- Leonard EJ, Yoshimura T. Neutrophil attractant/activation protein-1 (NAP-1 [interleukin-8]). Am J Respir Cell Mol Biol. 1990;2(6):479–486.
- Matsushima K, Morishita K, Yoshimura T, et al. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of MDNCF mRNA by interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167(6):1883–1893.
- Wendelbo O, Nesthus I, Sjo M, et al. Functional characterization of T lymphocytes derived from patients with acute myelogenous leukemia and chemotherapy-induced leukopenia. Cancer Immunol Immunother. 2004;53(8):740–747.
- Le Dieu R, Taussig DC, Ramsay AG, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. 2009;114(18):3909–3916.
- Krupka C, Kufer P, Kischel R, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood. 2014;123(3):356–365.
- Notta F, Zandi S, Takayama N, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351(6269):aab2116.
- Bill M, B van Kooten Niekerk P, S Woll P, et al. Mapping the CLEC12A expression on myeloid progenitors in normal bone marrow; implications for understanding CLEC12A-related cancer stem cell biology. J Cell Mol Med. 2018;22(4):2311–2318.
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28(8):1596–1605.
- Kenderian SS, Ruella M, Shestova O, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–1647.
- Cheadle EJ. MT-103 Micromet/MedImmune. Curr Opin Mol Ther. 2006;8(1):62–68.
- Klinger M, Brandl C, Zugmaier G, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119(26):6226–6233.