
1. Introduction
Cancer is a moving target, and targeted therapy must ultimately deal with the evolution of the disease. This is because evolution is the substrate for development of resistance to targeted therapy [Citation1]. A successful therapeutic strategy is one that can tackle and ultimately eliminate the evading phenotype. Thus, to steer cancer evolution for therapeutic purposes, one must first note that targeted therapy imposes selection pressure and resistant phenotypes prevail in a context of clonal heterogeneity. The quest for complete cure makes it imperative to control the evolutionary fate of the tumor.
We focus on the problem of cornering the evolving phenotype promoted by T cell checkpoint blockade [Citation2–Citation4]. These antibodies unleash the anti-tumor adaptive immune response by blocking an off-switch T-cell receptor whose natural ligand is secreted by the tumor. In this way, checkpoint blockers turn off the negative signal in tumor-induced immunosuppression. This immunotherapy constitutes possibly the most auspicious anticancer treatment to date. In this context, cancer evolution results in immunoediting, and the evolving phenotype may be steered by targeting signaling pathways that control the modulation of the adaptive immune response, as shown in this editorial.
2. Enhancing therapeutic impact by engineering drug-induced susceptibility to checkpoint blockade
Recent results revealed that checkpoint blockers such as pembrolizumab (Keytruda) may be used to treat almost any solid tumor provided the tumor has a sufficiently high mutational load [Citation3]. These immune-stimulating phenotypes typically stem from a DNA mismatch repair (MMR) deficiency that arises in rare genetic conditions, such as the Lynch syndrome [Citation4].
We recently observed that a drug-induced MMR deficiency, fostering susceptibility to checkpoint blockade, may be achievable [Citation5–Citation7], considerably enhacing the interest of such immunotherapies as generic cancer treatments. To induce susceptibility to a checkpoint blocker utilizing a kinase inhibitor [Citation5] requires that a drug is identified that impairs a major signaling pathway recruited to promote MMR [Citation6]. So it happens that the α-isoform of anticancer target phosphoinositide 3-kinase (PI3Kα) is also a key transducer in MMR-related signaling, playing a metabolic role so that its inhibition results in nucleotide depletion [Citation6]. This depletion is a consequence of reduced flux through glycolysis that results in a decrease in R5P needed for base ribosylation. This metabolic deficiency leads to ultimately compromising DNA synthesis in rapidly proliferating tumor cells that become subject to DNA replication stress as they enter the S phase. Thus, the metabolic effect of PI3Kα-inhibition results in error-prone DNA replication, likely to be specific to tumor cells. This is because tumor cells rely more heavily on de novo nucleotide biosynthesis than normal cells and because of their overloaded reliance on MMR. Tumor specificity as well as its central role in blocking cancer-related signaling pathways advocates for the therapeutic benefit of a PI3Kα inhibitor as inducer of susceptibility to checkpoint blockade [Citation5].
However effective the combination of targeted therapy and immunotherapy, a major caveat arises as the tumor undergoes immunoediting, which may ultimately result in an immune-evading phenotype [Citation8,Citation9]. As subsequently shown, addressing this problem requires engineering therapeutic conditions that can funnel immunoediting into a tumor phenotype susceptible to elimination in a second-line therapeutic intervention.
3. Therapeutic duality of MMR deficiency: immune stimulant and enabler of immune evasion
By significantly increasing antigen diversity, genetic [Citation3,Citation4] or drug-induced [Citation5] conditions that cause MMR deficiency enhance the stimulation of the adaptive immune response and thus promote the therapeutic susceptibility of the tumor. There is, however, an adverse effect of the MMR deficiency that mars its therapeutic value: By creating high mutational diversity in the tumor, the Lynch and similar conditions constitute human models for tumor immunoediting, an evolutionary outcome ultimately leading to the immune-evading phenotype [Citation8,Citation9]. In other words, immune surveillance of tumor cells with high mutational diversity imposes selection pressure upon them, enabling adaptive selection of tumor cell subpopulations that suppress or do not trigger T-cell activation. A key to complete cure is to funnel the immunoediting process toward a predictable subpopulation that can be eliminated with a second-line therapy.
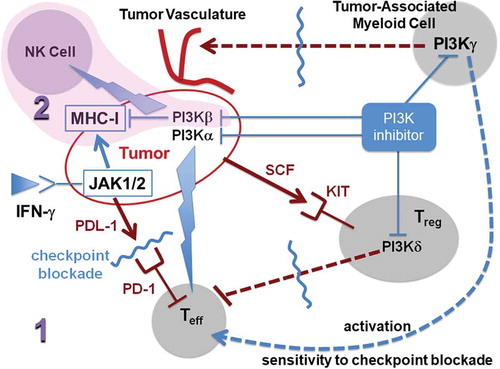
Once the hypermutated phenotype is induced through PI3Kα inhibition [Citation5] or epigenetic drugs [Citation7], the tumor has two broad avenues to curb the selection pressure imposed by the adaptive immune surveillance unleashed by the checkpoint blocker [Citation8,Citation9]: a) impairment or downregulated expression of the antigen-presenting apparatus in tumor cells, and b) recruitment of regulatory T cells (Tregs) and other modulators of the adaptive immune response (). Thus, we propose to block evasion route b through inhibition of the γ and δ isoforms of PI3K [Citation10], so immunoediting funnels the evading phenotype into the first route. Subsequently, the selected resistant subpopulation of tumor cells may get wiped out in a second-line treatment based on a pan-PI3K inhibitor [Citation11] ().
Figure 1. Schematic description of a two-stage (1, 2) sequential therapeutic strategy designed to steer the immunoedited tumor into a phenotype that evades checkpoint immunotherapy but becomes susceptible to elimination through NK-mediated lysis. The first-line treatment (1) combines blockade of checkpoint PD-1 receptor in effector T-cells (Teff) with inhibition of α, γ, and δ isoforms of phosphoinositide 3-kinase (PI3K). Inhibition of PI3Kα promotes mutational diversity enabling immunoediting of the tumor phenotype, while inhibition of the γ and δ isoforms guarantees that the route to evade adaptive immune surveillance would be one that leaves the tumor fully susceptible to innate immune attack by NKs. The first-line treatment 1 promotes a MHC-I-negative or MHC-I-deficient phenotype with an impaired antigen-presentation apparatus that evades the adaptive immune response unleashed by the checkpoint (PD-1) blocker. The selected immune-evading subpopulation may be subsequently eliminated by the second-line therapy (2, shaded) that stimulates the innate immune response through enhancement of NK-mediated lysis of the selected (MHC-I-deficient or MHC-I-negative) tumor cells. The stimulation of NK-mediated lysis is the result of inhibitory activity against PI3Kβ. The thunder symbol represents drug-induced stimulation. A tumor-induced suppression of Teff – activation occurs through secretion of PDL-1, the natural ligand of T-cell receptor PD-1. Tumor recruitment of regulatory T-cells (Tregs) becomes operative, for example, through secretion of stem-cell factor (SCF) the natural ligand of the KIT receptor expressed by Tregs.
4. Targeted therapy to corner the immune-evading tumor phenotype
To achieve complete cure, one needs to be able to channel the immunoediting process into a predictable state that may be eliminated. Within a therapeutic context where T cells are subject to checkpoint blockade, a highly immunogenic tumor resulting from treatment with a PI3Kα inhibitor will be under intense T-cell-mediated surveillance. The resulting selection pressure will eventually trigger immunoediting, leading to the selection of tumor cell subpopulations that evade the adaptive immune arm [Citation8,Citation9,Citation12]. As documented, the tumor may pursue two distinctive evasion strategies:
A direct strategy, where the major histocompatibility complex-class I (MHC-I) is impaired, dodging the adaptive immune attack through a compromise of the antigen-presentation apparatus [Citation9,Citation12]. This impairment may be achieved through mutation of beta-2 microglobulin, a key component of MHC-I [Citation8], or by impairing the interferon-gamma (IFN-γ)-mediated signaling that stimulates MHC-I expression and secretion of PDL-1 [Citation12]. The latter possibility is achieved through mutation of JAK1/2, key signal transducers in the IFN-γ stimulation of MHC-I expression.
An indirect strategy, whereby the tumor cell recruits immune-suppressing regulatory T cells (Tregs) and other modulators of the adaptive immune response () by promoting expression of regulatory hallmark genes such as Foxp3 in T lymphocytes [Citation8].
As now shown, by first administering a suitable isoform-sparing PI3K inhibitor [Citation10] in combination with the checkpoint blocker, it is possible to engineer a tumor immune microenvironment that will force immunoediting to adopt the direct evasion strategy. In this way, only MHC-I-impaired or MHC-I-negative tumor cells will prevail and then they will get eliminated by promoting susceptibility of this subpopulation to the innate arm of the immune system [Citation11].
The targeted therapy companion to checkpoint blockade is required to have strong anti-PI3Kα activity to promote susceptibility to checkpoint immunotherapy [Citation5]. To identify the other class-I isoforms of PI3K that should be targeted, we note the following [Citation10] ():
Inhibition of PI3Kδ in Tregs disrupts their suppressive activity on T cells and enables anti-tumor activation of CD8+ T cells.
Inhibition of PI3Kγ in tumor-associated myeloid cells (TAMCs) impedes induction of tumor angiogenesis and restores sensitivity to immune checkpoint blocking antibodies.
Inhibition of PI3Kβ in tumor cells promotes downregulation of MHC-I and upregulation of natural killer (NK)-activating ligands, generating a strong tumor-cell susceptibility to NK-mediated lysis and interferon-γ response [Citation11].
These observations inform us that to steer immunoediting toward selection of a tumor-cell subpopulation that adopts the direct immune-evasion strategy (selection of MHC-I-negative or MHC-I-deficient cells), we need a first-line therapy that combines the checkpoint blocker with a targeted therapy that curtails recruitment of immune-modulating components including TAMCs and Tregs. Thus, the targeted therapy in first-line treatment must be a β-isoform-sparing class I-PI3K inhibitor highly active (nanomolar or subnanomolar affinity) against the α, γ, and δ isoforms (p110α, p110γ, p110δ). A suitable first-line agent for such targeted therapy is taselisib (GDC0032) with differential high activity against the α, γ, and δ isoforms vis-à-vis the β-isoform [Citation10]. The second-line treatment is geared at eliminating the residual subpopulation representing the phenotype that directly evaded the adaptive immune response within the evolutionary context framed by the first-line combination treatment. As indicated, this may be achieved by promoting the NK-mediated lysis of the selected MHC-I-negative or MHC-I-deficient tumor cells [Citation11]. Thus, the second-line therapy consists of treatment with a pan-PI3K inhibitor with significant (nanomolar) activity against the PI3K β-isoform (p110β) [Citation11]. Viable inhibitor candidates for the second-line monotherapy include omipalisib (GSK2126458) [Citation10] or wortmannin [Citation11].
The rationale for the two-stage combination is based on the need to corner of the immune-evading phenotype into a state that promotes NK-mediated cytolysis. This requires that impairment of the antigen-presentation machinery, be it through beta-2 microglobulin or JAK1/2 mutation [Citation8,Citation12], becomes the sole avenue to avoid adaptive immune surveillance.
The combination of checkpoint immunotherapy with a pan-PI3K inhibitor would not yield satisfactory results in first-line treatment because activity against β-isoform has a net effect of downregulating MHC-I expression and, thus, renders fruitless the stimulus on the adaptive arm of the immune system promoted by inhibition of PI3Kα. Thus, complete elimination of the tumor demands a two-stage strategy (), with β-isoform-sparing PI3K inhibitor in combination with checkpoint blocker as first-line treatment, followed by a pan-PI3K inhibitor operative only after immunoediting enabled evasion from the adaptive arm of immune surveillance.
The success of the evolution-steering therapeutics outlined is contingent on the fulfillment of basic requirements on the tumor immunological microenvironment that would guarantee its susceptibility to the specified drug treatment, most notably:
The development of highly potent and selective (isoform-sparing) PI3K inhibitory drugs capable of blocking TAMCs, Tregs and MHC-I expression in accord with the therapeutic schedule proposed
A tumor microenvironment with immune modulatory properties predominantly arising from recruitment of TAMCs and Tregs
The availability of innate effector cells (especially NKs) capable of infiltrating the tumor microenvironment
A lack of resistance of innate immune effectors to PI3Kβ inhibitors so that an encounter with an MHC-I-negative tumor cell is not futile because of a lack of effector activation or inability to be recruited or proliferate
5. Expert opinion
In one of the most exciting developments in anticancer therapy, checkpoint blockers have been shown to be effective against almost any solid tumor expressing the checkpoint ligand PDL-1 provided it harbors a high immune-stimulating mutational load [Citation3,Citation4]. This hypermutability arises either naturally from genetic conditions that harbor a DNA mismatch repair (MMR) deficiency or may get generically induced by a PI3Kα inhibitor [Citation5] or an epigenetic drug [Citation7]. Yet, the checkpoint blocker alone or combined with the targeted therapy cannot always provide complete cure because immunoediting is likely to arise, providing a route of evasion for a selected tumor-cell subpopulation [Citation8,Citation9,Citation12]. Thus, the MMR deficiency becomes a double-edged sword that stimulates the adaptive immune system but also enables the onset of the immune-evading phenotype arising from natural selection.
As shown [Citation5], the combination of a PI3Kα-inhibitor with a checkpoint blocking antibody holds huge potential as a universal first-line treatment for most solid tumors. Other combinations of immune checkpoint blockers and isoform-specific PI3K inhibitors, in particular the PI3Kγ inhibitor suppressing immune-modulating myeloid cells, have also shown considerable promise [Citation13]. However, cancer evolves and resistance develops, and so the ultimate goal to achieve complete cure is to identify a second-line treatment to tackle the immune-evading phenotype. More precisely, complete cure requires elimination of the selected tumor-cell subpopulation that is resistant to the adaptive immune response unleashed by the checkpoint blockade.
To achieve this over-arching goal, a bioinformatics and systems analysis is required to identify the evolutionary routes of immune evasion that the tumor has at its disposal. The elimination of the immune-evading phenotype remains a standing challenge, and the biggest hurdle is to learn how to steer the evading phenotype toward a state susceptible to elimination. This steering of immunoediting may be achieved by targeting signaling pathways implicated in tumor-induced suppression of the immune response. To that effect, we rationalized a strategy to steer cancer evolution toward a predictable outcome with a significant chance of being eliminated. We show that a first-line therapy combining the checkpoint blocker with a β-sparing PI3K inhibitor against α, γ, δ isoforms funnels the immunoedited tumor into a state susceptible to elimination by a second-line therapy, wherein a pan-PI3K inhibitor promotes the innate immune attack on the selected MHC-I-deficient or MHC-I-negative tumor cells.
Further analysis of evolutionary immune evasion will be required first based on humanized mice models with good compatibility between tumor and stroma and capable of faithfully reproducing key components of the human tumor microenvironment [Citation14,Citation15]. These studies should inspire and complement clinical evaluations of the kinase inhibitor-checkpoint blocker combinations proposed.
The field of humanized mice models may well offer the technological platform for drug discovery essential to hit the moving targets arising as cancer develops resistance to immunotherapy.
Declaration of interest
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Turajlic S, Swanton C. Implications of cancer evolution for drug development. Nature Revs Drug Dis. 2017;16:441–442.
- Jacquelot N, Roberti MP, Enot DP, et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nature Commun. 2017;8:592.
- Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520.
- Le DT, Durham JN, Smith KN, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413.
- Fernández A. Engineering tumor hypersusceptibility to checkpoint immunotherapy. Trends Cancer. 2017;3:675–677.
- Juvekar A, Hu H, Yadegarynia S, et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc Natl Acad Sci USA. 2016;113:4338–4447.
- Jin B, Robertson KD. DNA methyltransferases (DNMTs), DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3–29.
- Echterdiek F, Janikovits J, Staffa L, et al. Low density of FOXP3-positive T cells in normal colonic mucosa is related to the presence of beta2-microglobulin mutations in Lynch syndrome-associated colorectal cancer. Oncoimmunol. 2016;5:e1075692.
- Garrido F, Aptsiauri N, Doorduijn EM, et al. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51.
- Pons-Tostivint E, Thibault B, Guillermet-Guibert J. Targeting PI3K signaling in combination cancer therapy. Trends Cancer. 2017;3:454–469.
- Bommarito D, Martin A, Forcade E, et al. Enhancement of tumor cell susceptibility to natural killer cell activity through inhibition of the PI3K signaling pathway. Cancer Immunol Immunother. 2016;65:355–366.
- Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829.
- De Henau O, Rausch M, Winkler D, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443–447.
- Rongvaux A, Willinger T, Martinek J, et al. Development and function of human innate immune cells in a humanized mouse model. Nature Biotechnol. 2014;32:364–372.
- Wang M, Yao LC, Cheng M, et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2017. Published online Nov 16. doi:10.1096/fj.201700740R