
1. Introduction
Pancreatic cancer, mainly pancreatic ductal adenocarcinoma (PDA), is currently the third-leading cause of cancer-related death in the United States and has a dismal prognosis, which is partially due to its resistance to conventional chemotherapy and radiotherapy. Immune therapy has been demonstrated to be effective in many malignancies, but its efficacy in treating PDA has been disappointing. One important reason for this lack of efficacy is the immunosuppressive tumor microenvironment (TME) that prevents effector T cells from infiltrating, being activated in, and eliminating tumor cells [Citation1]. Researchers are investigating various signaling pathways to identify potential therapeutic targets that can convert the TME to immunoresponsive status to improve immune therapy for PDA.
2. STING in innate immunity
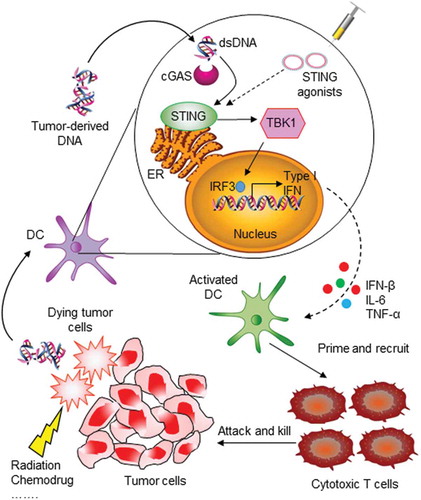
Innate immune sensing and type I interferon (IFN) production in the TME are required for tumor-induced T-cell priming and tumor cell killing. The discovery of the stimulator of interferon genes (STING) controlled innate immune pathway has made it an attractive therapeutic target for cancer immune therapy. STING is a transmembrane protein localized to the endoplasmic reticulum (ER) that is activated by direct binding of exogenous or endogenous cyclic dinucleotides (CDNs). STING activation triggers downstream signaling cascades, such as the TANK-binding kinase 1/interferon regulatory factor 3 (TBK1/IRF3), nuclear factor-κB (NF-κB), and signal transducer and activator of transcription 6 (STAT6) signaling pathways, which result in robust production of type I IFNs and other proinflammatory cytokines that participate in host immune defense. The STING pathway has a conserved host-protection mechanism that protects against pathogen infection. However, in the tumor therapeutic setting, the double-stranded DNA released by dying tumor cells can be taken up by stromal cells, mainly dendritic cells (DCs) and macrophages. These cytosolic DNA fragments will be transduced to structurally distinct CDNs by a host cyclic GMP-AMP synthase (cGAS), which functions as a cytosolic receptor for pathogen- or mislocated self-DNA. Consequently, activation of the cGAS-STING mediated pathway will produce large amount of type I IFNs and cytokines to prime antigen-presenting cells (APCs) and macrophages, and finally promote adaptive T-cell response against pathogen infection or tumor cells (). Significantly, STING can directly transmit retinoic acid inducible gene-I (RIG-I)-mitochondrial antiviral-signaling protein (MAVS)-mediated signals in cytosolic RNA sensing, making it playing a central role in the crosstalk between cytoplasmic RNA and DNA sensing pathways [Citation2]. Accordingly, RIG-I agonists have also been exploited for antiviral and antitumor immune therapies [Citation3–Citation5]. Moreover, since cGAS is the upstream enzyme of STING, direct modulation of cGAS activity may be an efficient approach to regulate cGAS-STING signaling, and significant efforts have been made to identify cGAS activators or inhibitors for cancer immunotherapy or autoimmune disease therapy [Citation6,Citation7].
Figure 1. STING pathway in innate immune sensing of tumor and induction of antitumor immunity. When tumor cells are killed by external influences, double-stranded DNA (dsDNA) fragments are released and then taken up by dendritic cells (DCs). Cytosolic DNA is recognized and processed by cyclic GMP-AMP synthase (cGAS) and then transduced to activate STING in the endoplasmic reticulum (ER). STING activation leads to phosphorylation of TBK1, which in turn phosphorylates transcription factor IRF3 in the cell nucleus. This results in production of type I IFN and other chemokines, which activate antigen-presenting cells and promote T-cell priming. Finally, tumor-specific T cells are recruited to tumor sites and kill tumor cells. The STING pathway can also be activated by therapeutic administration of STING agonists.
3. STING agonists and immune therapy for PDA
To activate the STING pathway for immune therapy, tremendous efforts have been made on screening and developing various STING agonists (). The small molecule 5,6-dimethylxanthenone-4-acetic acid (DMXAA) initially showed potent antitumor activity in many mouse tumor models, but it failed in clinical trials when combined with chemotherapy. Recent structure studies showed that DMXAA is a direct mouse STING activator but it cannot bind to human STING protein, which explains its lack of clinical activity in humans [Citation8]. In addition, unlike in the mouse genome, polymorphism exists in human STING alleles, which results in different STING genotypes in the general population. Therefore, in the design and screening of STING agonists, these findings should be taken into consideration.
Table 1. Representative STING agonists.
The natural CDNs, such as bis-(3ʹ-5ʹ)-cyclic dimeric guanosine monophosphate (cyclic di-GMP) and bis-(3ʹ-5ʹ)-cyclic dimeric adenosine monophosphate (cyclic di-AMP), were first explored for their anticancer immune potential as direct activators of the STING signaling pathway. Cyclic di-GMP, used as an adjuvant for cancer vaccination, showed improved antitumor effect in melanoma and breast cancer models [Citation9,Citation10]. Intratumoral administration of cyclic di-GMP to glioma-bearing mice enhanced type I IFN response and prolonged mice survival [Citation11]. The intravenous administration of liposome-encapsulated cyclic di-GMP into mice significantly induced type I IFN production and activation of natural killer cells in a lung metastatic melanoma mouse model and resulted in an antitumor effect [Citation12].
Due to human STING allele polymorphism, certain STING variants are refractory to bacterial CDNs with canonical 3ʹ-5ʹ phosphate bridge linkages. Researchers screened many synthetic CDN derivatives and found that some noncanonical mammalian CDNs with 2ʹ-5ʹ linkages or mixed 2ʹ-5ʹ, 3ʹ-5ʹ linkages may have better binding affinity for human STING and elevated immunostimulatory capacity. Synthetic 2ʹ,3ʹ-cGAMP analogs were explored as vaccine adjuvants in cancer immune therapy. When coformulated with an irradiated granulocyte–macrophage colony-stimulating factor (GM-CSF)-secreting whole-cell vaccine in the form of STINGVAX, the synthetic CDNs increased the antitumor efficacy in several tumor models tested, including a murine Panc02 metastatic pancreatic cancer model [Citation13].
Given that STING agonist treatment can significantly improve the immunosuppressive TME and help to generate or potentiate T-cell checkpoint therapy induced systemic antitumor immunity in preclinical models of different types of cancers [Citation14,Citation15], there have been attempts to combine STING agonistic immune therapy with other therapies for pancreatic cancer, and these attempts have had promising results. STINGVAX-treated mice were characterized by enhanced tumor-infiltrating CD8+ IFNγ+ T cells along with marked up-regulation of programmed death ligand 1 (PD-L1). STINGVAX combined with PD-1 blockade induced regression of established tumors that did not respond to PD-1 blockade alone [Citation13]. RP, RP dithio 2ʹ-3ʹ CDN molecules combined with CT-guided radiotherapy showed synergistic anticancer immune response in local and distal tumors in a murine pancreatic cancer model. This synergistic effect produces a two-phase response, in which T-cell–independent and TNFα-dependent hemorrhagic necrosis happens early in the treatment, followed later by CD8+ T-cell–dependent control of residual disease [Citation16]. In another study, researchers used a biopolymer scaffold to co-deliver chimeric antigen receptor engineered T cells (CAR T cells) and STING agonists directly to pancreatic tumors in mice [Citation17]. Data showed that combined delivery of CAR T cells and STING agonists resulted in synergistic activation of host APCs and robust tumor-specific host immune responses, which eliminate local tumors and trigger systemic host antitumor immunity powerful enough to prevent untreated distant metastases.
The STING pathway appears to be orthogonal to certain immune regulatory pathways, which may indicate a potential additive effect of combined STING agonist with other immune therapy. For example, a study demonstrated that CD40 stimulation caused a clonal expansion of T cells in a KPC mouse pancreatic cancer model, and the addition of chemotherapy optimized myeloid activation and T-cell function [Citation18]. Interestingly, the efficacy of CD40 and chemotherapy does not require STING or Toll-like receptors in innate immune pathways. Thus, combined CD40 and STING agonistic immune therapy await further studies.
4. Expert opinion
Immune therapy is a rapidly growing field and represents a paradigm shift in cancer treatment. However, the efficacy of immune therapy for pancreatic cancer is not yet satisfactory. Understanding the low immunogenic nature of pancreatic cancer and finding therapeutic targets to increase tumor-specific immune responses are urgently needed to improve outcomes in patients with PDA.
An ideal therapeutic strategy to propel efficient host antitumor immunity should bring together three key components: (1) a proper method, e.g. irradiation, chemotherapy, or cytotoxic T cells, that destroys tumor cells; (2) a large amount of tumor antigens released from damaged and killed cancer cells, which become available for APCs to present; and (3) high concentrations of stimulants that activate these APCs along with other tumor-reactive immune cells to create an activated immune microenvironment that recruits more effector T cells to tumor sites. STING activation–induced type I IFN and cytokine production make the bridge between the innate immune sensing of cancer and an adaptive immune response that controls tumor growth.
Although STING agonists showed promising therapeutic effects in many preclinical studies, there are still issues to be addressed before translating the findings to clinical use. The first issue is how to determine the appropriate dosage of STING agonists. Considering the natural role of the STING pathway, overactivation or sustained activation of this pathway can induce severe adverse effects, including autoimmunity, uncontrolled inflammation, and direct tissue toxicity. For example, a recent study showed that STING activation in the pancreas promoted inflammation in a mouse acute pancreatitis model [Citation19]. Another study showed that intratumoral CDN/checkpoint inhibitor combined therapy invoked substantial ulceration at the injection site and that reduced CDN dosing could preserve tissue integrity [Citation14]. These data indicate the importance of understanding the detailed mechanisms and spatiotemporal regulation of the STING pathway in cancers.
The second issue is to understand the status of STING polymorphisms and select proper STING agonists. Analysis of single nucleotide polymorphisms in STING revealed that R71H-G230A-R293Q (HAQ) occurs in 20.4%, R232H in 13.7%, G230A-R293Q (AQ) in 5.2%, and R293Q in 1.5% of the human population [Citation20]. STING variants have been reported to influence STING-dependent innate immune signaling and susceptibility to pathogen infection, contribute to severe inflammatory disorders, and affect the responsivity to various STING agonists [Citation21], highlighting the rationale for personalized STING immune therapy.
The third issue is how to proper formulate and deliver STING agonists. Due to the low molecular weight of common CDNs, locally administered unformulated CDNs are rapidly distributed to the bloodstream instead of being transported to draining lymph nodes, where they are taken up by immune cells. Therefore, the cytosolic drug concentration is low and a systemic inflammatory toxicity develops. Using a proper formulation and delivery system to deliver CDNs into the cytosol of immune cells may overcome this problem. One study used lipid nanoparticles as carriers to deliver cyclic di-GMP and gained increased drug concentration in lymph node APCs and enhanced immune responses [Citation22]. Another study tested an implantable biopolymer device to co-deliver CAR T cells and STING agonists directly to the surface of mouse pancreatic tumors. Results showed efficient immune responses to elimination of tumor cells [Citation17]. With the progress of biotechnology and biomaterial, new delivery strategies may be developed to increase the potency of STING agonists and decrease their toxicity.
The fourth issue is the stromal effects on immune therapy. The desmoplastic stroma is a key component of the immunosuppressive TME and a physical barrier to effective therapies for pancreatic cancer. Using stroma modulators to remodel the TME may improve the efficacy of immune therapy. Pegylated recombinant human hyaluronidase (PEGPH20), currently in clinical trial, can degrade stromal hyaluronic acid to improve intratumoral blood flow and drug delivery [Citation23]. Vitamin D is reported to modulate both innate and adaptive immune systems [Citation24], and vitamin D receptor (VDR) is expressed on stromal cells. VDR activation in the pancreas reduced fibrosis and inflammation and improved the response of PDA to gemcitabine in preclinical models [Citation25]. Thus, a combination of stroma modulator and immune regulator(s) such as STING agonists, may represent a future direction of immune therapy for PDA.
In summary, owning to its central role in innate immune pathways, STING has emerged as a promising target for immune therapy for various cancers, including pancreatic cancer. However, more studies are needed to advance this novel therapy to clinical application
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
We thank Tamara Locke from the Department of Scientific Publications for her critical reading of, and valuable input to, this manuscript.
Additional information
Funding
References
- Stromnes IM, Hulbert A, Pierce RH, et al. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol Res. 2017;5:978–991.
- Zevini A, Olagnier D, Hiscott J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017;38:194–205.
- Bhoopathi P, Quinn BA, Gui Q, et al. Pancreatic cancer-specific cell death induced in vivo by cytoplasmic-delivered polyinosine-polycytidylic acid. Cancer Res. 2014;74:6224–6235.
- Elion DL, Jacobson ME, Hicks DJ, et al. Therapeutically active RIG-I agonist induces immunogenic tumor cell killing in breast cancers. Cancer Res. 2018.
- Liu Y, Goulet ML, Sze A, et al. RIG-I-mediated STING upregulation restricts herpes simplex virus 1 infection. J Virol. 2016;90:9406–9419.
- Hall J, Brault A, Vincent F, et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017;12:e0184843.
- Vincent J, Adura C, Gao P, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750.
- Conlon J, Burdette DL, Sharma S, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190:5216–5225.
- Chandra D, Quispe-Tintaya W, Jahangir A, et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901–910.
- Wang Z, Celis E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol Immunother. 2015;64:1057–1066.
- Ohkuri T, Ghosh A, Kosaka A, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. 2014;2:1199–1208.
- Nakamura T, Miyabe H, Hyodo M, et al. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J Control Release. 2015;216:149–157.
- Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52.
- Ager CR, Reilley MJ, Nicholas C, et al. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. 2017;5:676–684.
- Sallets A, Robinson S, Kardosh A, et al. Enhancing immunotherapy of STING agonist for lymphoma in preclinical models. Blood Adv. 2018;2:2230–2241.
- Baird JR, Friedman D, Cottam B, et al. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res. 2016;76:50–61.
- Smith TT, Moffett HF, Stephan SB, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest. 2017;127:2176–2191.
- Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 2016;15:2719–2732.
- Zhao Q, Wei Y, Pandol SJ, et al. STING signaling promotes inflammation in experimental acute pancreatitis. Gastroenterology. 2018;154:1822–1835 e2.
- Yi G, Brendel VP, Shu C, et al. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One. 2013;8:e77846.
- Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–770.
- Hanson MC, Crespo MP, Abraham W, et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125:2532–2546.
- Wong KM, Horton KJ, Coveler AL, et al. Targeting the tumor stroma: the biology and clinical development of pegylated recombinant human hyaluronidase (PEGPH20). Curr Oncol Rep. 2017;19:47.
- Szymczak I, Pawliczak R. The active metabolite of vitamin D3 as a potential immunomodulator. Scand J Immunol. 2016;83:83–91.
- Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93.