
Triple-negative breast cancer (TNBC) is the most aggressive subtype of breast cancer, characteristically associated with worse outcomes compared to other breast cancer subtypes. Cytotoxic chemotherapy combinations are the mainstay treatment for TNBC but paradoxically, following initial high rates of response to chemotherapy, many patients relapse within 2.5 years with refractory cancers. TNBC is a complex heterogeneous disease that can be further categorized into six subtypes, with a diverse number of genetic drivers that occur at low frequencies. The key to developing successful targeted therapies in the future may be in focusing on drug targets that transcend TNBC heterogeneity. In contrast to ER+ and HER2+ breast cancer, in which kinase inhibitors such as palbociclib and trastuzumab, respectively, have had remarkable success in prolonging patient survival, no kinase inhibitor has been approved for the treatment of TNBC. However, the mitogen-activated protein kinase (MAPK) signaling pathway is highly activated in TNBC and promising data now indicate that p90 ribosomal S6 kinase 2 (RSK2), a downstream member of the MAPK signaling pathway, is a validated target for TNBC. The challenge ahead is to develop RSK inhibitors that have favorable pharmacokinetic properties, which will allow for their evaluation in clinical trials for patients with TNBC.
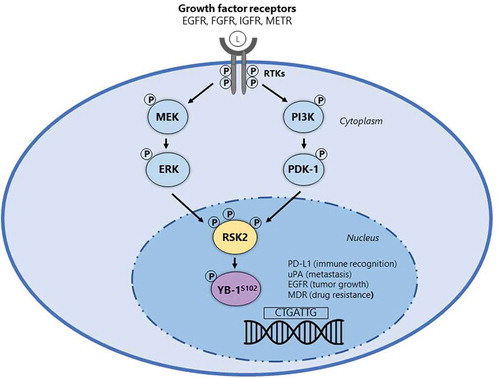
TNBC is characterized histologically by a lack of ER, PR and HER2 expression and a high degree of heterogeneity with characteristics overlapping with basal-like breast cancer (BLBC) [Citation1]. As such, TNBC and BLBC are often terms that are used interchangeably. The functional dependency of TNBC on RSK2 was discovered through unbiased kinome-wide screens across a heterogeneous panel of 34 breast cancer cell lines [Citation2]. This siRNA screen of more than 700 genes identified RSK2 as the most important drug target for TNBC relative to other breast cancer subtypes. The critical role of RSK2 in TNBC was then validated through work by Stratford et al. (2012) in which siRNA was used to silence RSK2, leading to TNBC growth inhibition and apoptosis induction in vitro and tumor growth suppression in mice [Citation3]. Pharmacological inhibition of RSK2 with the RSK inhibitor SL0101 further validated RSK2 as a TNBC target in xenografts in mice [Citation4].The four RSK isoforms are highly conserved Ser/Thr downstream effector kinases in the MAPK cascade, that have important overlapping and unique roles in diverse cellular functions. In particular, RSK2 is a uniquely positioned kinase because it intersects two major signaling cascades: the MAPK pathway and the PI3K pathway (). The reason that RSK2 is so important for TNBC may have to do with its ability to control transcription factors through Ser/Thr phosphorylation events. External growth factor ligands initiate signaling through ERK and PDK1 to activate RSK2 (). Following activation, RSK2 phosphorylates a wide range of cytoplasmic and nuclear substrates with critical roles in cell proliferation and regulation, including the transcription/translation factor YB-1, which is associated with tumorigenesis and has a negative prognostic role in numerous cancers [Citation5,Citation6]. Activated RSK2 subsequently translocates into the nucleus and it is this nuclear localization of RSK2 hypothesized as the event that triggers the transformation of normal cells to malignant cells, specifically in breast cancer [Citation7]. Indeed, a majority of TNBC patient tumors have activated nuclear RSK2. Thus, activated RSK2, visualized by immunohistochemical staining of nuclear RSK2 in tumor tissues, could be used as a novel biomarker for a TNBC companion diagnostic and phosphorylation of YB-1, a direct downstream substrate of RSK2, has utility as a biomarker of response to RSK therapy.
Figure 1. MAPK and PI3K signaling cascades converge on the activation of RSK and YB-1. Simplified schematic of the convergence of the MAPK and PI3K pathways showing components downstream that are involved in phosphorylation (P) of RSK2 and its downstream targets. Following binding of growth factor ligands (L) to receptors on the cell surface, both the MAPK and PI3K pathways are activated leading to a cascade of phosphorylation events on downstream substrates in the cytoplasm. Activated RSK2, which is phosphorylated on Ser/Thr residues on multiple phosphorylation sites, phosphorylates YB-1 on S102 and both translocate into the nucleus, where pYB-1S102 regulates the transcription of genes involved in a wide range of cellular functions including PD-L1 (immune recognition), uPA (metastasis), EGFR (tumor cell growth) and MDR (drug resistance).
Both MEK1/2 and RSK2 are associated with TNBC, but functionally they are very different kinases, with differences in their signaling processes (). Brough et al (2011) elegantly showed that TNBC is functionally dependent on RSK2, but not MEK, PI3K or PDK1 [Citation2]. Disrupting this specific point in signal transduction also leads to cell death [Citation3]. Interestingly, inhibition of MEK suppresses tumor growth in BLBC, but does not lead to apoptosis [Citation8]. In addition, following treatment with MEK inhibitors, there is increased activation of Akt and compensatory stimulation of the PI3K/mTOR pathway in TNBC cells [Citation4,Citation9]. In contrast to MEK inhibitors, RSK2 inhibition does not affect phospho- Akt levels and RSK2 inhibition induces apoptosis in TNBC [Citation8,Citation10]. Typically, phospho- Akt, PTEN and K-Ras are used as pharmacodynamic biomarkers of response to MEK1/2 inhibitors, whereas phospho- YB-1 (a direct substrate) is used to monitor RSK2 inhibition. Based on this data, it is clear that RSK2 has distinct advantages over MEK as a promising molecular target for TNBC therapy (). RSK inhibitors are thus positioned to short circuit TNBC cell signaling, that ultimately leads to cancer cell death.
Table 1. Key similarities and differences of MEK1/2 and RSK2.
Despite the established role for the MAPK pathway in cancer, MEK1/2 inhibitors are associated with toxicities that limit their clinical use. In patients, class effects of MEK inhibitors include severe gastrointestinal, ocular and cardiac toxicities [Citation4,Citation11]. Although the precise mechanisms for these toxicities are unknown, one can speculate that due to the importance of the MAPK pathway in cellular processes, key regulators of kinase activity, including ERK, have a larger number of downstream substrates than RSK2. Consequently, inhibiting MEK has potentially more unintended effects than inhibiting RSK2, which is further downstream in the signaling cascade [Citation4]. Knockout mouse models also show us that loss of MEK1 and MEK2 leads to embryonic lethality [Citation12], whereas RSK2 knockout does not [Citation11] (). It has therefore been proposed that RSK2 inhibitors offer a safety advantage over MEK1/2 inhibitors [Citation4,Citation11].
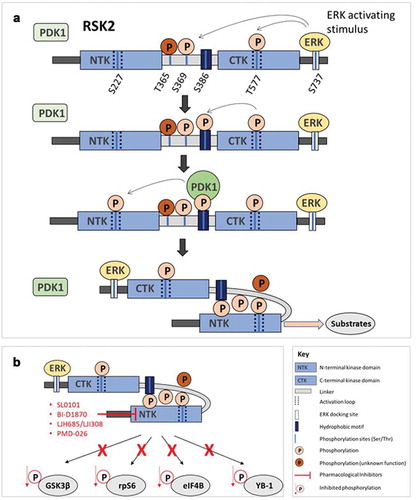
RSK has a unique structure with two separate kinase domains, each with distinct functions. The process of RSK activation is complex, involving sequential phosphorylation at multiple Ser/Thr residues ()). The purpose of the C-terminal kinase domain (CTKD) of RSK is to activate the N-terminal kinase domain (NTKD) via autophosphorylation. FMK is an irreversible inhibitor of RSK1/2, that binds covalently with the CTKD, however the CTKD is not critical for RSK signaling [Citation5] and therefore it is a poor target for inhibition. Hence, the majority of RSK specific inhibitors developed have targeted the NTKD of the protein ()).
Figure 2. (a) Model of RSK2 activation. ERK binds to a C-terminal MAP-kinase-docking site and phosphorylates residues in the linker, including Ser369 and the activation loop of CTK at Thr577. Phosphorylation of Thr577 then activates the CTK, which phosphorylates Ser386. Phosphorylation of Ser386 allows docking of PDK1, which stimulates PDK1 to phosphorylate the NTK at Ser227 in the activation loop. PDK1 then dissociates and pSer386 binds a phosphate-binding site in the NTK, coordinating with pSer227 in the activation loop to stabilize the NTK in an active conformation, synergizing to stimulate kinase activity. Activated RSK2 phosphorylates a wide range of cytoplasmic and nuclear substrates. (b) Inhibition of RSK2 signaling with RSK inhibitors. Inhibitors bind to the NTK domain of RSK2, blocking the activation of downstream substrates, including substrates that are involved in proliferation (GSK3β), translational regulation (rpS6; eIF4B) and transcriptional/translational regulation (YB-1). These particular RSK2 substrates are also recognized as having significance in cancer biology.
The first RSK NTKD specific inhibitor, SL0101, was isolated and characterized from botanical extracts. Early work by Smith et al. (2005) with this compound uncovered the crucial role that RSK plays in the proliferation of breast cancer, specifically compared to normal breast cells [Citation13]. While highly selective for RSK, SL0101 has low potency and despite modifications of the structure to produce analogs with enhanced activity in TNBC, has low cell permeability with a short half-life in mice from intravenous dosing of less than 30 minutes [Citation4]. Similarly, BI-D1870, another widely used experimental RSK inhibitor, is more potent than SL0101, but has only been administered intravenously in rodents and is nonspecific for the RSK isoforms. The RSK inhibitors LJH685 and LJI308 potently inhibit RSK and phosphorylation of its downstream substrate YB-1. Out of 21 different cancer cell lines, TNBC was the most sensitive to RSK inhibition by LJH685 and LJI308, when screened under anchorage independent conditions [Citation10]. However, despite the promising selectivity profile of LJH685, the in vivo half-life following intravenous administration in rats was only 13 minutes and exposure following oral administration was poor [Citation14]. Therefore, LJH685 is limited to use as an in vitro tool.
To conclude, RSK2 is a promising molecular target in TNBC because tumor cell survival hinges on this key kinase. RSK2 has been validated using siRNA and several small molecules each targeting the NTKD. The challenge up to this point has been in the development of a RSK2 inhibitor that has favorable pharmacological and pharmacokinetic properties required for oral delivery.
Expert opinion
Dysregulated MEK/ERK/RSK signaling plays an important role in TNBC. Comprehensive work from a number of independent laboratories have validated RSK2 as a critical component in MAPK/PI3K signaling pathways and its role in TNBC, therefore RSK2 has been recognized as an excellent target for TNBC therapy. One of the greatest challenges is to find durable treatments for all subtypes of TNBC. Current targeted therapies such as PARP inhibitors for BRCA mutated tumors and immunotherapy for PD-L1 expressing neoplasms, are only able to treat a fraction of TNBC patients with advanced disease. RSK is highly expressed in the majority of TNBC and is also a critical driver for the disease, thus RSK inhibition has distinct advantages as an effective therapy that transcends TNBC heterogeneity. In addition to tumor response with single agent RSK inhibition, an enduring response in refractory disease could also be accomplished through combinations with conventional chemotherapy or with the aforementioned targeted agents. Furthermore, given the high rates of relapse in TNBC, another challenge is to find effective treatments for refractory residual disease. A number of reports have observed increased activation of RSK signaling and increased YB-1 activity in treatment resistant cancer cells, thus providing a rationale for RSK inhibition in managing refractory TNBC. In addition to the central role of RSK in driving TNBC growth, emerging evidence suggests that RSK inhibition could work to stimulate the tumor immune microenvironment. RSK inhibition suppresses PD-L1, leading to the infiltration of tumor-infiltrating lymphocytes, thereby potentially turning ‘cold’ tumors, that do not respond to immunotherapy, into ‘hot’ tumors, thus revealing the potential of an effective treatment combination with RSK inhibitors and immunotherapy. Small molecule RSK inhibitors, SL0101, BI-D1870, LJH685 and LJI308, all potent ATP-competitive inhibitors that bind to the NTKD of RSK, have been developed. However, due to shortcomings in their pharmacokinetic properties, none of them have advanced into the clinic. As various research groups work toward advancing the pre-clinical development of these RSK inhibitor analogs, a recent patent describes novel small molecule compounds that are potent and specific inhibitors of RSK2, effectively suppressing TNBC tumor growth in mice following oral dosing [Citation15]. The structure of these new RSK inhibitors were designed to bind with high specificity to the kinase binding site with additional structural elements that serve to maximize solubility and cell permeability of the molecule. Therefore, the advantage of these new compounds over current RSK inhibitors is that they demonstrate suitable pharmacokinetics, are orally bioavailable and are effective across heterogeneous TNBC tumor models [Citation15]. An Investigational New Drug application for their lead compound, a first-in-class RSK inhibitor, has been approved by the FDA and is the first RSK inhibitor to enter clinical trials with a focus on TNBC.
Declaration of interest
M. Huynh, A. Jayanthan and S.E. Dunn are employed by Phoenix Molecular Designs and are co-inventors on the patent referenced in this publication. MR. Pambid is employed by Phoenix Molecular Designs. G. Los is contracted to Phoenix Molecular Designs as CSO. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Reviewers Disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Turner NC, Reis-Filho JS. Tackling the diversity of triple-negative breast cancer. Clin Cancer Res. 2013;19(23):6380–6388.
- Brough R, Frankum JR, Sims D, et al. Functional viability profiles of breast cancer. Cancer Discov. 2011;1(3):260–273.
- Stratford AL, Reipas K, Hu K, et al. Targeting p90 ribosomal S6 kinase eliminates tumor-initiating cells by inactivating Y-box binding protein-1 in triple-negative breast cancers. Stem Cells. 2012;30(7):1338–1348.
- Ludwik KA, Campbell JP, Li M, et al. Development of a RSK inhibitor as a novel therapy for triple-negative breast cancer. Mol Cancer Ther. 2016;15(11):2598–2608.
- Carriere A, Ray H, Blenis J, et al. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci. 2008;13:4258–4275.
- Stratford AL, Fry CJ, Desilets C, et al. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 2008;10(6):R99.
- Davies AH, Reipas KM, Pambid MR, et al. YB-1 transforms human mammary epithelial cells through chromatin remodeling leading to the development of basal-like breast cancer. Stem Cells. 2014;32(6):1437–1450.
- Hoeflich KP, O’Brien C, Boyd Z, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15(14):4649–4664.
- Risom T, Langer EM, Chapman MP, et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun. 2018;9(1):3815.
- Aronchik I, Appleton BA, Basham SE, et al. Novel Potent and selective inhibitors of p90 ribosomal S6 kinase reveal the heterogeneity of RSK function in MAPK-driven cancers. Mol Cancer Res. 2014;12(5):803–812.
- Ludwik KA, Lannigan DA. Ribosomal S6 kinase (RSK) modulators: a patent review. Expert Opin Ther Pat. 2016;26(9):1061–1078.
- Scholl FA, Dumesic PA, Barragan DI, et al. Mek1/2 MAPK kinases are essential for mammalian development, homeostasis, and raf-induced hyperplasia. Dev Cell. 2007;12(4):615–629.
- Smith JA, Poteet-Smith CE, Xu Y, et al. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005;65:1027–1034.
- Jain R, Mathur M, Lan J, et al. Discovery of potent and selective RSK inhibitors as biological probes. J Med Chem. 2015;58(17):6766–6783.
- Dunn SE, Jayanthan A, Nagireddy JR, et al. Carboxamide derivatives useful as rsk inhibitors. WO2017141116A1; 2017.