
ABSTRACT
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating disease with a lifetime risk of approximately 1:400. It is incurable and invariably fatal. Average survival is between 3 and 5 years and patients become increasingly paralyzed, losing the ability to speak, eat, and breathe. Therapies in development either (i) target specific familial forms of ALS (comprising a minority of around 10% of cases) or ii) emanate from (over)reliance on animal models or non-human/non-neuronal cell models. There is a desperate and unmet clinical need for effective treatments. Deciphering the primacy and relative contributions of defective protein homeostasis and RNA metabolism in ALS across different model systems will facilitate the identification of putative therapeutic targets.
Areas covered
This review examines the putative common primary molecular events that lead to ALS pathogenesis. We focus on deregulated RNA metabolism, protein mislocalization/pathological aggregation and the role of glia in ALS-related motor neuron degeneration. Finally, we describe promising targets for therapeutic evaluation.
Expert opinion
Moving forward, an effective strategy could be achieved by a poly-therapeutic approach which targets both deregulated RNA metabolism and protein dyshomeostasis in the relevant cell types, at the appropriate phase of disease.
1. Introduction
ALS is relentlessly progressive and uniformly fatal. It is characterized by progressive motor neuron (MN) degeneration. There exists significant clinical heterogeneity in ALS, which has been categorized in a variety of ways including the clinical site of onset (upper limb, lower limb or bulbar), cellular substrate (upper MN, lower MN or both) and rate of progression [Citation1]. The current standard of care in ALS is essentially supportive: a feeding tube is inserted into the stomach to maintain nutrition, noninvasive ventilation is needed to support breathing and a disease modifying drug (Riluzole) is prescribed, which can increase life expectancy by approximately 3 months. ALS is multifactorial with contributions from genetic and environmental factors, and aging [Citation2]. There is a desperate need to understand disease mechanisms in order to guide the development of effective therapies.
Although different forms of neurodegeneration are frequently considered as predominantly protein aggregation disorders or defects in RNA metabolism, this taxonomy is potentially facile and misleading. Indeed, accumulating evidence suggests that both processes are fundamentally implicated in ALS (reviewed in [Citation3]). The common molecular denominators in ALS are ribonucleoproteins (RNPs). An RNP is a complex of RNA and RNA-binding proteins (RBPs). These multifunctional complexes play key roles in regulating gene expression and RNA metabolism. Eukaryotic cells possess myriad strategies to mitigate a diverse range of stressors, which generally lead to the assembly of RNPs into ‘stress granules’ (non membrane delimited organelles), likely through tightly regulated prion-like polymerization of RBPs together with RNAs. Indeed, genetic evidence strongly implicates aberrant RBPs in ALS, including disease-causing mutations in genes encoding RBPs called fused in sarcoma/translocated in liposarcoma (FUS) and transactive response DNA-binding protein 43 (TDP-43) [Citation4–6]. Increased formation and persistence of such granules may contribute to the pathogenesis of ALS, although further investigation is required to comprehensively examine this issue. Aberrant phase transition of RBPs with low-complexity domains may also drive the formation of pathologically-relevant intracellular inclusions, a process regulated by interaction with RNAs [Citation7,Citation8]. More generally, evidence for RNA binding proteinopathy in ALS fundamentally implicates deregulation of both protein homeostasis and RNA metabolism. The integrated investigation of both processes is therefore crucial in the elucidation of therapeutically targetable underlying disease mechanisms.
Beyond such molecular considerations, the role of non-neuronal cells in ALS has become an increasingly important area of investigation. Until approximately 25 years ago, the consensus view regarding ALS pathogenesis was that selective injury to motor neurons is mechanistically cell autonomous. This ‘neuron-centric’ view has been increasingly challenged beginning with important mice-chimera studies using lineage-specific expression of mutant superoxide dismutase 1 (SOD1) [Citation9,Citation10]. These, and subsequent, studies have confirmed major non cell-autonomous roles for astrocytes, microglia, and oligodendrocytes, reviewed in [Citation11–13]. Increasing recognition of glial involvement in ALS raises the prospect of targeting these cells to increase neuroprotective capacity and/or to reduce acquired neurotoxic attributes.
Through integrated and multi-modal approaches to modeling ALS combined with key technological advances, recent discoveries are changing the landscape of therapeutic possibility. Here, we review a range of potential opportunities in this context and provide a personal view on the most promising approaches toward developing new therapies.
2. Defects in pre-mRNA splicing
Several studies have now examined splicing changes in ALS (or the related disorder frontotemporal lobar degeneration (FTLD), which shares pathogenetic features with ALS). For example loss of nuclear TDP-43 protein has been associated with cryptic splice site usage, which might ultimately induce RNA degradation via nonsense mediated RNA decay and thereby reduce abundance of (correctly spliced) mRNAs for translation [Citation14,Citation15]. Another recent notable study found aberrant splicing of the microtubule-associated stathmin-2 (STMN2) transcript caused by TDP-43 depletion in human neurons [Citation16,Citation17]. TDP-43 levels are regulated via a negative feedback loop, where TDP-43 binds to its own transcripts and destabilizes them, thereby reducing their translation and controlling the levels of TDP-43 protein. Perturbed TDP-43 autoregulation can lead to accumulation of cytoplasmic TDP-43 protein and subsequent neurodegeneration. We recently analyzed RNA sequencing data from human iPSC-derived MNs from ALS patients carrying mutations in valosin containing protein (VCP), SOD1 and FUS genes and discovered aberrant intron retention as a unifying molecular hallmark in these diverse genetic causes of ALS () [Citation18]. Intron retention is an understudied type of alternative splicing whereby mature polyadenylated transcripts retain one or more introns. Indeed intron retention is increasingly recognized as a fundamental mechanism for myriad cellular homeostatic processes [Citation19–22], reinforcing the importance of understanding its role in ALS. We additionally found evidence in support of a model whereby the RBPs splicing factor proline and glutamine rich (SFPQ) and FUS bind avidly to retained introns and are transported out of the nucleus by intron-retaining transcripts [Citation18]. It follows that silencing intron retention may offer a tractable therapeutic target as removing the aberrant (intron-retaining) transcripts may then prevent nuclear displacement of their avidly bound RBPs (). Importantly, by targeting the intronic sequence, this should leave intact the correctly spliced transcripts. Noting that >150 ALS-related intron retention transcripts have been identified, it would be important to prioritize therapeutically tractable events by their abundance in the cytoplasm, intron length and affinity for binding RBPs that are implicated in ALS. Depending on the approach for intervention adopted, considering polytherapy, targeting several aberrant transcripts, may be required to induce cellular remission. However, importantly, these aberrant intron retention events have not yet been proven to be pathogenic, which clearly argues for further evaluation in in vitro and in vivo models before considering progressing any individual candidates as potential therapeutic targets.
Figure 1. Aberrant splicing alterations in ALS. Loss of nuclear RNA binding proteins (RBP), including splicing factors, may lead to perturbed pre-mRNA splicing such as aberrant intron retention. Aberrant intron-retaining transcripts are exported to the cytoplasm, which may lead to mislocalization of their bound/cognate RBPs (e.g. SFPQ and FUS) from the nucleus to the cytoplasm in ALS. It follows that ASOs targeting aberrant splicing events might influence RBP nucleocytoplasmic distribution, but more experiments are required to demonstrate that these two phenomena are causally related. Figure created with BioRender.com
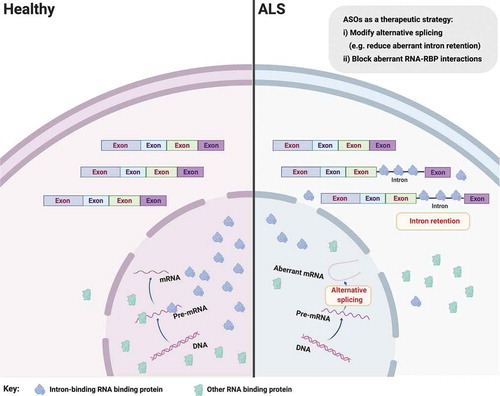
In the case that one or more aberrant intron retention events are found to be pathogenic, one approach to selectively targeting these transcripts is the use of antisense oligonucleotides (ASOs), which are short stretches of synthetic DNA that hybridize with complementary RNA. Within ASO biology, there exist an increasing number of possible chemical modifications to the oligonucleotide allowing target binding with predictable functional outcomes ranging from mRNA degradation (by activation of endogenous RNase H) to blocking RBP binding by steric hindrance on otherwise intact mRNAs). The ability to predictably manipulate the life cycle of targeted RNAs in this manner has already proved transformational in clinical trials for an FDA-approved ASO in patients with spinal muscular atrophy [Citation23,Citation24]. SMN2 specific alternative splicing modifier, Nusinersen, significantly improves motor function and event-free survival of patients with spinal muscular atrophy [Citation25]. Eteplirsen, another ASO that induces exon skipping within the dystrophin gene, has also been assessed to be both safe and effective in patients with Duchenne muscular dystrophy [Citation26]. Several ASOs are currently being developed for genetic causes of ALS, including a first-in-human ALS ASO against SOD1 [Citation27] and others against the intronic hexanucleotide repeat expansion in C9ORF72 [Citation28]. Furthermore, noting that the majority of ALS patients are sporadic, ASO-based therapy might be extended to modulate transversal biological pathways involved in the pathogenesis of ALS, including the aforementioned deregulated splicing events or RBP mislocalization directly.
3. RBP mislocalization and nucleocytoplasmic compartmentalization
The pathological hallmark in >95% of all ALS cases is nuclear-to-cytoplasmic mislocalization of the RBP TDP-43, where it becomes abnormally phosphorylated, cleaved and forms insoluble protein inclusions [Citation29] (). This occurs in all sporadic cases and in most familial cases, including those where protein function is not directly linked to RNA metabolism. However, SOD1 and FUS ALS-causing mutations do not generally exhibit TDP-43 mislocalization [Citation30], thus representing a conundrum for ALS researchers striving to identify common mechanisms across the full ALS spectrum. This is further reinforced by the fact that patients with SOD1 or FUS mutations are largely clinically (phenotypically) indistinguishable from other forms of familial or sporadic ALS. To this end, we recently described 2 further molecular hallmarks in ALS: nuclear loss of RBPs SFPQ and FUS, which we found in human induced pluripotent stem cell (hiPSC) models and validated in both mouse transgenic tissue and human postmortem tissue from sporadic cases [Citation18,Citation31]. Of these hallmarks, nuclear loss of SFPQ was observed in all models studied, including familial (VCP and SOD1 mutants) and sporadic tissue, and therefore seemingly represents a universal hallmark of ALS [Citation18]. Historical bias toward studying the constituents of aggregates themselves – rather than nuclear cytoplasmic ratio or the concentration of unaggregated cytoplasmic protein for example – likely explains why these findings have evaded detection until now. The importance of perturbed nucleocytoplasmic compartmentalization is exemplified by the fact that impaired FUS nuclear import correlates with the disease severity in ALS [Citation32–34]. Indeed, deletion of its nuclear localization signal (NLS) leads to dramatic FUS cytoplasmic mislocalization and an earlier age of onset [Citation35]. Two possible pathogenic mechanisms emerge from the nuclear-to-cytoplasmic mislocalization of RBPs: i) a nuclear loss-of-function causing aberrant pre-mRNA processing (e.g. intron retention, discussed above) or ii) a toxic gain of cytoplasmic function. Importantly, these mechanisms are not mutually exclusive and may occur simultaneously or sequentially in ALS pathogenesis.
Figure 2. RNA binding protein mislocalization and aggregation in ALS. Defective compartmentalization of RBPs leads to their cytosolic accumulation and nuclear depletion in ALS, including TDP-43, SFPQ and FUS. RBPs can undergo liquid-liquid phase separation, however some RBPs abnormally form distinct conformations of amyloid cross β sheets and aggregates, inducing native monomers to misfold in ALS. Aggregation-prone RBPs can spread from cell to cell in a prion-like fashion. Therapeutic approaches perturbing protein mislocalization and misfolding, promoting the clearance of abnormally oligomerized proteins, or preventing the intercellular spread of these proteins are promising therapeutic strategies in ALS. Figure created with BioRender.com
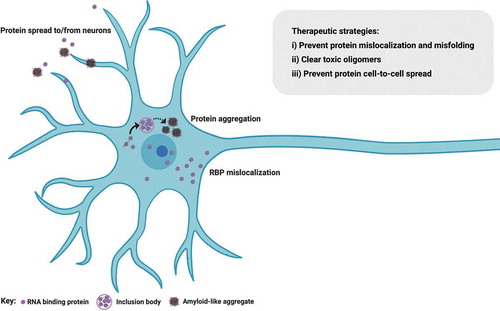
Perturbed cellular compartmentalization of molecular constituents (RBPs and/or RNA) also raises the important issue of nucleocytoplasmic transport defects. Nuclear transport deregulation is a common theme in the majority of ALS cases. Recognizing that aging is the major risk factor for ALS, it is noteworthy that cellular aging also induces changes in nucleocytoplasmic transport. Protein complexes of the nuclear pore have long half lives and therefore will not be replenished in postmitotic cells such as neurons [Citation36]. Therefore, these cells become ‘leaky’ with aging, likely secondary to cumulative oxidative damage and structural dysfunction at the level of the nuclear pore [Citation37]. Likewise, the importin receptors, which transport NLS-containing protein cargo through the nuclear pore, are downregulated with aging [Citation38,Citation39]. Whilst evidence of a mechanistic relationship between ALS pathogenic events and aging is emerging, nuclear-to-cytoplasmic mislocalization of TDP-43 in most cases together with other recent studies implicate perturbed nuclear transport. However, the temporal relationship between TDP-43 mislocalization, nuclear transport dysfunction, and neuronal loss remains unclear. An age-related defect in nuclear transport might cause mislocalization and cytoplasmic aggregation of RBPs [Citation40]. Indeed, aging cells accumulate misfolded and aggregated proteins due to compromised protein homeostasis [Citation41]. Conversely, cytoplasmic aggregation of RBPs might drive a secondary defect in nuclear transport. In support of this hypothesis, accumulation of cytoplasmic aggregates (e.g. C-terminal fragments of TDP-43), cause a partial dislocation of nuclear pore complex proteins to the cytoplasmic inclusions [Citation42]. Of course these two possibilities are not mutually exclusive and may occur simultaneously or sequentially. Promoting nuclear import of mislocalized TDP-43, FUS or more generally stimulating the canonical nuclear import pathway mediated by the importin β family of proteins has beneficial effects in ALS models. Conversely, decreasing nuclear import of these RBPs results in neurodegeneration, reviewed in [Citation43]. Histopathological studies of C9ORF72-associated cases have raised the hypothesis that dipeptide repeat (DPR) pathology may precede TDP-43 mislocalization [Citation44–46]. In animal models, C9ORF72 hexanucleotide repeat expression leads to TDP-43 nuclear loss and pathological aggregation in the cytoplasm. Therefore, therapeutic approaches correcting the deficits of nuclear transport may be an important consideration in ALS.
The prospect of relocalizing RBPs to the nucleus as a therapeutic strategy might also be realized through pharmacological targeting of their post-translational modifications. For example, the C-terminal NLS of FUS is juxtaposed to an arginine/glycine-rich region where several arginines and the NLS together are recognized by karyopherin β2 (also termed transportin-1) to regulate FUS localization. However, ALS-associated mutations within or near the NLS alter its interaction with karyopherin β2 (either directly through the mutation or disrupted methylation). Arginine methyltransferase inhibitors have been shown to ameliorate the cytoplasmic mislocalization of FUS [Citation47]. It follows that factors associated with nucleocytoplasmic transport (e.g. nuclear importins or exportins, transport-partners) may represent tractable therapeutic targets. Indeed, overexpressing importin α, karyopherin β1 or β2 has been shown to decrease and even reverse aberrant fibrillization of TDP-43, FUS, TAF15, EWSR1, hnRNPA1, and hnRNPA2 through interaction with their NLS [Citation48]. Karyopherin β2 can also dissolve aberrant fibril-containing hydrogels, prevent the RBP accumulation into stress granules and restore nuclear localization of misplaced RBPs [Citation49–51]. Selective inhibitors of the nuclear export receptor CRM1 (SINE compounds) have proved effective in ameliorating TDP-43 mediated locomotor defects in neuronal cells and animal models [Citation52,Citation53]. However the effect on TDP-43 localization could not be reproduced when using a neuroprotective concentration by a subsequent study, suggesting that RBP export from the nucleus operates through a more complex mechanism including CRM1-independent receptor-mediated or passive diffusion pathways [Citation53–55].
4. RBP aggregation – prion-like characteristics
RBP mislocalization from the nucleus to the cytoplasm is also associated with misfolding and cytoplasmic aggregation in ALS. Initiating events of this process possibly relate to deregulated liquid-liquid phase separation (LLPS). From the viewpoint of RBP aggregation, the ALS field has benefited from studies in the prototypic protein misfolding disorders, the prion diseases. It is widely accepted that misfolded proteins underlie the cellular pathogenesis and cell-to-cell propagation in classic prion diseases by forming distinct conformations of amyloid cross β sheets, which then serve as self-templates recruiting native monomers to misfold. Indeed, analogous ‘seeding’/propagation phenomena have been demonstrated experimentally for different ALS proteins including TDP-43 [Citation56,Citation57], FUS [Citation58] and SOD1 [Citation59]. These experimental models usually rely on protein overexpression in non-human or non-neuronal cell lines. However, we have recently demonstrated seeded aggregation in hiPSC-derived motor neurons treated with serially passaged sarkosyl-insoluble extract from sporadic ALS postmortem tissue. In this work, we also demonstrated that TDP-43 oligomers are at least part of the toxic principle in ALS [Citation60]. This raises the possibility of designing therapeutics that target these toxic oligomers. Broadly, three main strategies can be considered here: i) perturbing the formation of protein aggregates within the cell; ii) promoting their clearance from affected cells and iii) preventing their uptake into other cells. These approaches are discussed in more detail below.
ALS-associated RBPs with prion-like domains, such as TDP-43, FUS, TAF15, EWSR1, and hnRNPA1, are prone to form pathological aggregates [Citation61–65]. Multiple studies have suggested that FUS, hnRNPA1, and TIA-1 can form dynamic liquid droplets in vitro that, over time, form more stable hydrogels and pathological fibrils, resembling the behavior of protein aggregates in ALS [Citation66–70]. Protein aggregates observed in ALS are likely the consequence of overwhelmed cellular machinery coping with aberrant phase separation and protein misfolding. Therapeutic approaches to either enhance the endogenous regulation of RNP-granule disassembly or intervene in RBP recruitment to granules are therefore potential candidates in ALS therapy [Citation71]. These approaches essentially target aberrant RBP phase transitions, which may then play a key role in preventing pathological aggregation [Citation72,Citation73]. Somewhat paradoxically, recent studies have suggested that blocking the formation of stress granules may actually facilitate pathological inclusion formation and/or toxicity [Citation7,Citation8]. Taken together, it is clear that further investigation is required here. The precise consequences of stress granule formation are likely determined in a context-specific fashion by factors such as disease chronicity and cell type(s) involved. The aforementioned studies at least demonstrate that pathological inclusion formation can occur independently of stress granules but are likely the consequence of deregulated LLPS.
Heat shock proteins, or chaperones, can refold misfolded proteins into native functional conformations. Small molecules harnessing the disaggregase potential of heat shock proteins hold therapeutic significance for aggregate clearance. Arimoclomol (BRX-345), a potent activator of heat shock transcription factor 1 (HSF1), has been shown to increase the expression of Hsp70 and Hsp90, leading to reduced insoluble TDP-43 aggregate levels. Indeed, this compound has also shown promising results in the phase II trial in patients with rapidly progressive familial SOD1-ALS (NCT00706147) [Citation74–78]. Potentiated chaperones, such as engineered Hsp104, also bear real significance for therapeutic strategy by reversing ALS-linked TDP-43 and FUS aggregation [Citation79–81]. Small molecules that enhance endogenous chaperone activity in cells or the introduction of de novo ‘designer’ chaperone proteins can effectively remodel misfolded proteins and maintain disaggregase activity to reverse aberrant phase separation [Citation82–86]. Additionally, specific kinases, such as DYRK3 and CK2, have been found to modify stress granule proteins and regulate granule disassembly [Citation87–89]. As the β-sheet structure of aggregates plays an important role in seeding capacity, TDP-43 seeding might potentially be abrogated by formic acid [Citation89,Citation90]. Tafamidis meglumine (Fx-1006A), the only FDA-approved anti-amyloidogenic drug, is a potential candidate to test in preventing monomer misfolding and aggregation of TDP-43 in ALS [Citation91]. Several further compounds have been identified that decrease the aggregation of TDP-43 in stress granules through an acridine-imidazole derivative (AIM4), 4-aminoquinoline derivatives, copper complexes, and other compounds [Citation92–95]. Using ASOs or small molecule inhibitors targeting molecular seeding factors, such as ataxin-2 and PAR, are further therapeutic strategies that have been used to reduce aberrant phase transition in several ALS models [Citation67,Citation96–98]. For example, using ASOs to downregulate ataxin-2 affected stress granule dynamics and decreased TDP-43 recruitment, which then improved the lifespan and motor function of TDP-43 transgenic mice [Citation96]. It is noteworthy that RNA can either function as a molecular seed of RBP-containing membraneless condensates or to counter aberrant phase separation, although the precise molecular mechanism(s) underlying these processes remain incompletely resolved. Recognition that a high concentration of RNA in the nucleus acts as a buffer to prevent RBP phase separation (e.g. FUS and TDP-43) may have therapeutic significance [Citation7,Citation99]. Indeed, delivery or induced expression of particular RNAs can rescue RBP aggregation pathology. Specifically, a high concentration of ribosomal RNA, tRNA, and a noncoding RNA (Neat1) that is known to bind to FUS, have individually been shown to solubilize FUS droplets [Citation99]. Furthermore, the solubility of TDP-43 increases with delivery of its cognate single-stranded DNA (ssDNA) or RNA (ssRNA) [Citation100], and overexpression of nontoxic short UGGAA repeat RNA can also suppress mutated RBP aggregation (TDP-43, FUS, and hnRNPA2B1) and toxicity in ALS drosophila models [Citation101]. On the other hand, blocking the RNA-binding ability of TDP-43 has been shown to enhance protein destabilization and ameliorate TDP43-dependent neurotoxicity probably through affecting transcripts encoding ribosome and oxidative phosphorylation components [Citation102]. Overall, the molecular ‘logic’ of how sequence specific RNAs can affect phase separation of particular RBPs is an exciting and intensely active field where much remains to be understood.
The cellular ubiquitin-proteasome system and autophagy pathways are also responsible for clearing misfolded and aggregated proteins. Compounds stimulating autophagy can improve TDP-43 clearance and localization in iPSC-derived neurons and astrocytes [Citation103]. Colchicine, a potent HspB8 inducer, can facilitate cellular autophagy to remove insoluble TDP-43 species, which now is under a phase II clinical trial in ALS (NCT03693781) [Citation104,Citation105]. The mTOR pathway inhibits autophagy and inhibitors of mTOR (rapamycin, resveratrol, BECN1, or calpastatin) can induce protective autophagy and restore neuronal homeostasis, which have been modeled and trialed in ALS, Alzheimer’s disease and Huntington’s disease treatments [Citation106–108]. However, there is some recognized concern over the efficacy of mTOR inhibitors in neurons, and so mTOR-independent autophagy activation may ultimately be a preferable strategy for the clearance of aggregated misfolded proteins in this context [Citation109,Citation110]. Beyond the clearance of accumulated aberrant protein aggregates, prion-like protein propagation can be also prevented by blocking cellular release and/or uptake, and promoting the degradation of misfolded proteins in the extracellular space. There is a unifying chaperone-dependent mechanism for the release of pathogenic proteins in various neurodegenerative diseases. DnaJC5, a heat shock protein co-chaperone, has been found to be associated with the release of TDP-43, α-synuclein and Tau. Small molecule inhibitors preventing the interaction between DnaJC5 and other heat shock protein chaperones may hold therapeutic promise [Citation111]. There is also a unifying mechanism involved in cellular uptake whereby protein aggregates bind heparan sulfate proteoglycans (HSPGs) on the surface of recipient cells. Therefore blocking the HSPGs pharmacologically by adding a synthetic heparin mimetic may prevent cell-to-cell uptake [Citation112], but the overall effect of such a strategy on cellular homeostasis is clearly an important consideration here. Developing vaccines and passive immunization with antibodies to block misfolded proteins from spreading is also worthy of consideration. For instance, the development of vaccination or monoclonal antibodies against SOD1 has been shown to clear extracellular SOD1 mutant proteins, delay disease onset and prolong survival in SOD1 transgenic mice [Citation113–115]. A note of caution, however, is required here when considering the multiple failed drug trials using analogous approaches for other neurodegenerative disorders such as Alzheimer’s disease [Citation116,Citation117]. These studies together reinforce the importance of selecting tractable and high confidence therapeutic targets that are orthogonally validated across a range of models, and which carefully take into account clinical heterogeneity, phase of disease, toxicity, cellular and molecular pathophysiology together with careful trial design.
5. The contribution of glia in ALS
Ourselves and others have demonstrated that hiPSC-derived patient-specific astrocytes exhibit cell autonomous and non-cell autonomous pathology in ALS () [Citation118–122]. We have also recently demonstrated seeded aggregation in human iPSC-derived motor neurons and astrocytes when they are treated with serially passaged sarkosyl-insoluble extract from sporadic ALS postmortem tissue. We showed that neurons are generally more vulnerable to this process compared with astrocytes. Additionally, we found that astrocytes are neuroprotective to seeded aggregation within motor neurons by reducing (mislocalized) cytoplasmic TDP-43, TDP-43 aggregation and cell toxicity [Citation60]. These findings raise the prospect of invoking this endogenous reparative potential as a therapeutic option. However, it is likely that this initial protective capacity is eroded as the disease progresses, with the astrocytes undergoing a deleterious pro-inflammatory reactive transformation, as suggested by recent studies [Citation11,Citation123]. Indeed, neuroinflammation induced centrally by activated microglia, astrocytes and infiltrating T lymphocytes is increasingly recognized to play a key role in ALS pathophysiology and in fact may possibly be a primary event preceding the neurodegeneration cascade, reviewed in [Citation124]. The importance of this inflammatory component in ALS is exemplified by mass spectrometry of human ALS plasma showing a significant increase of ficolin-3 which results in an increased complement activation potential [Citation125]. Some attributes of this deleterious glial reactive transformation can also be observed upon normal aging [Citation126]. Clearly the ability to predictably manipulate astrocyte and/or microglial reactive states across this inflammatory continuum, back to being more neuroprotective, represents a large therapeutic opportunity for ALS ().
Figure 3. Glial involvement in ALS. Glia exhibit cell autonomous and non-cell autonomous neurodegeneration in ALS. As the disease progresses, astrocytes and microglia likely transition from an initial neuroprotective state to a toxic pro-inflammatory state in ALS with fewer neurotrophic factors and more neurotoxic factors secretion, such as inflammatory cytokines (TNF, IL-1, etc.), prostaglandin D2 PGD2 [Citation120,Citation127]. Reduced expression and activity of the astrocytic glutamate transporter EAAT2 influences motor neuron excitability. Oligodendrocytes fail to support axons of motor neurons through the disruption of lactate transport and normal myelination. Upon aging, glia-specific genes, but not neuron-specific genes, shift strikingly their regional expression patterns. A viable therapeutic approach might be to invoke the neuroprotective capacity of astrocytes or microglia. Similarly, cellular (glial) transplants may be a tractable therapy in ALS by promoting survival of juxtaposed motor neurons. Figure created with BioRender.com
![Figure 3. Glial involvement in ALS. Glia exhibit cell autonomous and non-cell autonomous neurodegeneration in ALS. As the disease progresses, astrocytes and microglia likely transition from an initial neuroprotective state to a toxic pro-inflammatory state in ALS with fewer neurotrophic factors and more neurotoxic factors secretion, such as inflammatory cytokines (TNF, IL-1, etc.), prostaglandin D2 PGD2 [Citation120,Citation127]. Reduced expression and activity of the astrocytic glutamate transporter EAAT2 influences motor neuron excitability. Oligodendrocytes fail to support axons of motor neurons through the disruption of lactate transport and normal myelination. Upon aging, glia-specific genes, but not neuron-specific genes, shift strikingly their regional expression patterns. A viable therapeutic approach might be to invoke the neuroprotective capacity of astrocytes or microglia. Similarly, cellular (glial) transplants may be a tractable therapy in ALS by promoting survival of juxtaposed motor neurons. Figure created with BioRender.com](/cms/asset/3f979417-a02a-433e-8289-d99cda654fe4/iett_a_1805734_f0003_oc.jpg)
Several studies have focused on glutamate transporter EAAT2 expression and the glutamate uptake capacity of astrocytes, primarily because excessive glutamate stimulation can cause excitotoxicity in surrounding motor neurons. Translational activation of EAAT2 (e.g. through compounds such as LDN/OSU-0212320) has been demonstrated to protect neurons from glutamate-mediated excitotoxic injury, which extended the lifespan of SOD1 transgenic mice [Citation128]. Neuroimmunophilins (e.g. tacrolimus) also induced the expression of EAAT2 in astrocytes, which was shown to protect motor neurons in vitro and prolongthe lifespan of SOD1 transgenic mice [Citation129]. However, two further studies where upregulation of astrocytic EAAT2 was achieved through HDAC inhibitors or β-lactam antibiotics (e.g. ceftriaxone) failed to show efficacy in ALS, suggesting that restoration of glutamate uptake alone may not sufficient for ALS therapy [Citation130–133]. Targeting activation of astrocytes has been tested as a possible treatment in ALS. Anti-oxidative agent bromocriptine (BRC), cannabigerol quinone derivative VCE-003.2, or cyclic nitroxides (e.g. tempol), have been shown to reduce astrocyte activation, lower the level of inflammatory factors (TNF-α and IL-1β) and improve motor function in transgenic mice [Citation134–136]. Glucagon-like peptide-1 receptor (GLP1R) agonists, such as NLY01, have also been suggested as potential neuroprotective agents to inhibit the formation of deleterious reactive astrocytes through blocking the microglial activation [Citation137]. Another experimental drug, RNS60, has been shown to have anti-inflammatory and neuroprotective properties through inducing a protective state in astrocytes and microglia. RNS60 is currently in a phase II clinical trial (NCT03456882) [Citation138,Citation139].
Cellular implantation is an alternative potential therapeutic consideration (). Harnessing astrocytes as cellular material currently seems tractable as this bypasses the challenge of neuronal implantation (i.e. reconstructing highly complex connections over long distances between motor neurons and their distal muscle targets). Multiple clinical trials of cell transplantation have used neural progenitor cells to generate astrocytes and interneurons, which can release growth factors and/or reduce inflammation to protect surrounding motor neurons [Citation140–143]. Another study utilized human neural progenitor cells that had been genetically engineered ex vivo to produce glial cell line-derived neurotrophic factor (GDNF), a protein that promotes the survival of neurons and has proved beneficial in ALS rat models [Citation144,Citation145]. Transplantation of the GDNF-secreting cells into the spinal cord of ALS patients is now in phase I/IIa clinical trials (NCT02943850). Another FDA-approved cell transplantation trial using highly purified glial-restricted progenitors is also planned [Citation146]. Notably, grafting human iPSCs-derived neural progenitor cells leads to astrocyte differentiation and improvement in the lifespan of rodents [Citation147,Citation148], underlining the utility of this source of cellular material for transplantation in ALS therapy. Cumulatively, astrocytes represent an underexplored therapeutic opportunity in ALS but it remains possible that different phases of disease may necessitate distinct therapeutic approaches.
Concordant with the perspective that neuroinflammation is relevant to ALS, microglia are the immune-competent sentinels of the CNS and are activated in ALS especially around the site of MN degeneration [Citation149–152] (). Increased microglial pathology is associated with the severity of degeneration of upper MNs in the motor cortex and with disease progression [Citation150,Citation153]. Studies using transgenic mouse models showed that the number of activated microglia increases during ALS disease progression with phenotypic transformation from M2 anti-inflammatory to M1 pro-inflammatory microglial states [Citation154–156]. At the presymptomatic stage, microglia exhibit an anti-inflammatory profile with overexpression of IL-10 and attenuated TLR2 responses [Citation157]. During disease onset, the M2 markers Ym1 and CD206 are upregulated in microglia. In later phases, microglia exhibit enhanced ROS production and secretion of inflammatory cytokines such as TNF-α, IL-1, and IL-6, which coincide with a decrease in neurotrophic/anti-inflammatory factors such as IGF-1, IL-4, and IL-10, and the expression of high levels of NOX2, together considered an M1 phenotype [Citation158,Citation159]. Isolated early disease stage M2 microglia enhance MN survival in co-culture, whereas end-stage M1 microglia are neurotoxic [Citation155]. Activated microglia also induce A1 reactive astrocytes through secreting cytokines and other factors, including IL-1α, TNF and C1q [Citation123]. Microglia derived from SOD1 transgenic mice exhibit an upregulation of neurotoxic factors, consistent with the recognized non cell-autonomous effect of microglia in ALS pathogenesis [Citation154,Citation160]. Ablating mutant SOD1 in microglia efficiently maintained tissue homeostasis and prolonged survival in the same mouse model [Citation161]. Transplantation of wide-type donor-derived bone marrow to replenish microglia in SOD1G93A/PU.1−/− mice, a model unable to generate lymphoid and myeloid cells (e.g. CNS microglia), both slowed MN loss and disease progression [Citation162]. In a sporadic ALS-like mouse model expressing hTDP43∆NLS, reactive microgliosis was associated with pathological TDP-43 clearance and motor recovery [Citation163]. These data suggest that microglia have temporally-regulated dynamic roles in ALS disease progression.
A potential therapeutic approach would be to maintain anti-inflammatory and neuroprotective functions of microglia (). Nuclear factor-kappa β (NF-κβ) protein is upregulated in mouse models and ALS patients, which may play an important role in the regulation of microglial inflammation. Selective inhibition of NF-κβ signaling in microglia rather than astrocytes rescued MN loss in vitro and delayed disease progression in vivo by preventing pro-inflammatory conversion of microglia, while the constitutive activation of NF-κβ in wild-type microglia induced gliosis and MN death [Citation164]. Insulin-like growth factor (IGF1)-mediated suppression of NF-κB activation is a potential novel therapeutic avenue, as intrathecal injection of scAAV9-hIGF1 in SOD1 transgenic mice at presymptomatic and symptomatic stages inhibited the inflammatory response and prolonged the lifespan of mice [Citation165]. Histamine plays a role in regulating the release of pro-inflammatory factors (e.g. TNFα, IL-6) from activated microglia partially through histamine H1 and H4 receptors and NF-κB signaling pathway [Citation166]. Clemastine (also known as meclastin), an histamine H1 antagonist, can reduce microgliosis, modify microglial inflammatory parameters (e.g. downregulation of NOX2), and enhance MN survival [Citation167]. Cromolyn sodium treatment increased MN survival and decreased the denervation of neuromuscular junctions in SOD1 transgenic mice through inducing a shift in microglial activation states from pro-inflammatory to anti-inflammatory [Citation168]. In the same mouse model where inflammatory cytokine IL-1 was upregulated with the acquisition of a pro-inflammatory phenotype, treatment with IL-1 receptor antagonist attenuated inflammatory pathology and extended survival [Citation169]. A recent study reported immunotherapy targeting poly-GA dipeptide repeat proteins in a C9ORF72 mouse model vaccinated by ovalbumin-(GA)10 can inhibit the activation of microglia and improve motor function [Citation170]. Colony stimulating factor 1 receptor (CSF1R) and the ligand (CSF1) regulate the proliferation and activation of microglia in SOD1 transgenic mice and treatment with a selective CSF1R inhibitor GW2580 rescued MN death, slowed down disease progression and extended survival [Citation171]. Masitinib, a selective oral tyrosine kinase inhibitor, which can prevent CSF1-induced microgliosis, cell migration, and the inflammatory response [Citation172], is in a phase III trial (NCT03127267). Treatment of SOD1 transgenic mice with tempol can also reduce the level of microglial reactivity and, as alluded to above, the expression of pro-inflammatory cytokines (IL-1β and TNF-α) at the initial stage of symptoms and delay disease onset [Citation136]. Ibudilast, which can attenuate inflammation in the CNS by preventing the production of pro-inflammatory factors from microglia [Citation173,Citation174], is now being evaluated in two trials (NCT02714036, NCT02238626).
There is accumulating evidence that oligodendrocytes also contribute to ALS pathogenesis (). Degeneration of oligodendrocytes has been demonstrated in ALS mice before disease onset and observed in ALS patients, resulting in progressive demyelination in the motor cortex and spinal cord [Citation175,Citation176]. Genetic deletion of mutant SOD1 from oligodendrocytes substantially delayed the disease onset and extended survival in mice [Citation175]. Oligodendrocytes support axons of MNs partially through the transport of lactate, however the lactate transporter MCT1 in oligodendrocytes is reduced in ALS and associates with axon damage and neuron loss in mouse models and patients [Citation177]. In a zebrafish model, selectively expressing mutant SOD1 in mature oligodendrocytes induced disruption of myelin sheath and downregulation of MCT1, which resulted in MN degeneration [Citation178]. In a human co-culture system, oligodendrocytes derived from familial and sporadic ALS patient-derived iPSCs induced MN hyperexcitability and death, while early downregulation of the misfolded SOD1 in progenitor cells resulted in MN rescue in all ALS cases (except for those carrying C9ORF72 repeat expansions) [Citation179]. A genome-wide association analyses with more than ten thousands ALS patients identified myelin-associated oligodendrocyte basic protein (MOBP) as a new ALS-related risk locus, the mutation of which may disturb the intercellular communication between oligodendrocytes and MNs [Citation180]. Taken together, glia represent attractive cellular targets in ALS and a better understanding of regionally encoded functional heterogeneity [Citation181], glial-glial crosstalk and their interplay with aging will be important to highlight further therapeutic opportunities.
6. Conclusion
Approximately 200 clinical trials examining drugs with varied mechanisms of action have been conducted across > 50 clinical research centers. However, no therapy to halt or reverse disease progression has been identified. Indeed, in the UK only 1 approved therapy of modest efficacy (Riluzole) is licensed. Reasons underlying this failure in translation may include over reliance on animal studies and/or non-human or non-neuronal models and possibly issues with trial design. Furthermore, in preclinical studies, the delivery of treatments has often been commenced prior to disease onset/establishment, which biases efficacy toward largely non clinically representative outcomes. Recognizing the phenotypic variability of ALS patients, treatment approaches would likely benefit from being tailored toward specific rates of disease progression. Against this background, accurately understanding cellular and molecular pathophysiology in human experimental models, validation of key phenotypes using orthogonal models and the establishment of universal biomarkers will be essential for guiding drug trials. These biomarkers will ideally indicate not only the presence of disease, but also the rate of progression and inform on pathogenetic subtypes of ALS to help target the optimal therapeutic approaches to the correct patient cohorts.
7. Expert opinion
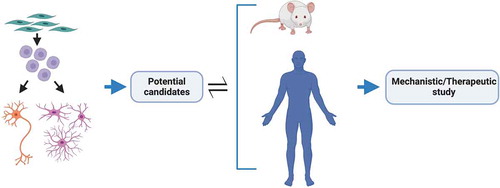
An integrated approach to modeling is crucial to yield high confidence findings of translational value by reducing inherent biases of each particular model system. Human iPSCs can be predictably manipulated into becoming any human cell type without the need for artificial overexpression or knock down [Citation182–185], whilst faithfully recapitulating human cell type-specific properties of ALS [Citation119,Citation186–188]. A recent important example here is aberrant splicing of the microtubule-associated stathmin-2 (STMN2) transcript caused by TDP-43 depletion in human neurons [Citation16,Citation17]. Although hiPSC-derivatives essentially represent a fetal maturational state [Citation189], strategies are now emerging to induce aging or preserve aging of the donor cell (reviewed in [Citation190]). It follows that primary discovery in hiPSCs and secondary validation in mouse transgenic models and human postmortem tissue is a particularly powerful combination to identify promising candidates for further mechanistic/therapeutic evaluation (). Such ‘cross-modal’ validation will yield candidates that may then be rigorously evaluated for their utility as therapeutic targets in relevant cell types. This approach can also be strengthened by moving beyond just motor neurons; indeed, potential candidates should be shown to be at least neutral – but ideally also to exert a positive effect – in astrocytes, microglia and oligodendrocytes. It is worth then evaluating the drug in co-culture paradigms that can undergo stepwise increases in sophistication once cell autonomous effects are confidently established. Initial discovery science in hiPSCs also allows elucidation of the primacy of molecular pathogenic events in clinically relevant target cell types. This can be followed by only necessary in vivo testing. Such an integrated approach arguably helps to ensure relevance to human target cell types, whilst also reducing animal experimentation. Beyond evaluating the effect of one potential drug on multiple cell types (therapeutic and toxicity assays [Citation186]), we feel it is also crucial to consider a poly-therapeutic approach where distinct salient mechanisms can be targeted in specific cell types by simultaneously employing different therapies.
Figure 4. Integration of models for ALS therapeutic study. An integrated approach for the discovery of high confidence therapeutic candidates for ALS. Patient iPSC-derived region-specific neurons and glia for primary discovery with mouse transgenic models and human postmortem tissue for secondary validation to identify promising candidates for further mechanistic and/or therapeutic evaluation. Insights gained from in vivo studies are then used to improve or modify in vitro model systems to better portray the disease and/or focus on different targets. Figure created with BioRender.com
Neurodegenerative disorders including ALS have long been considered as protein misfolding diseases characterized by the formation of (cytoplasmic > nuclear) protein aggregation. Defective RNA metabolism is rapidly becoming acknowledged as playing crucial roles in ALS. The molecular mechanisms by which these defects in cellular homeostasis conspire to cause motor neuron degeneration is a key issue to resolve. Therefore, it is our view that the most promising therapeutic approaches will consider targeting both deregulated RNA metabolism and protein dyshomeostasis. Implicit within this goal is to resolve both the primacy and relative contributions of defective protein homeostasis and RNA metabolism within different cell types and at different phases of the disease. Noting that ALS is an asynchronous disease, therapeutic intervention at the time of diagnosis may indeed lead to significant prevention of neurological disability. However, for this promise to be realized, we first require robust identification of cellular and molecular therapeutic targets, taking account the spatio-temporal heterogeneity of ALS pathogenesis within individual patients.
Article highlights
ALS is a devastating neurodegenerative disease in which further investigation of molecular and cellular events is crucial to elucidate therapeutically targetable mechanisms.
Aberrant splicing (including intron retention) and nuclear loss of RNA binding proteins (RBPs) are observed in ALS.
Protein misfolding (including toxic oligomerization) underlies ALS pathogenesis to some degree, including aggregation and a prion-like cell-to-cell propagation.
Astrocytes and microglia undergo deleterious reactive transformation in ALS, which perturbs their neuroprotective capacity.
Elucidation of primary pathogenic events in motor neurons and glia through integration of different ALS models is crucial and will allow the identification of high confidence therapeutic targets.
Considering a poly-therapeutic approach will likely advance our efforts in ultimately discovering the most effective therapeutic strategies.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Acknowledgments
The work of the authors is supported by the Francis Crick Institute which receives its core funding from Cancer Research UK 65 (FC010110), the UK Medical Research Council (FC010110), and the Wellcome Trust (FC010110). R.P. holds an MRC Senior Clinical Fellowship [MR/S006591/1].
Additional information
Funding
References
- Balendra R, Patani R. Quo vadis motor neuron disease? World J Methodol. 2016;6:56–64.
- Pandya VA, Patani R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: a cellular perspective. Brain. 2019. Internet. DOI:10.1093/brain/awz360.
- Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206.
- Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672.
- Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211.
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268.
- Mann JR, Gleixner AM, Mauna JC, et al. RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron. 2019;102:321–338.e8.
- Gasset-Rosa F, Lu S, Yu H, et al. Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron. 2019;102:339–357.e7.
- Nagai M, Re DB, Nagata T, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons [Internet]. Nat Neurosci. 2007;10:615–622.
- Yamanaka K, Chun SJ, Boillee S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253.
- Serio A, Patani R. Concise review: the cellular conspiracy of amyotrophic lateral sclerosis. Stem Cells. 2018;36:293–303.
- Tyzack G, Lakatos A, Patani R. Human stem cell-derived astrocytes: specification and relevance for neurological disorders. Curr Stem Cell Rep. 2016;2:236–247.
- Tognatta R, Miller RH. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology. 2016;110:539–547.
- Ling JP, Pletnikova O, Troncoso JC, et al. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349:650–655.
- Humphrey J, Emmett W, Fratta P, et al. Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med Genomics. 2017;10:38.
- Klim JR, Williams LA, Limone F, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22:167–179.
- Melamed ZE, López-Erauskin J, Baughn MW, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22:180–190.
- Luisier R, Tyzack GE, Hall CE, et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat Commun. 2018;9:2010.
- Yap K, Lim ZQ, Khandelia P, et al. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev. 2012;26:1209–1223.
- Wong JJ-L, Ritchie W, Ebner OA, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013;154:583–595.
- Buckley PT, Lee MT, Sul J-Y, et al. Cytoplasmic intron sequence-retaining transcripts can be dendritically targeted via ID element retrotransposons. Neuron. 2011;69:877–884.
- Braunschweig U, Barbosa-Morais NL, Pan Q, et al. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014;24:1774–1786.
- Wadman M. Updated: FDA approves drug that rescues babies with fatal neurodegenerative disease [Internet]. Science. 2016. DOI:10.1126/science.aal0476
- Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study [Internet]. Lancet. 2016;388:3017–3026.
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723–1732.
- Kinane TB, Bernard Kinane T, Mayer OH, et al. Long-term pulmonary function in duchenne muscular dystrophy: comparison of eteplirsen-treated patients to natural history [Internet]. J Neuromuscul Dis. 2018;5:47–58.
- Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12:435–442.
- Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90:535–550.
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133.
- Mackenzie IRA, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434.
- Tyzack GE, Luisier R, Taha DM, et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain. 2019;142:2572–2580.
- Chiò A, Restagno G, Brunetti M, et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging. 2009;30:1272–1275.
- Bosco DA, Lemay N, Ko HK, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–4175.
- Dormann D, Rodde R, Edbauer D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. Embo J. 2010;29:2841–2857.
- DeJesus-Hernandez M, Kocerha J, Finch N, et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010;31:E1377–E1389.
- Savas JN, Toyama BH, Xu T, et al. Extremely long-lived nuclear pore proteins in the rat brain. Science. 2012;335:942.
- D’Angelo MA, Raices M, Panowski SH, et al. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295.
- Mertens J, Paquola ACM, Ku M, et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell. 2015;17:705–718.
- Lu T, Pan Y, Kao S-Y, et al. Gene regulation and DNA damage in the ageing human brain [Internet]. Nature. 2004;429:883–891.
- Kim HJ, Taylor JP. Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron. 2017;96:285–297.
- Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. [Internet. 2011; 3. DOI:10.1101/cshperspect.a004440.
- Woerner AC, Frottin F, Hornburg D, et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351:173–176.
- van Blitterswijk M, Rademakers R. Neurodegenerative disease: c9orf72 repeats compromise nucleocytoplasmic transport. Nat Rev Neurol. 2015;11:670–672.
- Vatsavayai SC, Yoon SJ, Gardner RC, et al. Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain. 2016;139:3202–3216.
- Proudfoot M, Gutowski NJ, Edbauer D, et al. Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability [Internet]. Acta Neuropathol. 2014;127:451–458.
- Baborie A, Griffiths TD, Jaros E, et al. Accumulation of dipeptide repeat proteins predates that of TDP-43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathol Appl Neurobiol. 2015;41:601–612.
- Fujii S, Takanashi K, Kitajo K, et al. Treatment with a global methyltransferase inhibitor induces the intranuclear aggregation of ALS-linked FUS mutant in vitro. Neurochem Res. 2016;41:826–835.
- Guo L, Kim HJ, Wang H, et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell. 2018;173:677–692.e20.
- Yoshizawa T, Ali R, Jiou J, et al. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell. 2018;173:693–705.e22.
- Hofweber M, Hutten S, Bourgeois B, et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell. 2018;173:706–719.e13.
- Qamar S, Wang G, Randle SJ, et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell. 2018;173:720–734.e15.
- Chou -C-C, Zhang Y, Umoh ME, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21:228–239.
- Archbold HC, Jackson KL, Arora A, et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep. 2018;8:4606.
- Ederle H, Funk C, Abou-Ajram C, et al. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci Rep. 2018;8:7084.
- Pinarbasi ES, Cağatay T, Fung HYJ, et al. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci Rep. 2018;8:7083.
- Furukawa Y, Kaneko K, Watanabe S, et al. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J Biol Chem. 2011;286:18664–18672.
- Smethurst P, Newcombe J, Troakes C, et al. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol Dis. 2016;96:236–247.
- Nomura T, Watanabe S, Kaneko K, et al. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem. 2014;289:1192–1202.
- Chia R, Tattum MH, Jones S, et al. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS One. 2010;5:e10627.
- Smethurst P, Risse E, Tyzack GE, et al. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain. 2020;143:430–440.
- Kim HJ, Kim NC, Wang Y-D, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473.
- Sun Z, Diaz Z, Fang X, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9:e1000614.
- Johnson BS, Snead D, Lee JJ, et al. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339.
- Couthouis J, Hart MP, Shorter J, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A. 2011;108:20881–20890.
- Couthouis J, Hart MP, Erion R, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–2911.
- Molliex A, Temirov J, Lee J, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133.
- Patel A, Lee HO, Jawerth L, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077.
- Mackenzie IR, Nicholson AM, Sarkar M, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron. 2017;95:808–816.e9.
- Chuang E, Hori AM, Hesketh CD, et al. Amyloid assembly and disassembly. J Cell Sci. Internet 2018;131: jcs189928. .
- Zhang P, Fan B, Yang P, et al. Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. Elife. 2019;8. Internet. DOI:10.7554/eLife.39578.
- Gomes E, Shorter J. The molecular language of membraneless organelles. J Biol Chem. 2019;294:7115–7127.
- Li YR, King OD, Shorter J, et al. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–372.
- Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736.
- Kieran D, Kalmar B, Dick JRT, et al. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405.
- Kalmar B, Lu C-H, Greensmith L. The role of heat shock proteins in amyotrophic lateral sclerosis: the therapeutic potential of arimoclomol. Pharmacol Ther. 2014;141:40–54.
- Wang P, Wander CM, Yuan C-X, et al. Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat Commun. 2017;8:82.
- Chen H-J, Mitchell JC, Novoselov S, et al. The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain. 2016;139:1417–1432.
- Benatar M, Wuu J, Andersen PM, et al. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS [Internet]. Neurology. 2018;90:e565–e574.
- Jackrel ME, DeSantis ME, Martinez BA, et al. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell. 2014;156:170–182.
- Jackrel ME, Shorter J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis Model Mech. 2014;7:1175–1184.
- Yasuda K, Clatterbuck-Soper SF, Jackrel ME, et al. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J Cell Biol. 2017;216:1015–1034.
- Shorter J. Engineering therapeutic protein disaggregases. Mol Biol Cell. 2016;27:1556–1560.
- Jackrel ME, Shorter J. Engineering enhanced protein disaggregases for neurodegenerative disease. Prion. 2015;9:90–109.
- Mack KL, Shorter J. Engineering and evolution of molecular chaperones and protein disaggregases with enhanced activity [Internet]. Front Mol Biosci. 2016;3. DOI:10.3389/fmolb.2016.00008
- Jackrel ME, Shorter J. Reversing deleterious protein aggregation with re-engineered protein disaggregases. Cell Cycle. 2014;13:1379–1383.
- Shorter J. Designer protein disaggregases to counter neurodegenerative disease [Internet]. Curr Opin Genet Dev. 2017;44:1–8.
- Wippich F, Bodenmiller B, Trajkovska MG, et al. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 2013;152:791–805.
- Rai AK, Chen J-X, Selbach M, et al. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature. 2018;559:211–216.
- Reineke LC, Tsai W-C, Jain A, et al. Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol Cell Biol. 2017;37. Internet. DOI:10.1128/MCB.00596-16.
- Nonaka T, Masuda-Suzukake M, Arai T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–134.
- Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629–9634.
- Prasad A, Raju G, Sivalingam V, et al. An acridine derivative, [4,5-bis(N-carboxy methyl imidazolium)methylacridine] dibromide, shows anti-TDP-43 aggregation effect in ALS disease models [Internet]. Sci Rep. 2016;6. DOI:10.1038/srep39490.
- Boyd JD, Lee P, Feiler MS, et al. A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J Biomol Screen. 2014;19:44–56.
- Cassel JA, McDonnell ME, Velvadapu V, et al. Characterization of a series of 4-aminoquinolines that stimulate caspase-7 mediated cleavage of TDP-43 and inhibit its function. Biochimie. 2012;94:1974–1981.
- Parker SJ, Meyerowitz J, James JL, et al. Inhibition of TDP-43 accumulation by bis(thiosemicarbazonato)-copper complexes. PLoS One. 2012;7:e42277.
- Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544:367–371.
- Elden AC, Kim H-J, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075.
- McGurk L, Gomes E, Guo L, et al. Poly(ADP-Ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell. 2018;71:703–717.e9.
- Maharana S, Wang J, Papadopoulos DK, et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360:918–921.
- Huang Y-C, Lin K-F, He R-Y, et al. Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS One. 2013;8:e64002.
- Ishiguro T, Sato N, Ueyama M, et al. Regulatory role of RNA chaperone TDP-43 for RNA misfolding and repeat-associated translation in SCA31. Neuron. 2017;94:108–124.e7.
- Flores BN, Li X, Malik AM, et al. An intramolecular salt bridge linking TDP43 RNA binding, protein stability, and TDP43-dependent neurodegeneration. Cell Rep. 2019;27:1133–1150.e8.
- Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677–685.
- Rusmini P, Cristofani R, Galbiati M, et al. The role of the heat shock protein B8 (HSPB8) in motoneuron diseases. Front Mol Neurosci. 2017;10:176.
- Mandrioli J, Crippa V, Cereda C, et al. Proteostasis and ALS: protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open. 2019;9:e028486.
- Towers CG, Thorburn A. Therapeutic targeting of autophagy [Internet]. EBioMedicine. 2016;14:15–23.
- Mandrioli J, D’Amico R, Zucchi E, et al. Rapamycin treatment for amyotrophic lateral sclerosis: protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine (Baltimore). 2018;97:e11119.
- Wang I-F, Guo B-S, Liu Y-C, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 2012;109:15024–15029.
- Fox JH, Connor T, Chopra V, et al. The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington’s disease. Mol Neurodegener. 2010;5:26.
- Tsvetkov AS, Miller J, Arrasate M, et al. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–16987.
- Fontaine SN, Zheng D, Sabbagh JJ, et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. Embo J. 2016;35:1537–1549.
- Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–E3147.
- Urushitani M, Ezzi SA, Julien J-P. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104:2495–2500.
- Takeuchi S, Fujiwara N, Ido A, et al. Induction of protective immunity by vaccination with wild-type apo superoxide dismutase 1 in mutant SOD1 transgenic mice [Internet]. J Neuropathol Exp Neurol. 2010;69:1044–1056.
- Gros-Louis F, Soucy G, Larivière R, et al. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010;113:1188–1199.
- Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015 [Internet]. Expert Opin Investig Drugs. 2017;26(6):735–739.
- Huang L-K, Chao S-P, Hu C-J. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. 2020;27:18.
- Hall CE, Yao Z, Choi M, et al. Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell Rep. 2017;19:1739–1749.
- Tyzack GE, Hall CE, Sibley CR, et al. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat Commun. 2017;8:1164.
- Serio A, Bilican B, Barmada SJ, et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2013;110:4697–4702.
- Giorgio FPD, Di Giorgio FP, Carrasco MA, et al. Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model [Internet]. Nat Neurosci. 2007;608–614. DOI:10.1038/nn1885.
- Giorgio FPD, Di Giorgio FP, Boulting GL, et al. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation [Internet]. Cell Stem Cell. 2008;3:637–648.
- Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487.
- Liu J, Wang F. Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications [Internet]. Front Immunol. 2017. DOI:10.3389/fimmu.2017.01005
- Xu Z, Lee A, Nouwens A, et al. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:362–376.
- Clarke LE, Liddelow SA, Chakraborty C, et al. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 2018;115:E1896–E1905.
- Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016;37(9):608–620.
- Kong Q, Chang L-C, Takahashi K, et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest. 2014;124:1255–1267.
- Ganel R, Ho T, Maragakis NJ, et al. Selective up-regulation of the glial Na+-dependent glutamate transporter GLT1 by a neuroimmunophilin ligand results in neuroprotection. Neurobiol Dis. 2006;21:556–567.
- Fischer A, Sananbenesi F, Mungenast A, et al. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–617.
- Lapucci A, Cavone L, Buonvicino D, et al. Effect of Class II HDAC inhibition on glutamate transporter expression and survival in SOD1-ALS mice. Neurosci Lett. 2017;656:120–125.
- Cudkowicz ME, Titus S, Kearney M, et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:1083–1091.
- Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77.
- Tanaka K, Kanno T, Yanagisawa Y, et al. Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis [Internet]. Exp Neurol. 2011;232:41–52.
- Rodríguez-Cueto C, Santos-García I, García-Toscano L, et al. Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1 transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem Pharmacol. 2018;157:217–226.
- Chiarotto GB, Cartarozzi LP, Perez M, et al. Tempol improves neuroinflammation and delays motor dysfunction in a mouse model (SOD1G93A) of ALS [Internet]. J Neuroinflammation. 2019;16. DOI:10.1186/s12974-019-1598-x.
- Yun SP, Kam T-I, Panicker N, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease [Internet]. Nat Med. 2018;24:931–938.
- Vallarola A, Sironi F, Tortarolo M, et al. RNS60 exerts therapeutic effects in the SOD1 ALS mouse model through protective glia and peripheral nerve rescue. J Neuroinflammation. 2018;15:65.
- Paganoni S, Alshikho MJ, Luppino S, et al. A pilot trial of RNS60 in amyotrophic lateral sclerosis. Muscle Nerve. 2019;59:303–308.
- Mazzini L, Gelati M, Profico DC, et al. Human neural stem cell transplantation in ALS: initial results from a phase I trial. J Transl Med. 2015;13:17.
- Glass JD, Boulis NM, Johe K, et al. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells. 2012;30:1144–1151.
- Feldman EL, Boulis NM, Hur J, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol. 2014;75:363–373.
- Glass JD, Hertzberg VS, Boulis NM, et al. Transplantation of spinal cord-derived neural stem cells for ALS: analysis of phase 1 and 2 trials. Neurology. 2016;87:392–400.
- Suzuki M, McHugh J, Tork C, et al. GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One. 2007;2:e689.
- Nichols NL, Gowing G, Satriotomo I, et al. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis [Internet]. Am J Respir Crit Care Med. 2013;187:535–542.
- Lepore AC, O’Donnell J, Kim AS, et al. Human glial-restricted progenitor transplantation into cervical spinal cord of the SOD1 mouse model of ALS. PLoS One. 2011;6:e25968.
- Kondo T, Funayama M, Tsukita K, et al. Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Reports. 2014;3:242–249.
- Sareen D, Gowing G, Sahabian A, et al. Human induced pluripotent stem cells are a novel source of neural progenitor cells (iNPCs) that migrate and integrate in the rodent spinal cord. J Comp Neurol. 2014;522:2707–2728.
- Corcia P, Tauber C, Vercoullie J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One. 2012;7:e52941.
- Turner MR, Cagnin A, Turkheimer FE, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–609.
- Kawamata T, Akiyama H, Yamada T, et al. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691–707.
- Engelhardt JI, Appel SH. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol. 1990;47:1210–1216.
- Brettschneider J, Toledo JB, Van Deerlin VM, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012;7:e39216.
- Chiu IM, Morimoto ETA, Goodarzi H, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4:385–401.
- Liao B, Zhao W, Beers DR, et al. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS [Internet]. Exp Neurol. 2012;237:147–152.
- Ohgomori T, Yamada J, Takeuchi H, et al. Comparative morphometric analysis of microglia in the spinal cord of SOD1G93Atransgenic mouse model of amyotrophic lateral sclerosis [Internet]. Eur J Neurosci. 2016;43:1340–1351.
- Gravel M, Béland L-C, Soucy G, et al. IL-10 controls early microglial phenotypes and disease onset in ALS caused by misfolded superoxide dismutase 1. J Neurosci. 2016;36:1031–1048.
- Beers DR, Henkel JS, Zhao W, et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134:1293–1314.
- Moisse K, Strong MJ. Innate immunity in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1083–1093.
- Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581.e9.
- Boillée S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392.
- Beers DR, Henkel JS, Xiao Q, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026.
- Spiller KJ, Restrepo CR, Khan T, et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy [Internet]. Nat Neurosci. 2018;21:329–340.
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023.
- Hu H, Lin H, Duan W. Intrathecal Injection of scAAV9–hIGF1 prolongs the survival of ALS model mice by inhibiting the NF-kB pathway. Neuroscience. 2018;381:1–10.
- Dong H, Zhang W, Zeng X, et al. Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol Neurobiol. 2014;49:1487–1500.
- Apolloni S, Fabbrizio P, Parisi C, et al. Clemastine confers neuroprotection and induces an anti-inflammatory phenotype in SOD1G93A mouse model of amyotrophic lateral sclerosis [Internet]. Mol Neurobiol. 2016;53:518–531.
- Granucci EJ, Griciuc A, Mueller KA, et al. Cromolyn sodium delays disease onset and is neuroprotective in the SOD1 mouse model of amyotrophic lateral sclerosis. Sci Rep. 2019;9:17728.
- Meer F, Molawi K, Zychlinsky A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc Natl Acad Sci U S A. 2010;541:13046–13050.
- Zhou Q, Mareljic N, Michaelsen M, et al. Active poly-GA vaccination prevents microglia activation and motor deficits in a C9orf72 mouse model. EMBO Mol Med. 2020;12:e10919.
- Martínez-Muriana A, Mancuso R, Francos-Quijorna I, et al. CSF1R blockade slows the progression of amyotrophic lateral sclerosis by reducing microgliosis and invasion of macrophages into peripheral nerves. Sci Rep. 2016;6:25663.
- Trias E, Ibarburu S, Barreto-Núñez R, et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation. 2016;13:177.
- Suzumura A, Ito A, Yoshikawa M, et al. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999;837:203–212.
- Mizuno T, Kurotani T, Komatsu Y, et al. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46:404–411.
- Kang SH, Li Y, Fukaya M, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579.
- Philips T, Bento-Abreu A, Nonneman A, et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis [Internet]. Brain. 2013;136:471–482.
- Lee Y, Morrison BM, Li Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448.
- Kim S, Chung A-Y, Na JE, et al. Myelin degeneration induced by mutant superoxide dismutase 1 accumulation promotes amyotrophic lateral sclerosis. Glia. 2019;67:1910–1921.
- Ferraiuolo L, Meyer K, Sherwood TW, et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc Natl Acad Sci U S A. 2016;113:E6496–E6505.
- van Rheenen W, Shatunov A, Dekker AM, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043–1048.
- Clarke BE, Taha DM, Tyzack GE, et al. Regionally encoded functional heterogeneity of astrocytes in health and disease: A perspective [Internet]. Glia. 2020. DOI:10.1002/glia.23877.
- Zirra A, Wiethoff S, Patani R. Neural conversion and patterning of human pluripotent stem cells: a developmental perspective. Stem Cells Int. 2016;2016:8291260.
- Patani R. Generating diverse spinal motor neuron subtypes from human pluripotent stem cells. Stem Cells Int. 2016;2016:1036974.
- Maffioletti SM, Sarcar S, Henderson ABH, et al. Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 2018;23:899–908.
- Patani R. Human stem cell models of disease and the prognosis of academic medicine. Nat Med. 2020;26:449.
- Devine H, Patani R. The translational potential of human induced pluripotent stem cells for clinical neurology: the translational potential of hiPSCs in neurology. Cell Biol Toxicol. 2017;33:129–144.
- Kelley KW, Ben Haim L, Schirmer L, et al. Kir4.1-dependent astrocyte-fast motor neuron interactions are required for peak strength. Neuron. 2018;98:306–319.e7.
- Simone R, Balendra R, Moens TG, et al. G-quadruplex-binding small molecules ameliorate FTD/ALS pathology and. EMBO Mol Med. 2018;10:22–31.
- Patani R, Lewis PA, Trabzuni D, et al. Investigating the utility of human embryonic stem cell-derived neurons to model ageing and neurodegenerative disease using whole-genome gene expression and splicing analysis. J Neurochem. 2012;122:738–751.
- Ziff OJ, Patani R. Harnessing cellular aging in human stem cell models of amyotrophic lateral sclerosis. Aging Cell. 2019;18:e12862.