
ABSTRACT
Introduction
Genomic studies have allowed to identify molecular predictors for chronic lymphocytic leukemia (CLL) treatment tailoring. TP53 disruption is the strongest predictor of chemo-refractoriness and its assessment is the first decisional node in the disease treatment algorithm.
Areas covered
The review covers the p53 biological pathway, its genetic alterations and clinical implications in CLL, and its druggable targets. The potential therapeutic options for TP53 disrupted patients are described, including: i) agents circumventing TP53 disruption; ii) targeted therapies restoring the physiological function of mutant p53; and iii) medicines potentiating p53 function.
Expert opinion
The key approach to improve CLL outcome is treatment tailoring in individual patients. BCR and BCL2 inhibitors have significantly improved CLL survival, however TP53 disrupted patients still have a less favorable outcome than wild type cases, possibly because these novel drugs do not directly target p53 and do not restore the function of the disrupted p53 pathway. Emerging innovative molecules in cancer are able to restore the p53 mutant protein and/or potentiate the activity of the p53 wild type protein. If these compounds were confirmed as efficacious also for CLL, they would represent another step forward in the care of high risk CLL patients with TP53 abnormalities.
1. Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in Western countries, representing approximately 30% of all adult leukemias [Citation1]. Although the median survival of patients with CLL is around 10 years, the disease has an extremely variable clinical course [Citation2]. The extensive body of molecular studies in CLL has allowed to better understand the disease pathogenesis and also to identify molecular predictors to select the most suitable therapy for every single patient [Citation3]. Among these molecular predictors, TP53 currently represents the pivotal and initial decisional node for treatment tailoring in CLL. TP53 disrupted patients, due to their chemorefractoriness, are treated with biological drugs whose mode of action (MoA) is independent of TP53 and are able, at least in part, to circumvent the refractoriness imposed by TP53 disruption [Citation3]. These biological agents include: i) B-cell receptor inhibitors (BCRi) that inhibit the BCR signaling pathway essential for survival and growth of malignant B cells, and ii) BCL2 inhibitors (BCL2i) that inhibit the anti-apoptotic protein BCL2 whose overexpression is a hallmark of CLL. BCRi and BCL2i are changing the natural history of CLL [Citation2,Citation3]. However, BCRi and BCL2i do not directly target TP53, but act in a TP53 independent manner. Recently, innovative drugs directly targeting p53 and its pathway are emerging and entering clinical trials for several human cancers [Citation4].
In the present review we discuss the TP53 gene from both a biological and a clinical point of view. We then focus on the different therapeutic strategies in CLL that are capable of circumventing TP53 disruption, as well as on novel promising compounds that may restore the function of the mutated p53 protein or potentiate the activity of wild type p53.
1. The TP53 gene
TP53 is a member of a broader gene family including also TP63 and TP73, of which only TP53 has well documented tumor suppressive capabilities. TP53 maps to the most distal band of chromosome 17 (17p13.1) and encodes the tumor-suppressor protein p53, a DNA binding protein that regulates transcription of numerous cellular activities including cell cycle, apoptosis and promotion of DNA repair in response to cellular stress signals such as DNA damage, hypoxia, and ribosomal stress, that are often encountered during tumor development and malignant progression [Citation5]. Following DNA damage, p53 can trigger either apoptosis or G1 cell-cycle arrest until the cell has completed DNA repair processes, thereby preventing replication of potentially harmful genetic abnormalities [Citation5,Citation6]. Because of its central role in the DNA damage response (DDR), p53 is often referred to as the ‘guardian of the genome’.
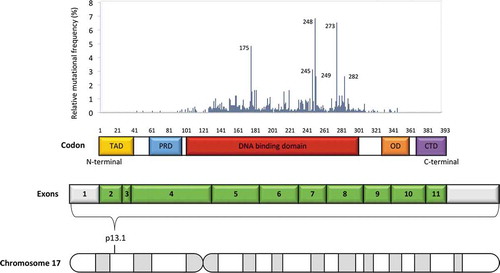
The p53 protein consists of different domains, each of which harbors unique characteristics that allow its correct function () [Citation7]. The two tandem transcription activation domains (TADs) located at the amino terminus region are needed for proper p53 target gene induction in response to DNA damage and for exploiting the p53 tumor suppressor function [Citation8]. Immediately close to the TADs is a proline-rich domain that contributes to transcription activation and is necessary for restricting cell growth [Citation9]. The region adjacent to the proline-rich domain of the p53 protein includes the DNA-binding domain and represents the core of p53 function [Citation7]. Consistently, most mutations that impair p53 function are located in this domain. The p53 protein ends at the C-terminal region with the oligomerization domain. Physiologically, p53 binds to its DNA target site as a tetramer (dimer of dimers) and, although the core domain can tetramerize on its own, the oligomerization domain facilitates these interactions [Citation10,Citation11]. Also, p53 post-translational modifications (PTMs), such as acetylation, seem to have a role in regulating the strength of DNA binding, target-gene selection, stability and overall p53 function [Citation12].
Figure 1. Schematic p53 domain structure and mapping of tumor associated mutations. The TP53 gene maps at the p13.1 locus on the short arm of chromosome 17 and comprises 11 exons. Coding exons are represented in green and non-coding exons (or parts of exons) in gray. p53 is a transcription factor containing several well defined domains, including: an amino-terminal transactivation domain (TAD), a proline rich domain (PRD), a central sequence-specific DNA-binding core, an oligomerization domain (OD) and a carboxy-terminal domain (CTD). While 95% of gene mutations occur within the DNA binding domain (codons 100–300, exons 4–8), a few gene mutations, albeit with a lower prevalence, have been detected in almost every codon
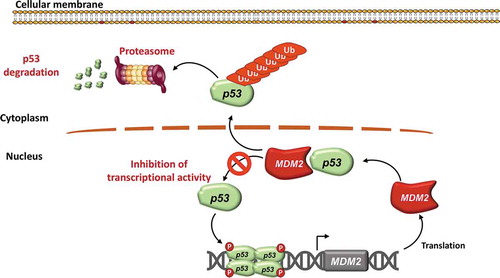
Under basal conditions, p53 protein in normal cells is maintained at low levels by a series of regulators such as mouse double minute 2 (MDM2), an E3 ubiquitin protein ligase that facilitates ubiquitin mediated degradation () [Citation13]. Conversely, in response to cellular stress, p53 is phosphorylated, rendering it insensitive to MDM2 degradation and leading to p53 accumulation in cells. When activated, p53 binds strongly to several specific DNA sequences via the two transactivation domains and thus starts the p53 transcriptional response () [Citation14]. The absolute levels of p53 protein matter for the choice of cellular outcome in response to DNA damage, but they are not the only determinants, since changes in p53 levels over time also affect cell fate: temporal regulation of p53 protein levels is therefore emerging as an additional modulator of cell fate control by p53 [Citation14].
Figure 2. MDM2 regulates p53 protein levels and function. p53 is a transcription factor that exerts its function as a tetramer. MDM2 is a ubiquitin protein ligase that physiologically catalyzes p53 ubiquitination. Upon ubiquitination, p53 is then degraded in the proteasome. MDM2 is also a transcriptional target gene of TP53, thus creating a negative autoregulatory feedback
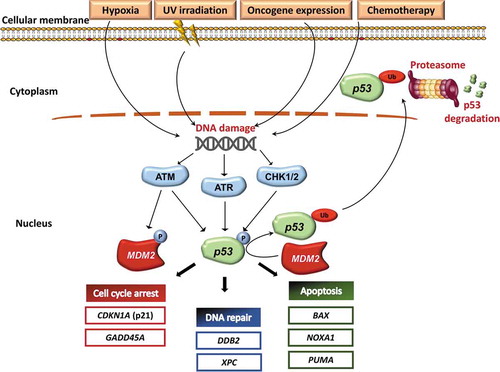
The genes induced by p53 activation span multiple biological functions and include: i) DDR (DDB2 and XPC); ii) cell cycle arrest (CDKN1A encoding p21 and GADD45A); and iii) apoptosis (NOXA1, PUMA and BAX) () [Citation7,Citation14]. Therefore, in cells with nonfunctional or dysfunctional p53, DNA damage cannot induce cell cycle arrest or DNA repair, enabling cells to tolerate and accumulate substantial levels of DNA alterations, that increase genomic instability and thereby lead to the emergence of cells with additional genetic mutations that can drive malignant progression [Citation15,Citation16]. A paradigmatic example is the Li-Fraumeni syndrome, where the presence of a germ-line TP53 mutation induces predisposition to early onset cancers [Citation17]. Indeed, it has been shown how the presence of mutant TP53 compromises cellular responses to therapeutic agents in vitro, closely reflecting the association of TP53 mutations with reduced responses to chemotherapeutic agents [Citation18].
Figure 3. Activation of the p53 pathway. Oncogene induction, stress signals, UV irradiation and chemotherapy may activate the p53 signaling pathway. As a consequence, ATM, ATR, CHK1 and CHK2 phosphorylate both MDM2 and p53. ATM, ATR, CHK1 and CHK2 are kinase proteins that are activated by DNA damage and play a pivotal role in activating the DNA damage response machinery. On the one hand, phosphorylation of MDM2 inhibits its normal activity. Inhibition of MDM2 activity, in turn, prevents p53 ubiquitination and proteasome degradation. On the other hand, p53 phosphorylation stabilizes the protein thus upregulating the transcription of TP53 target genes. p53 target genes include genes that negatively regulate the cell cycle (CDKN1A, GADD45A), genes involved in the DNA repair machinery (DDB2 and XPC), and proapoptotic genes (BAX, NOXA1 and PUMA). Therefore, activation of the pathway by an array of stimuli induces the accumulation of the p53 protein in the nucleus by two distinct biochemical mechanisms, represented by reduced protein degradation and increased protein stabilization. In turn, nuclear accumulation of p53 leads to inhibition of cell cycle, enhanced DNA repair, and increased apoptosis
In CLL, the DDR pathway is also disrupted by lesions of the ATM gene, including both 11q22–23 deletions and ATM mutations [Citation3]. Deletions in the 11q22-23 chromosomal region are found in ~20% of patients with CLL at diagnosis, and more than one-third of 11q deleted CLL cases carry mutations in the remaining ATM allele [Citation3]. Similar to cases with TP53 disruption, also ATM disrupted CLL are associated with genomic instability, the acquisition of additional genetic lesions and chemoresistance [Citation19–21]. Due the fact that the TP53 and ATM gene products exert their function in the same biological pathway, namely DDR, genetic lesions of TP53 and ATM are usually mutually exclusive [Citation3].
TP53 is the most frequently disrupted tumor suppressor gene and overall the most frequently mutated gene in cancer. The function of p53 in cancer may be lost by various mechanisms, including 17p13 deletion and mutations within the TP53 gene. Tumor associated mutations in TP53 are predominantly point mutations in more than 90% of cases that result in single amino-acid substitutions of p53 [Citation22,Citation23]. Among the reported tumors that harbor TP53 mutations, missense substitutions are the most common variants representing approximately 75% of TP53 mutations. Approximately 1,500 different missense TP53 variants have been identified, ranging from several hotspots (at positions 175, 248, or 273) to infrequent variants detected at very low frequencies [Citation22,Citation23]. Based on available data, 95% of mutations target the DNA binding domain of the protein (amino acids 100–300), and only 5% of TP53 mutations are found in the regulatory domains (amino terminus, amino acids 1–99; carboxyl terminus, amino acids 301–393) () [Citation22,Citation23]. TP53 missense mutations produce a mutant p53 protein that cannot activate the p53 tumor suppressive transcriptional response and, at least in some instances, lead to dominant-negative inhibition of wild-type p53 function [Citation24,Citation25]. Moreover, TP53 mutations affecting the DNA binding domain may also lead to a functional phenotype that goes beyond the sole inactivation of p53 function [Citation26]. For instance, since p53 indirectly regulates BCR signaling in CLL, p53 aberrant B cells fail to limit BCR activation after DNA damage induced by chemotherapy [Citation27].
Loss of function of TP53 is therefore considered a major event in tumorigenesis and is also linked to chemotherapy resistance and a poor prognosis in many cancers. Although hematological cancers possess these mutations at a lower rate than solid tumors, CLL displays an unusually high number of TP53 aberrations with well-documented evidence for their role in disease pathogenesis [Citation28–30].
2. TP53 analysis in CLL
Chronic lymphocytic leukemia displays a highly heterogeneous clinical behavior, and TP53 gene defects represent a key decision-making biomarker in the algorithm for CLL treatment. In fact, chromosomal 17p13 deletion and TP53 gene mutations are consistently associated with adverse disease outcome due to resistance to chemoimmunotherapy (CIT) [Citation2,Citation3,Citation31]. Since TP53 mutational status has a major impact on treatment decisions, international collaborations, such as the European Research Initiative on CLL (ERIC), have developed a network of TP53 reference centers in various countries aiming to provide standardization and harmonization of diagnostic strategies for TP53 mutational assessment [Citation32]. Assessment of TP53 aberrations is mandatory for patients requiring therapy [Citation31,Citation32]. Importantly, TP53 status must be reassessed before the initiation of any subsequent line of therapy since clonal evolution can occur [Citation29,Citation31,Citation32]. Consistently, the frequency of TP53 abnormalities in patients requiring first-line treatment is 10 to 12%, rises to 40% in fludarabine-refractory patients and to 60% in patients with Richter syndrome [Citation3,Citation33,Citation34].
For most CLL patients, peripheral blood is an appropriate starting material for TP53 mutation analysis. One exception is represented by patients with a disease characterized predominantly by lymphadenopathy with few circulating clonal cells (i.e. small lymphocytic lymphoma), for which bone marrow or lymph node biopsies may be an alternative option [Citation32]. The techniques frequently used for assessing TP53 status in CLL include fluorescent in situ hybridization (FISH) for 17p13 deletion, as well as Sanger sequencing and next-generation sequencing (NGS) for TP53 mutations. Because TP53 mutations are associated with a poor prognosis independent of the presence of 17p13 deletion, it is important to assess both types of genetic alterations [Citation31,Citation32]. The region of the TP53 gene subjected to DNA sequencing must include at least exons 4–10, that correspond to the DNA-binding domain (codons 100–300) and to the oligomerization domain (codons 323–356) of the protein [Citation32]. Different studies have shown that mutations can also occur in exons outside the DNA binding domain, albeit with low prevalence. Therefore, the analysis of the whole coding region of TP53 (exons 2–11) is desirable in order to have a comprehensive view of TP53 mutational status [Citation32].
Sanger sequencing is a relatively simple sequencing approach that has been commonly used to assess TP53 mutation status in CLL [Citation19,Citation32]. The ERIC TP53 Network provides the opportunity to analyze Sanger sequencing data via a web-based software called GLASS, that has been created in order to assist with the assessment of somatic gene variations and to provide a standardized variant output as recommended by the Human Genome Variation Society (HGVS) [Citation32,Citation35]. The major limitation of Sanger sequencing is its sensitivity, with a detection threshold for mutated alleles of 10%, so that minor subclones cannot be detected. Conversely, targeted NGS has been demonstrated to detect all variants detected by Sanger and in addition to have a higher sensitivity compared to Sanger sequencing, allowing the detection also of TP53 mutated minor subclones [Citation32,Citation36]. The clinical importance of TP53 mutated minor subclones, however, is still uncertain, and has not been confirmed in the UK LRF CLL4 trial [Citation37]. Although retrospective studies have demonstrated that TP53 mutated minor subclones appear to carry the same unfavorable prognostic impact as clonally represented TP53 mutations, recent recommendations from ERIC have concluded that there is not yet sufficient evidence to make therapeutic decisions based on mutations whose intraclonal representation, independent of the sequencing strategy utilized, is below the sensitivity threshold of Sanger sequencing (i.e. 10% of variant allele frequency, usually termed as VAF) [Citation32]. This, however, is an evolving field and results from clinical trials analyzing the impact of minor TP53 subclones are highly desirable.
To describe genetic variants, it is recommended to use the HGVS nomenclature, reporting all variants in terms of genomic DNA, coding DNA and RNA sequences, including the entire gene with the promoter and the 5ʹ and 3ʹ untranslated region in the genomic reference sequence [Citation22,Citation23,Citation32]. Therefore, variant descriptions in terms of protein and RNA should be indicated in parenthesis if effects at the RNA and protein level are predicted [Citation22].
In addition to FISH and DNA sequencing analysis, functional read‐outs of the ATM/TP53 axis might provide clinically relevant information on the actual DDR and chemo‐sensitivity of the leukemia, as well as important biological insights on the signaling capacity of the axis [Citation27,Citation38–40]. Several p53‐function assays for CLL have been developed, that measure the expression of responsive targets at the (micro‐)RNA (eg: miR-34a) or protein level [Citation27,Citation38–40]. For instance, the miR-34 family has been discovered to be a direct p53 transcriptional target and mediates some of the p53-dependent effects, including the induction of cell cycle arrest by p21 and apoptosis induction. miR-34a levels are up-regulated after DNA damage in the presence of functional p53, but not in cases with TP53 disruption, and therefore may represent a surrogate to better evaluate p53 function [Citation40].
3. TP53 as a molecular predictor for treatment tailoring in CLL
In CLL, the prevalence of TP53 abnormalities, including 17p deletion and TP53 mutations, varies across the different phases of the disease. In early stage asymptomatic CLL patients and in CLL patients requiring treatment, TP53 abnormalities are detected in approximately 5–7% and 10%, respectively [Citation3,Citation30,Citation33,Citation34,Citation41]. As previously described, p53 plays a pivotal role in the DDR induced by chemotherapy [Citation5–7,Citation14]. When TP53 is disrupted by mutation and/or deletion, chemotherapy fails to induce apoptosis in CLL cells conferring a growth advantage to the clones harboring TP53 disruption [Citation7]. Consistently, the frequency of TP53 disruption reaches 30–35% in chemorefractory patients, in whom TP53 disrupted cells progressively emerge as the dominant clone [Citation33,Citation42].
Several retrospective and prospective studies have substantiated the notion that patients with TP53 disruption are characterized by a poor outcome when treated with chemotherapy or chemoimmunotherapy (CIT) [Citation43–46]. Importantly, the negative prognostic impact of TP53 disruption has been confirmed with both low-intensity CIT, such as chlorambucil-obinutuzumab, as well as with higher intensity CIT such as fludarabine-cyclophosphamide-rituximab [Citation44–46]. In the era of CIT, the unmet clinical need of TP53 disrupted patients has fostered the development of novel drugs that are effective in this setting of patients.
Recent evidence supports also the notion that TP53 abnormalities might impact on the pharmacokinetics of anti-CD20 monoclonal antibodies (i.e. rituximab) [Citation47,Citation48]. In fact, pharmacokinetic studies have documented that rituximab clearance is faster in TP53 disrupted patients than in wild type patients, suggesting an implication of TP53 non only in the DDR, but also in the effectiveness of anti-CD20 monoclonal antibodies [Citation47].
One important aspect concerns the significance of TP53 disruption in CLL patients with an early stage according to the Binet and Rai staging systems [Citation2]. The prevalence of TP53 abnormalities in this group of CLL patients is low and, especially in the presence of mutated IGHV genes, does not predict a worse outcome [Citation49,Citation50]. Recently, the IPS-E (International Prognostic Score-Early) new prognostic model has identified three different subgroups of treatment naïve Binet A CLL patients with a high risk of early treatment requirement according to the combination of lymphocyte count >15,000/μl, palpable lymph nodes and unmutated IGHV genes [Citation41]. Interestingly, in this model, TP53 abnormalities did not sort out as an independent predictor of shorter time to first treatment, in line with the notion that TP53 disruption interacts with the selective pressure exerted by treatment but not with a watch and wait strategy [Citation41]. Conversely, TP53 abnormalities are an important component of the CLL-IPI (CLL International Prognostic score) that is designed using overall survival as primary endpoint, and therefore may be affected by treatment exposure [Citation51].
4. Drugs that circumvent TP53 disruption
Before the introduction of BCRi and BCL2i, alemtuzumab was the only drug that demonstrated efficacy in TP53 disrupted patients, including patients with mutations affecting the DNA binding domain [Citation52]. BCRi and BCL2i have been changing the natural history of CLL and are able to circumvent, at least in part, the negative prognostic impact imposed by TP53 disruption. However, neither BCRi nor BCL2i directly target p53, but rather act in a TP53 independent manner.
Results from phase 1 and phase 2 clinical trials with a 5-year follow-up of the BCRi ibrutinib used as single-agent have highlighted the high efficacy of the drug and the long lasting durability of the response [Citation53,Citation54]. Because the MoA of ibrutinib circumvents the DNA damage response, the drug is also effective in patients harboring TP53 disruption. Despite this, however, the presence of a disrupted TP53 affects CLL progression-free survival (PFS) also in the context of ibrutinib treatment when compared to TP53 wild type patients. For example, in the ibrutinb arm of the RESONATE trial, comparing ibrutinib versus ofatumumab in relapsed/refractory CLL, the median PFS of TP53 mutated patients was 40.7 months but was not reached in wild type patients (HR 1.4, 95% C.I 0.8–2.4) () [Citation55]. Nevertheless, the PFS of TP53 disrupted patients treated with ibrutinib is longer compared to historical cohorts of CLL patients treated with CIT [Citation53–55]. Similar to ibrutinib, BCL2i inhibitors have demonstrated high efficacy also in patients with TP53 disruption in phase 1 and phase 2 clinical trial, although TP53 status remains a negative prognostic factor also in patients treated with BCL2i () [Citation56–58].
Table 1. Impact of TP53 gene mutations in CLL clinical trials with BCRi and BCL2i
The positive results of the above mentioned phase 1 and 2 clinical trials prompted the design of phase 3 clinical trials comparing CIT vs BCRi or BCL2i [Citation59–65]. The trial comparing bendamustine plus rituximab versus ibrutinib versus ibrutinib plus rituximab in treatment naïve patients has demonstrated an improved PFS for patients with 17p deletion when treated with ibrutinib, independent of the addition of rituximab [Citation59]. Similarly, also in the trial comparing ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab (iLLUMINATE trial), the hazard ratio for patients with TP53 disruption when treated with ibrutinib was 0.11, 95% C.I (0.3–0.38) [Citation60]. Acalabrutinib, a selective and covalent BTK inhibitor, has also demonstrated high efficacy alone or in combination with obinutuzumab in TP53 disrupted patients compared to chlorambucil plus obinutuzumab in the context of the ELEVATE-TN trial [Citation64]. Compared to the chlorambucil plus obinutuzumab arm, the HR for PFS in TP53 disrupted patients was 0.10 (0.03–0.34) and 0.23 (0.09–0.61) in the acalabrutinib plus obinutuzumab and in the acalabrutinib single agent arms, respectively [Citation64]. Idelalisib, a small molecule that targets the delta isoform of phosphoinositol 3-kinase, thus inhibiting the BCR signaling with another mode of action compared to ibrutinib and acalabrutinib, has demonstrated high efficacy in TP53 disrupted patients, but its toxicity profile limits its use to patients who are not candidate to ibrutinib or acalabrutinib [Citation65].
These randomized trials have documented that BCRi are more effective than CIT in patients with TP53 disruption. However, till now, no data are available regarding the impact of TP53 disruption within the ibrutinib containing arms. A limitation of some of these studies is represented by the fact that TP53 disrupted patients currently are not candidate for CIT and that the inclusion of TP53 disrupted patients in randomized trials of CIT vs BCRi might have reduced the survival of the CIT arm.
Regarding BCL2i, the phase 3 trial comparing venetoclax-obinutuzumab versus chlorambucil-obinutuzumab has demonstrated a higher efficacy of venetoclax-obinutuzumab in patients with either 17p deletion or TP53 mutations, or both, with a HR of 0.33, 95% C.I (0.12–0.89) and 0.35, 95% C.I (0.14–0.89), respectively [Citation58]. Despite the improvement of PFS for patients with TP53 disruption in the venetoclax-obinutuzumab arm compared to the CIT arm, the trial results show that TP53 disruption maintains a relatively poor prognostic role also in the context of venetoclax-obinutuzumab treatment with a HR of 3.39 (p = 0.03) [Citation58]. The CLL14 trial treatment schedule consists of a fixed duration therapy of both chlorambucil-obinutuzumab and venetoclax-obinutuzumab. The worse outcome of TP53 disrupted patients also in the venetoclax-obinutuzumab arm may suggest that a fixed duration strategy is not optimal for these high risk patients, who might require a more prolonged pharmacological control of the disease. Future clinical trials are essential to confirm this hypothesis.
All the aforementioned results on the efficacy of BCRi and BCL2i in TP53 disrupted patients are derived from subgroup analysis that are not statistically powered, and future phase 3 randomized trials are needed to confirm these findings.
5. Targeted therapies restoring the physiological function of mutant p53 protein
BCRi and BCL2i do not directly target the p53 protein, but instead exert their antileukemic action through a p53 independent pathway that may circumvent, at least in part, the chemorefractoriness conferred by TP53 disruption. Historically, p53 has been difficult to target with drugs for a number of reasons: i) lack of an accessible and hydrophobic pocket allowing the binding by small molecules with high affinity; ii) lack of an enzymatic activity that can be targeted by selective inhibitors; iii) nuclear localization, preventing targeting with high molecular weight drugs such as monoclonal antibodies; iv) not all mutations are functionally equivalent [Citation4].
Drug research in the cancer field has generated many inhibitors of proto-oncogenes, but few ‘activators’ or ‘correctors’ of tumor suppressor proteins that have lost their physiological function due to mutations. In this respect, a promising model can be drawn from the example of a non-neoplastic disease, i.e. cystic fibrosis, an autosomal recessive disorder caused by mutations that disrupt the function of an ion channel, namely the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The introduction of new molecules that are able to restore the function of the CFTR mutated protein has been changing the natural history of the disease [Citation66]. Based on their MoA, these agents may be distinguished into correctors and potentiators. Correctors are small-molecules that improve the processing and trafficking of CFTR thus increasing its cell-surface expression, whereas potentiators are small-molecules that augment channel gating [Citation67–70].
A drug that specifically targets the mutant p53 protein should be able to revert the mutated protein to its normal function. Because several TP53 mutations, namely the so called ‘conformational’ mutations, alter the configuration of the wild type p53 protein, restoring the normal conformation would be expected to induce mutant p53 to regain its normal properties and tumor suppressor activity [Citation4]. In the field of cancer, many efforts have been devoted to the development of new molecules that are able to restore the physiological function of TP53 mutated proteins. APR-017 (PRIMA-1) and its methylated form APR-246 (PRIMA-1 MET; Eprenetapopt) are first-in-class drugs, identified by drug screening, that selectively induce apoptosis in cancer cells with mutant TP53 [Citation4,Citation71]. Mechanistically, APR-246 is a prodrug that is transformed into its active form, namely methylene quinuclidinone (MQ). MQ is a Michael acceptor that can react covalently with nucleophiles, such as cytidine. APR-246 can bind to the cysteine residue 124 and 277 in the DNA binding domain of p53 and, in an in vitro model, is able to restore the physiological p53 conformation and function in cell lines with TP53 missense mutations [Citation72]. Interestingly, both unfolded mutant TP53 and unfolded wild-type TP53 can be refolded by these compounds [Citation72–75]. In addition, MQ can also induce oxidative stress in cells and is able to reduce the cell export of platinum based chemotherapeutic agents, suggesting a synergistic effect of APR-246 with chemotherapy [Citation58].
APR-246, alone or in combination with other drugs, has shown antitumor activity in different solid and hematological tumors. In myelodysplastic syndromes (MDS), APR-246 has demonstrated high efficacy especially in combination with azacytidine (AZA) [Citation76]. Based on these promising results, a phase III international trial comparing AZA alone and AZA + APR 246 is ongoing in TP53 mutated MDS (NCT03745716). Recently, the Food and Drug Administration has granted Fast Track and Orphan Drug Designation to APR-246 for the treatment of MDS carrying a TP53 mutation. In CLL, the activity of APR-246 has been only tested in pre-clinical models, in which the compound has demonstrated activity against TP53 mutant CLL cells [Citation73,Citation74,Citation77]. An ongoing phase I clinical trial is planned to start in the second half of 2020 for patients with TP53 mutant relapsed/refractory non-Hodgkin lymphoma, including CLL/SLL (NCT04419389).
Approximately 10% of TP53 mutations are nonsense mutations leading to the insertion of a premature stop codon that causes the translation of a truncated protein [Citation26,Citation27]. Importantly, APR-246 acts by refolding mutant p53 protein but is not effective in cases of truncated proteins caused by nonsense mutations [Citation78]. Nonsense mutations, therefore, would require drugs with a MoA different from that of APR-246 or other drugs that revert conformational mutants to the wild type p53 configuration [Citation4]. In fact, when ribosome encounters a premature termination codon, release factor eRF1 and eRF3 bind to the nascent amino acid chain, and translation is terminated leading to the release of a truncated p53 protein. Readthrough induction allows the ribosome to continue translation until the physiological stop codon is encountered resulting in a full-length p53 protein [Citation78]. Different molecules are able to rescue nonsense mutated p53. Initial evidence has demonstrated that aminoglycosides (i.e. G418, gentamicin and amikacin) restore the function of a p53 truncated protein with a mechanism that is not completely understood but is probably related to the binding of aminoglycosides to the ribosomal proteins [Citation78,Citation79]. The development of novel readthrough inducer molecules is ongoing.
6. Targeted therapies that potentiate p53 function
The p53 protein is highly regulated by different molecules that control its activity. MDM2 is an E3 ubiquitin ligase that controls the half-life of p53 via ubiquitin-dependent proteasomal degradation () [Citation13,Citation80]. In response to cellular stress, the p53-MDM2 interaction is disrupted, and p53 can exert its function by activating the transcription of target genes that trigger cell cycle arrest and apoptosis [Citation81]. Since the discovery of the first selective MDM2 inhibitor, i.e. the Nutlin-3a small molecule, newer compounds have been developed with increased potency and improved bioavailability [Citation82,Citation83]. These non-genotoxic compounds bind to MDM2 in the p53-binding pocket and can release p53, leading to effective stabilization of the protein and activation of the Tp53 pathway () [Citation82,Citation83]. Initial preclinical and clinical studies have demonstrated promising efficacy of this class of drugs in a number of TP53 wild type adult and pediatric cancers, both as single agents and in combination with other targeted therapies [Citation84–90].
Figure 4. Inhibitors of MDM2 and XPO1 interfere with p53 degradation and nuclear export. The physiological function of p53 is exerted in the nucleus, where p53 binds to DNA and induces the transcription of its target genes, thus favoring cell cycle arrest, DNA repair, and apoptosis. Under basal conditions, the p53 protein in normal cells is maintained at low levels by MDM2, an E3 ubiquitin protein ligase that facilitates p53 ubiquitin-mediated degradation. MDM2 binds and ubiquitinates p53, that is then degraded in the proteasome. MDM2 inhibitors, such as RG7388, interfere with p53 binding to MDM2, and therefore inhibit p53 ubiquitination, promote p53 stability and enhance p53 function and transcriptional activity in the nucleus. XPO1 is a key nuclear export protein that mediates the export from the nucleus to the cytoplasm of different proteins, including p53. XPO1 binds to p53 and then mediates its delocalization from the nucleus to the cytoplasm. Once exported into the cytoplasm, p53 is no longer able to exert its normal function as a tumor suppressor gene. XPO1 is usually overexpressed in tumoral cells and may export p53 out of the nucleus with a greater extent, leading to the loss of the physiological p53 activity in the nucleus. XPO1 inhibitors are able to reduce the export of p53 out of the nucleus, thus increasing its nuclear concentration and enhancing its physiological activity
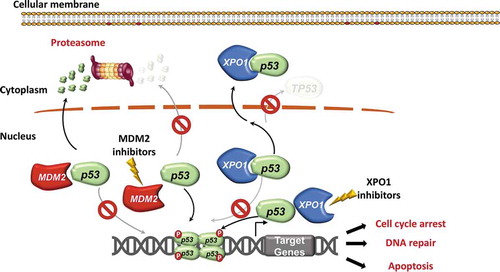
Recently, a strong rationale for the future evaluation of MDM2 inhibitors in CLL therapy has been demonstrated with RG7388, a second-generation MDM2 inhibitor [Citation91]. A pre-clinical study has shown that RG7388 enhances p53 function in CLL cells, thus inducing the characteristic p53 pro-apoptotic gene expression signature in the leukemic cells [Citation91]. No significant induction of transcriptional targets was observed in TP53 disrupted CLL cells, consistent with the specificity of RG7388 for TP53 wildtype cells [Citation91]. However, a CLL sample harboring a subclonal 17p deletion in 22% of nuclei showed functional activation of p53 and induction of cell death in response to RG7388 [Citation91]. This suggests that, in the presence of low subclonal levels of p53 loss, the predominant p53 functional cell population can still respond to non-genotoxic activation of p53 [Citation91]. These in vitro results prompt the hypothesis that patients with subclonal TP53 loss might still benefit from treatment with new-generation MDM2 inhibitors, especially in combination with other p53-independent targeted therapies [Citation91]. An obvious, but currently unexplored question, is whether MDM2 inhibition in patients with minor TP53 subclones may lead to the selection and to the subsequent expansion of the TP53 disrupted clones.
In addition to MDM2, other biological mechanisms regulate p53 expression and activity. Exportin-1 (XPO1), also called chromosome region maintenance 1 (CRM1), is a key nuclear export protein that mediates protein export from the nucleus to the cytoplasm through the nuclear pore complex [Citation92]. XPO1 is the primary nuclear exporter for a number of tumor suppressor and cell cycle regulatory proteins such as p53, p21, FOXO1, and cyclin B1/D1 () [Citation92]. Physiologically, XPO1 is usually deregulated in cancer by different mechanisms including gain of function mutations in codon E571 that increase the protein activity [Citation93]. Although the anti-tumoral activity of XPO1 inhibitors is not fully understood, a recent study has shown that the XPO1 inhibitor selinexor conceivably exerts its function by reducing the p53 export out of the nucleus, thus enhancing p53 nuclear retention () [Citation92]. This hypothesis has been also corroborated by the fact that selinexor upregulates the transcription of p53 target genes coding for anti-apoptotic proteins and for proteins that lead to cell cycle arrest such as p21 [Citation94].
7. Other mechanisms that enhance cell death in TP53 disrupted cells
Another strategy to overcome TP53 disfunction may stem from the concept of ‘synthetic lethality’ [Citation95,Citation96]. Synthetic lethality is based on the fact that p53 deficient cells lack a functional G1/S checkpoint and therefore fully depend on the remaining checkpoints, mainly controlled by the ataxia telangiectasia and Rad3-related (ATR) protein and by the checkpoint kinase 1 (CHK1) which can be utilized by targeted inhibition [Citation95,Citation96]. Preliminary results suggest that in p53 or ATM defective CLL cells, inhibition of ATR signaling by the AZD6738 small molecule leads to an accumulation of unrepaired DNA damage, which is carried through into mitosis because of defective cell cycle checkpoints, resulting in cell death by mitotic catastrophe [Citation95]. Similar results have also been obtained by the inhibition of the CHK1 protein [Citation96].
8. Conclusions
In CLL, as in many cancers, the key approach to improve clinical outcome is treatment tailoring in every individual CLL patient based on the clinical and biological features of the disease [Citation3,Citation97]. Whole genome and whole exome sequencing studies have allowed a better understanding of CLL pathogenesis and have led to the identification of genetic lesions with potential clinical relevance [Citation3,Citation97]. TP53 is mutated and/or deleted in approximately 10% of CLL patients at the time of first treatment requirement, and the frequency of this genetic lesion rises progressively upon subsequent relapses and reaches 60% in CLL that have transformed to Richter syndrome [Citation34]. Due to the chemorefractoriness imposed by TP53 disruption, the status of TP53 represents the pivotal decisional node for treatment tailoring in CLL and prompts upfront treatment with BCRi and BCL2i that, at least in part, circumvent TP53-mediated chemorefractoriness [Citation2,Citation3,Citation31].
BCRi and BCL2i, however, do not directly target the p53 protein and do not restore the function of the disrupted p53 pathway. The persistent lack of a fully functional p53 conceivably explains the observation that TP53 mutated patients have a poorer outcome compared to TP53 wild type patients even when treated with BCRi and BCL2i treatment, posing the need to explore novel avenues for optimizing therapy in this subset of CLL patients [Citation97,Citation98]. Importantly, TP53 disruption remains a poor biomarker also in patients treated with a fixed duration combination of a BCL2i and an anti-CD20 antibody, as it is the case with venetoclax-obinutuzumab in the CLL14 trial [Citation58]. This observation points also to the potential need of prolonged treatment as a novel concept in the management of TP53 disrupted CLL. Accordingly, overcoming TP53 disruption would require not only drugs that circumvent the DNA damage response, but also a prolonged exposure to those drugs until disease progression. This concept might represent a working hypothesis for the design of new trials dedicated to TP53 disrupted patients.
9. Expert opinion
Targeting p53 disruption with a precision medicine approach is an important task for the future in the field of CLL, in order to offer the possibility of improved outcomes also to these hard to treat patients. Because the molecular mechanisms of TP53 disruption in CLL, and in cancer in general, are heterogeneous, a true precision medicine approach may need to consider whether, in the individual patient, TP53 disruption is due to 17p deletion, loss-of-function missense mutations, gain of function missense mutations, nonsense mutations, or a combination of these alterations in the same clone (). Several avenues might be envisaged. Nuclear localization of p53 is a critical feature for its physiological function as a tumor suppressor gene. However, cancer cells may progressively acquire intracellular mechanisms for exclusion from the nucleus of tumor suppressor proteins [Citation7]. Remarkably, a fraction of CLL carries mutations of XPO1, that are known to enhance p53 export from the nucleus to the cytoplasm [Citation94]. Inhibiting XPO1, eg with Selinexor, may restore p53 localization to the nucleus and normal function. Activation of normal p53 function might also be achieved with non-genotoxic compounds that target and inhibit MDM2, thus stabilizing p53 and activating its transcriptional activity to promote apoptosis. Inhibition of XPO1 and MDM2 might be a strategy at least in cases with monoallelic deletion or monoallelic frameshift or nonsense mutations in which one p53 allele is fully functional [Citation91,Citation94]. In fact, inhibition of XPO1 or MDM2, by different mechanisms, might lead to an increase of the nuclear concentration of wild type p53 derived from the residual normal allele.
Figure 5. Putative models and future perspectives for p53 targeting according to the mutation and deletion status of the gene. (a) TP53 missense mutations reduce or abolish the normal activity of p53. APR-246 belongs to a new class of compounds that modulate p53 refolding and selectively induce apoptosis in cancer cells with mutant TP53. On these grounds, APR-246 may restore p53 configuration and function in cells carrying TP53 conformational missense mutations, thus leading to tumor regression. (b) In the case of TP53 nonsense mutations, APR-246 is not able to restore the physiological function of the p53 protein. Conversely, readthrough inducing molecules (e.g. G418, gentamicin, amikacin) may allow the ribosome to continue translation until the physiological stop codon is encountered resulting in a full-length p53 protein. Readthrough inducing molecules restore the function of a p53 truncated protein with a mechanism that is not completely understood. (c) In cells harboring monoallelic 17p deletion, inhibitors of MDM2 (eg: RG7388) and of XPO1 (eg: selinexor) might potentiate the activity of the residual normal p53 protein translated from the wild type allele
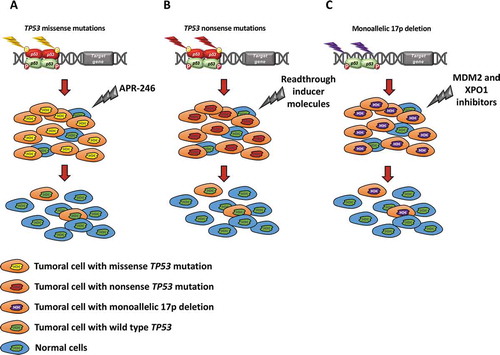
Patients carrying TP53 missense mutations altering the conformation of the p53 protein may pose a different challenge to the attempt of p53 targeting [Citation99]. In fact, simply promoting the nuclear accumulation of p53 might not be a reasonable strategy, since that would drive an even higher nuclear concentration of a misfunctional p53 protein that would enable further inactivation of other p53 family members, including p63 and p73. Rather, agents capable of restoring a normal function of p53 proteins carrying missense mutations might be, in principle, a rational approach [Citation4,Citation71]. The development of drugs restoring the function of mutated p53 proteins is ongoing, and at least one compound, eprenetapopt (APR-246), has shown promising activity in early phase clinical trials in hematological and solid cancers [Citation73–79]. If drugs restoring p53 function were confirmed as a viable therapeutic strategy also for CLL, a subsequent step to be envisaged might imply the combination of p53 restoring drugs with p53 potentiators, such as MDM2 or XPO1 inhibitors, or with other drugs already in use for CLL therapy and targeting other cellular mechanisms [Citation91,Citation94].
An alternative avenue will be to address other molecules, such as BIRC3, that are involved in conserving wild type p53 levels by avoiding NF-κB-mediated transcriptional and post-translational adaptations of MDM2 expression and function [Citation100]. Remarkably, genetic alterations of BIRC3 confer refractoriness to CIT in CLL with outcomes similar to those of patients carrying p53 disruption [Citation101,Citation102]. Since BIRC3 genetic alterations are mutually exclusive with TP53 mutations and deletion [Citation101,Citation102], analysis of the p53, MDM2 and NF-κB pathways in BIRC3 mutated CLL may offer novel insights for devising new therapeutic strategies. Finally, the issue of minor TP53 mutated subclones might also be relevant in a future perspective of p53 therapeutic targeting [Citation36]. At present, guidelines do not recommend to take clinical decisions based on the finding of TP53 mutated subclones with a VAF<10% [Citation32]. Results from prospective clinical trials will be important to unequivocally understand the true predictive and prognostic value of these minor subclones, and to understand when and how they deserve to be therapeutically targeted.
Article highlights
TP53 disruption is the strongest predictor of chemo-refractoriness in chronic lymphocytic leukemia and is the most important decisional node in the treatment algorithm of the disease
Several molecular mechanisms lead to TP53 disruption in CLL, including 17p deletion and missense, nonsense and frameshift mutations
BCR and BCL2 inhibitors circumvent TP53 mediated chemorefractoriness and confer markedly improved outcomes to TP53 disrupted CLL
However, the prognosis of TP53 disrupted CLL even with BCR and BCL2 inhibitors remains poorer compared to that of wild type CLL
Medicines that specifically target mutant p53 or potentiate physiological p53 function are emerging in other cancers and might represent a meaningful strategy to pursue also in CLL
This box summarizes key points contained in the article.
Declaration of interest
Gianluca Gaidano has to disclose roles in advisory boards or speakers’ bureaus of the following companies: Astra-Zeneca, Sunesys, Abbvie and Janssen. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.ll the other authors have nothing to disclose.
Reviewers disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Teras LR, DeSantis CE, Cerhan J, et al. 2106 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66:443–459.
- Hallek M, Shanafelt TD, Eichhorst B, et al. Chronic lymphocytic leukemia. Lancet. 2018;391(10129):1524–1537.
- Gaidano G, Rossi D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematology Am Soc Hematol Educ Program. 2017;2017(1):329–337.
- Duffy MJ, Crown J. Drugging “Undruggable” genes for cancer treatment: are we making progress?Int J Cancer. 2020 Online ahead of print.].
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanism of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370.
- Aylon Y, Oren M. The paradox of p53: what, how and why? Cold Spring Harb Perspect Med. 2016;6(10):a026328.
- Hafner A, Bulyk ML, Jambhekar A, et al. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210.
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405.
- Walker KK, Levine AJ. Identification of a novel p53 domain that is necessary for efficient growth suppression. Proc Natl Acad Sci USA. 1996;93(26):15335–15340.
- Weinberg RL, Veprintserv DB, Fersht AR, et al. Cooperative binding of tetrameric p53 to DNA. J Mol Biol. 2004;341(5):1145–1159.
- Clore GM, Ernst J, Clubb R, et al. Refined solution structure of the oligomerization domain of the tumor suppressor p53. Nat Struct Biol. 1995;2:321–333.
- Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol. 2019;11(7):564–577.
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13(1):49–58.
- Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604.
- Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9(5):573–580.
- Tarapore P, Horn HF, Tokuyama Y, et al. Direct regulation of the centrosome duplication cycle by the p53-p21 Waf1/Cip1 pathway. Oncogene. 2001;20(25):3173–3184.
- Valdez JM, Nichols KE, Kesserwan C. Li-fraumeni syndrome: a paradigm for the understanding of hereditary cancer predisposition. Br J Hematol. 2017:539–552.
- Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018;15(1):13–30.
- Spunarova M, Tom N, Pavlova S, et al. Impact of gene mutations and chromosomal aberrations on progression-free survival in chronic lymphocytic leukemia patients treated with front-line chemoimmunotherapy: clinical practice experience. Leuk Res. 2019;81:75–81.
- Best OG, Gardiner AC, Majid A, et al. A novel functional assay using etoposide plus nutlin-3a detects and distinguishes between ATM and TP53 mutations in CLL. Leukemia. 2008;22(7):1456–1459.
- Navrkalova V, Sebejova L, Zemanova J, et al. ATM mutations uniformly lead to ATM dysfunction in chronic lymphocytic leukemia: application of functional test using doxorubicin. Haematologica. 2013;98(7):1124–1131.
- Leroy B, Ballinger ML, Baran-Marszac F, et al. Recommended guidelines for validation, quality control, and reporting of TP53 variants in clinical practice. Cancer Res. 2017;77(6):1250–1260.
- Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat. 2014;35(6):672–688.
- De Vries A, Flores ER, Miranda B, et al. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc Natl Acad Sci USA. 2002;99(5):2948–2953.
- Pfister NT, Prives C. Trascriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb Perspect Med. 2017;7(2):a026054.
- Trbusek M, Smardova J, Malcikova J, et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol. 2011;29(19):2703–2708.
- Cerna K, Oppelt J, Chochola V, et al. MicroRNA miR-34a downregulates FOXP1 during DNA damage response to limit BCR signalling in chronic lymphocytic leukaemia B cells. Leukemia. 2019;33(2):403–414.
- Gaidano G, Ballerini P, Gong JZ, et al. P53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 1991;88:5413–5417.
- Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726.
- Catherwood MA, Gonzalez D, Donaldson D, et al. Relevance of TP53 for CLL diagnostics. J Clin Pathol. 2019;72(5):343–346.
- Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–2760.
- Malcikova J, Tausch E, Rossi D, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia - Update on methodological approaches and results interpretation. Leukemia. 2018;32(5):1070–1080.
- Rossi D, Cerri M, Deambrogi C, et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009;15:995–1004.
- Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131:2761–2772.
- Pal K, Bystry V, Reigl T, et al. European Research Initiative on CLL (ERIC)—TP53 Network, GLASS: assisted and standardized assessment of gene variations from Sanger sequence trace data. Bioinformatics. 2017;33(23):3802–3804.
- Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123:2139–2147.
- Blakemore SJ, Clifford R, Parker H, et al. Clinical significance of TP53, BIRC3, ATM and MAPK-ERK genes in chronic lymphocytic leukaemia: data from the randomised UK LRF CLL4 trial. Leukemia. 2020;34(7):1760–1774.
- Te Raa GD, Malčiková J, Mraz M, et al. Assessment of TP53 functionality in chronic lymphocytic leukaemia by different assays; an ERIC-wide approach. Br J Haematol. 2014;167(4):565–569.
- Carter A, Lin K, Sherrington PD, et al. Detection of p53 dysfunction by flow cytometry in chronic lymphocytic leukaemia. Br J Haematol. 2004;127(4):425–428.
- Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113(16):3801–3808.
- Condoluci A, Terzi Di Bergamo L, Langerbeins P, et al. International prognostic score for asymptomatic early-stage chronic lymphocytic leukemia. Blood. 2020;135(21):1859–1869.
- Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530.
- Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–1916.
- Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123:3247–3254.
- Rossi D, Terzi-Di-Bergamo L, De Paoli L, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood. 2015;126(16):1921–1924.
- Estenfelder S, Tausch E, Robrecht S, et al. Gene mutations and treatment outcome in the context of chlorambucil (Clb) without or with the addition of rituximab (R) or obinutuzumab (GA-101, G) - Results of an extensive analysis of the phase III study CLL11 of the German CLL study group. Blood. 2016;128:3227.
- Bagacean C, Tempescul A, Ternant D, et al. 17p deletion strongly influences rituximab elimination in chronic lymphocytic leukemia. J Immunother Cancer. 2019;7(1):22.
- Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174.
- Best OG, Gardiner AC, Davis ZA, et al. A subset of Binet stage A CLL patients with TP53 abnormalities and mutated IGHV genes have stable disease. Leukemia. 2009;23(1):212–214.
- Tam CS, Shanafelt TD, Wierda WG, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009;114(5):957–964.
- International CLL-IPI working group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol. 2016;17(6):779–790.
- Pettitt AR, Jackson R, Carruthers S, et al. Alemtuzumab in combination with methylprednisolone is a highly effective induction regimen for patients with chronic lymphocytic leukemia and deletion of TP53: final results of the national cancer research institute CLL206 trial. J Clin Oncol. 2012;30(14):1647–1655.
- Ahn IE, Farooqui MZH, Tian X, et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood. 2018;131:2357–2366.
- O’Brien S, Furman RR, Coutre S, et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood. 2018;131:1910–1919.
- Byrd JC, Hillmen P, O’Brien S, et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood. 2019;133(19):2031–2042.
- Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322.
- Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–778.
- Tausch E, Schneider C, Robrecht S, et al. Prognostic and predictive impact of genetic markers in patients with CLL treated with obinutuzumab and venetoclax. Blood. 2020;135(26):2402–2412.
- Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib Regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379(26):2517–2528.
- Moreno C, Greil R, Demirkan F, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:43–56.
- Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380:2225–2236.
- Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–443.
- Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107–1120.
- Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278–1291.
- Sharman JP, Coutre SE, Furman RR, et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J Clin Oncol. 2019;37(16):1391–1402.
- Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519–2531.
- Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672.
- Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N Engl J Med. 2015;373:220–231.
- Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor–ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377:2024–2035.
- Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381:1809–1819.
- Bykov VJN, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–288.
- Lambert JMR, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388.
- Nahi H, Lehmann S, Mollgard L, et al. Effects of PRIMA-1 on chronic lymphocytic leukaemia cells with and without hemizygous p53 deletion. Br J Haematol. 2004;127:285–291.
- Nahi H, Selivanova G, Lehmann S, et al. Mutated and non-mutated TP53 as targets in the treatment of leukaemia. Br J Haematol. 2008;141:445–453.
- Tessoulin B, Descamps G, Moreau P, et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. 2014;124:1626–1636.
- Maslah N, Salomao N, Drevon L, et al. Synergistic effects of PRIMA-1Met (APR-246) and Azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2020;105(6):1539–1551.
- Jaskova Z, Pavlova S, Malcikova J, et al. PRIMA-1MET cytotoxic effect correlates with p53 protein reduction in TP53-mutated chronic lymphocytic leukemia cells. Leuk Res. 2020;89:106288.
- Bykov VJN, Eriksson SE, Bianchi J, et al. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102.
- Lia Floquet C, Deforges J, Rousset J-P, et al. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011;39(8):3350–3362.
- Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299.
- Brown CJ, Lain S, Verma CS, et al. Awakening guardian angels: drugging the P53 pathway. Nat Rev Cancer. 2009;9(12):862–873.
- Vu B, Wovkulich P, Pizzolato G, et al. Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett. 2013;56:5979–5983.
- Ding Q, Zhang Z, JJ L, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–5983.
- Drakos E, Singh RR, Rassidakis GZ, et al. Activation of the p53 pathway by the MDM2 inhibitor nutlin-3a overcomes BCL2 overexpression in a preclinical model of diffuse large B-cell lymphoma associated with t(14;18)(q32;q21). Leukemia. 2011;25(5):856–867.
- Endo S, Yamato K, Hirai S, et al. Potent in vitro and in vivo antitumor effects of MDM2 inhibitor nutlin-3 in gastric cancer cells. Cancer Sci. 2011;102(3):605–613.
- Ray-Coquard I, Blay JY, Italiano A, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13(11):1133–1140.
- Andreeff M, Kelly KR, Yee K, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22(4):868–876.
- Verreault M, Schmitt C, Goldwirt L, et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin Cancer Res. 2016;22(5):1185–1196.
- Mascarenhas J, Lu M, Kosiorek H, et al. Oral idasanutlin in patients with polycythemia vera. Blood. 2019;134(6):525–533.
- Montesinos P, Beckermann BM, Catalani O, et al. MIRROS: a randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020;16(13):807–815.
- Ciardullo C, Aptullahoglu E, Woodhouse L, et al. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica. 2019;104(12):2429–2442.
- Azizian NG, Li Y. XPO1-dependent nuclear export as a target for cancer therapy. J Hematol Oncol. 2020;13(1):61.
- García-Santisteban I, Arregi I, Alonso-Mariño M, et al. A cellular reporter to evaluate CRM1 nuclear export activity: functional analysis of the cancer-related mutant E571K. Cell Mol Life Sci. 2016;73(24):4685–4699.
- Subhash VV, Yeo MS, Wang L, et al. Anti-tumor efficacy of Selinexor (KPT-330) in gastric cancer is dependent on nuclear accumulation of p53 tumor suppressor. Sci Rep. 2018;8(1):12248.
- Kwok M, Davies N, Agathanggelou A, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127(5):582–595.
- Boudny M, Zemanova J, Khirsariya P, et al. Novel CHK1 inhibitor MU380 exhibits significant single-agent activity in TP53-mutated chronic lymphocytic leukemia cells. Haematologica. 2019;104(12):2443–2455.
- Moia R, Patriarca A, Schipani M, et al. Precision medicine management of chronic lymphocytic leukemia. Cancers (Basel). 2020;12(3):642.
- Moia R, Patriarca A, Deambrogi C, et al. An update on: molecular genetics of high-risk chronic lymphocytic leukemia. Expert Rev Hematol. 2020;13(2):109–116.
- Zhu J, Sammons MA, Donahue G, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525(7568):206–211.
- Lau R, Niu MY, Pratt MA. cIAP2 represses IKK/-mediated activation of MDM2 to prevent p53 degradation. Cell Cycle. 2012;11(21):4009–4019.
- Diop F, Moia R, Favini C, et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica. 2020;105(2):448–456.
- Moia R, Patriarca A, Mahmoud AM, et al. Assessing prognosis of chronic lymphocytic leukemia using biomarkers and genetics. Expert Opin Orphan Drugs. 2020;8(9):329–342.