
ABSTRACT
Introduction
Tenosynovial giant cell tumor (TGCT) is a mono-articular, benign or locally aggressive and often debilitating neoplasm. Systemic therapies are becoming part of the multimodal armamentarium when surgery alone will not confer improvements. Since TGCT is characterized by colony-stimulating factor-1 (CSF1) rearrangements, the most studied molecular pathway is the CSF1 and CSF1 receptor (CSF1R) axis. Inhibiting CSF1-CSF1R interaction often yields considerable radiological and clinical responses; however, adverse events may cause treatment discontinuation because of an unfavorable risk-benefit ratio in benign disease. Only Pexidartinib is approved by the US FDA; however, the European Medicines Agency has not approved it due to a uncertain risk-benefit ratio. Thus, there is a need for safer and effective therapies.
Areas covered
Light is shed on disease mechanisms and potential drug targets. The safety and efficacy of different systemic therapies are evaluated.
Expert opinion
The CSF1-CSF1R axis is the principal drug target; however, the effect of CSF1R inhibition on angiogenesis and the role of macrophages, which are essential in the postoperative course, needs further elucidation. Systemic therapies have a promising role in treating mainly diffuse-type, TGCT patients who are not expected to clinically improve from surgery. Future drug development should focus on targeting neoplastic TGCT cells.
1. Introduction
Tenosynovial giant cell tumor (TGCT) is a monoarticular, proliferative lesion located in and around joints throughout the body[Citation1]. According to the most recent ‘World Health Organization Classification of Tumors: Soft tissue and Bone Tumors,’ TGCT comprises a family of lesions originating from the synovium of joints, bursae, and tendon sheaths[Citation2]. TGCT consists of two main subtypes: Localized-type TGCT (L-TGCT) and Diffuse-type TGCT (D-TGCT), previously referred to as Giant Cell Tumor of Tendon Sheath and Pigmented Villonodular Synovitis (PVNS), respectively[Citation3]. Although varieties of names have been used in the past, both subtypes are now unified under one denomination, sharing common pathogenesis and morphology [Citation4,Citation5].
1.1. Epidemiology
TGCT is considered an orphan disease, with reported incidence rates ranging from 30–39 and 5–8 per million person-years for L-TGCT and D-TGCT, respectively [Citation6,Citation7]. There is a female predilection (♂:♀ ratio 1:1.5) and both subtypes affect a relatively young population. TGCT is mainly diagnosed between 40 and 60 years; nevertheless, it can occur at all ages, even in children[Citation8]. L-TGCT is primarily located in the digits of the hand and feet (85%), while D-TGCT is more involved in the large joints, especially the knee[Citation9].
1.2. Clinical presentation
L-TGCT and D-TGCT behave differently and are categorized as separate clinical entities. Common experienced symptoms are pain, swelling, stiffness and limited range of motion. Other functional signs are instability, giving way and joint blockage[Citation10]. However, these nonspecific presentations can lead to a delay in diagnosis and visits to various medical professionals[Citation11]. Additionally, a decrease in quality of life (QoL) and interference with daily activities, leading to work loss and requiring domestic help, are often reported. TGCT’s associated burden on QoL and healthcare emphasize the need for an adequate treatment strategy [Citation12–14].
1.3. Diagnostics
Magnetic Resonance Imaging (MRI) is the state of art imaging technique for TGCT and demonstrates the extent of the disease, presence of joint effusion, and secondary degenerative changes. L-TGCT is characterized by a focal, well-demarcated lesion, whereas D-TGCT is classified as more than one, multilobulated lesions with synovial thickening, villous projections and hemosiderin depositions, often extending both intra- and extra-articular[Citation15]. Additionally, the diffuse-type is often associated with bone erosions, cartilage loss and osteophyte formation. In more progressive stages this leads to secondary osteoarthritis[Citation16]. Histological confirmation of the diagnosis is mainly obtained by either excisional biopsies, arthroscopic biopsies or core needle biopsies[Citation11]. However, since often no clear histological distinction can be made between the two main subtypes, L-TGCT and D-TGCT are mainly differentiated by a radiological distribution of tumor within the joint and clinical characteristics.
1.4. Pathogenesis
The pathogenesis of TGCT has been the subject of debate for a long period. In 1941, Jaffe et al. suggested that TGCT had a reactive or inflammatory origin[Citation17]. Several decades later, cytogenic studies revealed numerical and structural chromosomal alterations, indicating a clonal, neoplastic process [Citation18,Citation19]. In 2006, the main view regarding the pathogenesis changed after West et al. demonstrated the presence of recurrent translocations in several TGCT patients. These translocations involve region 1p11-13, on which the colony-stimulator factor 1 (CSF1) gene is located[Citation20]. CSF1, also known as macrophage colony-stimulating factor (M-CSF), regulates survival, proliferation, differentiation and function of macrophages and their precursors by binding to its receptor (CSF1R) [Citation21,Citation22]. Most cells in TGCT express CSF1R, while CSF1 (the ligand of CSF1R) is only present in a low percentage of cells (2–16%). These neoplastic TGCT cells produce elevated levels of CSF1 as a result of the translocation, leading to an increase in more neoplastic cells (autocrine loop) and an accumulation of non-neoplastic CSF1R-expressing cells of the macrophage lineage (paracrine loop). This is referred to as the landscape effect[Citation20]. The most common translocation partner of CSF1 is collagen type VI alpha-3 (COL6A3), located on chromosome 2p37 resulting in t(1;2)(p13;p37) [Citation23,Citation24]. However, recent studies showed that this fusion is only present in a subset of patients. Also, they showed the involvement of other fusion partners leading to additional underlying mechanisms for CSF1 upregulation [Citation24–27]. More specific, novel fusions result in the deletion of CSF1 exon 9, a negative regulator of CSF1 expression, and truncation of 3’-UTR region may account for CSF1 overexpression in more cases instead of overexpression of full-length CSF1 via promoter swapping [Citation28,Citation29]. Also, CBL (Cas-Br-Murine ecotropic retroviral transforming sequence) mutations are present in more than a third of TGCT cases, although not mutually exclusive to CSF1 fusions [Citation28,Citation29]. CBL is a multifunctional protein that associates with receptor tyrosine kinases and could be the driver event in some cases where CSF1 rearrangements are not present[Citation29]. A consensus regarding the effect of different fusions or truncations and levels of CSF1 overexpression and the role of CBL mutations on clinical behavior is still lacking [Citation30–32].
The gene expression profile of TGCT is consistent with apoptosis resistance, inflammation, and matrix degradation, leading to ongoing proliferation and joint destruction. Genes highly overexpressed include CD53, ALOX5AP, SPP1, MMPs 1 and 9[Citation33]. Contrarily, tumor suppressor genes, such as TP53, are downregulated[Citation33].
1.5. Histopathology
TGCT belongs to the so-called fibrohistiocytic tumors according to the 5th edition of the World Health Organization Classification of Tumors and the tumor microenvironment contains a heterogeneous cell population[Citation2]. Tumors are composed of a variable proportion of mononuclear cells, multinucleated (osteoclast-like) giant cells, foamy macrophages (xanthoma cells), inflammatory cells, and siderophages (hemosiderin-laden macrophages), with stromal hyalinization () [Citation34–36]. Two principle cell types are described, in variable proportion, within the mononuclear cell compartment: small histiocyte-like cells with round to oval nuclei often represent the main cellular component. In addition, larger epithelioid cells with abundant cytoplasm and larger nuclei are seen ()[Citation2]. To date, it remains unknown from which lineage these cells are derived. Immunohistochemically, the larger mononuclear cells express clusterin, podoplanin and to lesser extent desmin, highlighting dendritic processes [Citation2,Citation37]. The smaller histiocyte-like cells are positive for CD68, CD163 and CD45[Citation38]. Although osteoclast-like giant cells are the most distinctive histological feature of TGCT, giant cells may be sparse or absent[Citation39]. In the joint fluid, various inflammatory factors are present, such as Interleukine-1β (IL-1β) and Tumor necrosis factor-α (TNF-α), indicating the presence of a highly inflammatory microenvironment within joints affected by TGCT ()[Citation40]. Besides the two main subtypes, malignant TGCT is incidentally reported. This aggressive subtype has a high potential for metastasis, mainly in pulmonary and regional lymph nodes[Citation41]. Besides the typical TGCT histology, malignant TGCT displays increased mitotic activity, including atypical mitoses, necrosis, enlarged nuclei with nucleoli, spindling of mononucleated cells, and myxoid change[Citation2]. The malignant TGCT cells are suggested to be derived from clusterin-positive large mononuclear cells[Citation42]. The etiology of malignant TGCT is not well understood.
Figure 1. TGCT H&E stained tissue section showing characteristic histologic features such as small histiocyte-like cells with oval nuclei, larger epithelioid cells of which some contain hemosiderin depositions, multinucleated osteo-clast like giant cells, and stromal hyalinization.
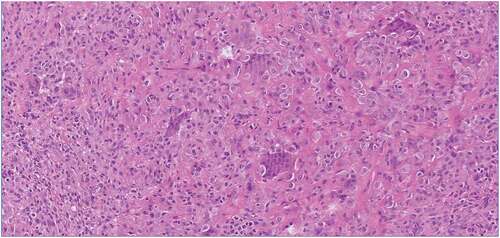
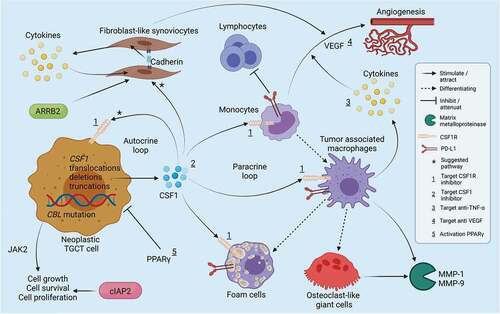
Figure 2. Simplified overview of pathways involved in TGCT chromosomal aberrations of the CSF1 gene lead to an overexpression of CSF1. Elevated levels of CSF1 are suggested to cause an increase in neoplastic TGCT cells (autocrine loop) and accumulate CSF1R presenting cells of the macrophage lineage (paracrine loop). CSF1 is also suggested to be involved with the proliferation of FLS. Monocytes/macrophages can differentiate into osteoclast-like giant cells and foam cells. Monocytes can produce VEGF resulting in angiogenesis and expressing PD-L1, leading to attenuation of lymphocytes. VEGF can also be induced by hypoxic stress by FLS amongst others. Macrophage produced cytokines can strengthen the effect of VEGF. Cadherin-11 can stimulate FLS to produce cytokines, while the cytokines can stimulate FLS through a positive loop. Additionally, the effect of proteins ARRB2, cIAP2, PPARγ and targets over several drugs are depicted. Created with BioRender.com.
1.6. Current gold-standard treatments
Complete excision of TGCT is the primary choice of treatment[Citation43]. L-TGCT is often removed relatively easily by either arthroscopy or open surgery, depending on the localization, with a recurrence rate around 10% [Citation44,Citation45]. Contrarily, adequate removal of D-TGCT can be challenging due to the localization in- and outside the joint and the non-clear-cut boundaries of the tumor. Additionally, D-TGCT has high recurrence rates and often requires re-excision [Citation46,Citation47]. Relapse-free survival rate is estimated at 40% at 10 years[Citation46]. The optimal surgical technique remains to be elucidated[Citation48]. Some specialists claim that open surgery leads to better overview and access, especially in widespread lesions. On the other hand, open surgery may lead to more iatrogenic morbidity, especially in patients having relapses.
The high rate of recurrences in D-TGCT underlines the demand for (neo)adjuvant or new stand-alone treatment approaches[Citation47]. Radiotherapy, consisting of radiosynoviorthesis (RSO) with the injection of intra-articular radioactive isotopes and external beam radiotherapy (EBR), is occasionally performed [Citation49–51]. Only a few studies are available with low-level evidence for both types of radiotherapy. Mixed results have been reported regarding the efficacy of RSO, but this is also associated with serious complications such as skin necrosis [Citation52–54]. A meta-analysis by Mollon et al. suggested that surgery combined with EBR leads to a reduced recurrence rate, but they also concluded that large long-term prospective multicenter studies are required to confirm these findings[Citation55]. Long-term findings are important since EBR is associated with radiation-induced malignancies, especially in a relatively younger patient population. This is regarded as an unacceptable outcome in a benign or intermediate disease[Citation56].
In 2008, Blay et al. were the first to report the effect of the drug imatinib (or imatinib mesylate) in a patient with TGCT, leading to complete remision[Citation57]. This implied a promising role for targeted therapies in TGCT, especially for patients not amenable to surgery. The interest in systemic therapies is increasing more recently, leading to the development and clinical testing of new and available drugs, providing enlargement of the current therapeutic armamentarium.
1.7. Cell line models
Patient-derived tumor cell lines are essential to investigate molecular mechanisms of TGCT pathogenesis and develop novel therapeutic strategies. Recently, a new cell line was established from TGCT tissue[Citation58]. Complimentary use of a culture can be helpful in in vitro studies to gain new insights and increase drug screening reliability[Citation58]. In addition, Tang et al. established the use of patient-derived tumor TGCT xenografts for drug validation in ex vivo models[Citation59].
2. Potential therapeutic targets
Systemic therapies provide a new avenue for patients with inoperable TGCT and are sometimes considered as a last resort[Citation49]. Various underlying molecular pathways have been explored as new therapeutic strategies () ().
Table 1. (Potential) TGCT drug targets
2.1. Therapeutic targets related to tumor cells
2.1.1. Colony stimulating factor 1
Since the discovery of CSF1 overexpression in TGCT patients, the CSF1-CSF1R axis has been the most studied target[Citation60]. Where in some patients genomic alterations cause overexpression of CSF1 in the neoplastic cells, in other patients, the origin of elevated levels of CSF1 and CSF1R remains unclear [Citation20,Citation26,Citation28]. Nonetheless, targeting the CSF1-CSF1R signaling pathway is suggested to have an anti-tumoral effect by blockade of CSF1R. Different approaches to CSF1R blockade have been investigated, such as CSF1R antibodies and tyrosine kinase inhibitors (TKIs)[Citation27].
2.1.2. Janus-kinase-2
Tsuda et al. found that CBL mutations in TGCT prolong the phosphorylation of tyrosine kinase Janus-Kinase-2 (JAK2), leading to enhanced cell proliferation[Citation28]. They also found that CBL mutations are associated with increased JAK2 expression and worse disease outcomes. Therefore, the authors suggest inhibiting JAK2 as a possible new therapeutic strategy. However, the CBL mutation is not mutually exclusive to CSF1 fusions [Citation28,Citation29]. In cases with the presence of CSF1 rearrangements and CBL mutations, combined treatment might be reasonable.
2.1.3. Cellular inhibitor of apoptosis 2
Cellular inhibitor of apoptosis 2 (cIAP2) is an anti-apoptotic protein, and deregulation of this protein is associated with tumor development and progress[Citation61]. Levels of cIAP2 are significantly higher in D-TGCT compared to the localized-type. Besides, overexpression of cIAP2 is related to ligament, cartilage and bone erosion, and tumor relapses[Citation62]. Since high levels of cIAP2 in TGCT patients were associated with poor prognosis, cIAP2 gene may have a promising role in prognosis prediction and targeted therapy in D-TGCT[Citation62].
2.1.4 β-Arrestin2
β-Arrestin2 (ARRB2) is associated with cell survival, apoptosis, migration, and proliferation in several tumor types. ARRB2 is also highly expressed in TGCT, leading to increased cell proliferation and inhibition of apoptosis through activation of the PI3K-Akt pathway[Citation63]. Cao et al. observed that knockdown of ARRB2 inhibited cell proliferation and increased apoptosis of FLS. They concluded that ARRB2 could be a potential molecular target in TGCT treatment[Citation63].
2.1.5. Proliferator-activated receptor gamma
Proliferator-activated receptor gamma (PPARγ) is a member of the nuclear receptor super family of transcription factors. It is expressed in high levels in adipose tissue and monocyte-derived macrophages and stimulates adipocyte and macrophage differentiation[Citation64]. Ligand activation of PPARγ in monocytes/macrophages inhibits inflammatory mediator and cytokine production. Also, PPARγ is expressed in a variety of cancer cells and specific ligands can induce growth inhibition and apoptosis[Citation65].
2.2. Therapeutic targets related to reactive myeloid cells
2.2.1. Tumor necrosis factor-α
Analysis of the gene expression pattern of TGCT showed elevated levels of macrophages and proinflammatory cytokines, such as TNF-α[Citation33]. Proinflammatory cytokines drive inflammation, which is associated with joint destruction[Citation66]. In addition, the synergistic paracrine loop between TNF-α and CSF1 contributes to monocyte, macrophage, and osteoclast proliferation and differentiation, resulting in tumorigenesis. TNF-α blockade is assumed to antagonize the inflammatory process predominantly[Citation67].
2.2.2. Vascular endothelial growth factor
Vascular endothelial growth factor (VEGF) promotes endothelial cell proliferation and new blood vessel formation. Angiogenesis is crucial for tumor development. In addition, CSF1 can activate multiple cell signaling pathways leading to VEGF production. VEGF inhibition is associated with reducing tumor growth by blocking angiogenesis and, therefore, could be a potential target[Citation68].
2.2.3. Receptor-activator of nuclear factor kappa-B ligand
Receptor-activator of nuclear factor kappa-B ligand (RANKL) is a cytokine involved in differentiation and activation of osteoclasts, causing bone resorption[Citation69]. The monoclonal antibody denosumab successfully inhibits RANKL in various diseases, such as giant cell tumors of the bone[Citation70]. Yamagishi et al. showed RANKL expression in several soft tissue tumors, for instance, TGCT[Citation69]. RANKL expression can be induced by proinflammatory cytokines, such as TNF-α and interleukins, among others[Citation71]. Although RANKL levels were lower compared to giant cell tumors of the bone, there may be a role for RANKL antibody treatment to inhibit giant cells, although this has not yet been under study [Citation69,Citation72]. However, multinucleated cells such as osteoclast-like giant cells from synovial tumors might represent a different entity than bone tumor-derived multinucleated cells, which may result in different efficacy of RANKL blockage. Additionally, since RANKL antibody treatment targets giant cells and not underlying mechanisms of pathogenesis in TGCT, such as the CSF1-CSF1R axis, this strategy remains uncertain.
2.2.4. Programmed cell death ligand 1
Programmed cell death ligand 1 (PD-L1) regulates immune responses by attenuating lymphocyte activation and stimulating tumor growth. PD-L1 was highly positive in 53% of CSF1-activated TGCT cases, expressed on mononuclear cells, multinucleated giant cells, and foam cells[Citation73]. In addition, a positive correlation was found between PD-L1 expression and larger tumor sizes. Based on their findings, Zheng et al. suggested that anti-PD-L1 immunotherapy in combination with other molecular targeted therapy may possibly improve outcomes of anti-tumor therapy in patients with CSF1/CSF1R signaling[Citation73]. Nonetheless, PD-L1 immunohistochemistry expression can be variable and thus the value of PD-L1 is questionable[Citation74].
2.2.5. Cadherin-11
Cadherin-11 mediates adhesion between FLS and contributes to the formation of the synovium lining layer. Additionally, cadherin-11 can stimulate FLS to produce inflammatory cytokines and MMP[Citation40]. Meanwhile, cytokines IL-1β and TNF-α in the joint fluid can increase cadherin-11 expression through the PI3K-Akt pathway, eventually promoting proliferation, migration, and invasion of FLS through a positive feedback loop. This underlying molecular mechanism can cause joint destruction, relapses, or even metastasis and, therefore, may be used as a prognostic molecular marker for D-TGCT. Finally, cadherin-11 inhibition could present a new promising treatment strategy by weakening the migration and invasion of FLS [Citation40,Citation64,Citation65].
3. Systemic therapies
In the last decade, the safety and efficacy of several drugs regarding TGCT have been investigated () (). The systemic therapies are categorized by drugs targeting the CSF1-CSF1R axis and other therapeutics and published peer-reviewed data is separated from studies whose results are awaited. Furthermore, studies are presented in chronological order of publication.
Table 2. Systemic treatment studies
3.1. CSF1-CSF1R targeted therapies
3.1.1. Imatinib
Imatinib is an inhibitor of a few tyrosine kinases, including Abelson proto-oncogene (Abl), breakpoint cluster region-Abl (Bcr-Abl) complex, c-Kit proto-oncogene (KIT), platelet-derived growth factor receptor (PDGFR) and CSF1R[Citation75]. Before imatinib was applied in TGCT, this oncogene-targeted therapy was already indicated for patients with chronic myeloid leukemia and gastrointestinal stromal tumors[Citation57]. The effect of imatinib on TGCT was first evaluated in one patient, in which complete remission was observed five months after initiation[Citation57]. After treatment interruption, TGCT relapsed both clinically and radiologically, but diminished again after treatment was resumed. After successful treatment of this single patient, a retrospective study reported the effect of imatinib in 29 patients with advanced or metastatic D-TGCT [Citation57,Citation75]. Imatinib is an oral drug, dosed at 400 mg daily. Imatinib led to an objective response rate (ORR) of 20% according to Response Evaluation Criteria in Solid Tumors (RECIST) and symptomatic/functional improvement in 74% of patients. Although a substantial activity against D-TGCT was observed, the effect was less when compared with GIST and dermatofibrosarcoma protuberans, probably due to a lower activity against CSF1-driven chemotaxis[Citation75]. Six patients had to stop due to toxicity (of which three had grade 3 or 4 toxicities), and four without any apparent medical reason[Citation75].
Subsequently, the same study group described the long-term efficacy of imatinib on the initial 29 patients and 33 additional patients. In this cohort, 31% had radiological response and 78% had clinical improvement. A drop-off rate of 59% within a year suggested an unfavorable efficacy/toxicity balance, with grade 3–4 toxicities occurring in 11%. All four patients with metastatic TGCT progressed rapidly on imatinib[Citation76].
Another research group observed partial response in 32% after imatinib use, consistent with the previous reports. Additionally, this study showed a significant decrease in the maximum standardized uptake value (SUV-max) on PET-CT after imatinib. However, 80% of the patients discontinued treatment with imatinib for poor response or intended surgery[Citation77].
Finally, Stachiotti et al. showed anti-tumor activity following imatinib in two patients resistant to prior nilotinib. They assumed that targeted agents with similar profiles could induce different clinical results. However, further research is required[Citation78].
3.1.2. Emactuzumab
Emactuzumab is a recombinant, humanized monoclonal Immunoglobulin 1 (IgG1) antibody directed against CSF1R[Citation79]. In a phase I trial, Cassier et al. determined the safety, tolerability, and clinical activity of emactuzumab in 28 patients[Citation80]. After a median follow-up of 12 months, 86% of the patients showed an objective response, of which 2 achieved complete response; one patient had a relapse. The most frequent adverse events were facial edema, asthenia, pruritus, and rash. Five serious adverse events were reported, of which three were grade 3, including lupus erythematosus twice. Overall, emactuzumab was well tolerated, but 20% of patients dropped out due to adverse events. Eight patients who were resistant to either imatinib or nilotinib or both before achieved objective tumor response. The authors hypothesized that the difference in activity between imatinib, nilotinib and emactuzumab could be attributed to the fact that imatinib and nilotinib are no strong inhibitors of CSF1R.
The same research group performed a subsequent study, including 63 patients, to evaluate the long-term clinical benefit and safety[Citation81]. The best ORR of 71% was observed, and responses were durable at one- and two years at 70% and 64%, respectively. Stable disease was achieved in 98%, which changed to 93% at one- and two-year follow-up. In addition, a significant improvement in the EQ-5D-3 L QoL and WOMAC was observed. Reported AEs were comparable with the previous study. Nine patients (14%) withdrew from the study due to AEs. Finally, a reduction in tumor-associated CD68/CD163-positive and CSF1R-positive cells was shown. Durable responses were seen despite a relatively short treatment duration, which can be interpreted as an advantage.
A third study estimated the optimal biological dose (OBD) based on the previous phase I study. They recommended an OBD of 1000 mg intravenously every two weeks. Dosing flexibility is possible by dosing emactuzumab once every three weeks[Citation82].
3.1.3. Nilotinib
Nilotinib was the first drug whose efficacy and safety was investigated in an open-label, phase II trial for non-resectable D-TGCT[Citation83]. Nilotinib inhibits tyrosine kinases Abl, KIT, PDGFR and CSF1R. Patients received 400 mg orally twice daily until treatment discontinuation or completion of 1 year of treatment. Of 51 evaluable patients, 49 (96%) were progression-free at 12 weeks and 46 (90%) at 24 weeks. After one year of treatment, the best objective response was partial response achieved in three patients (6%) and stable disease in 46 (90%). In total, ten (20%) patients had had disease progression during the 1-year study period. Six patients discontinued nilotinib due to disease progression; five patients due to toxicities. Headache, nausea, increased alanine aminotransferase (ALT) concentrations, fatigue and asthenia, were the most common AEs. Nine grade 3 AEs occurred, including hepatic disorders and toxicodermia. Post-hoc analysis showed progression-free survival in 57% at 48 months. However, secondary resection and nilotinib treatment duration had no additional effect on progression-free survival, although selection biases of patients for surgery are probable.
3.1.4. Pexidartinib
Pexidartinib is a novel TGCT targeting drug, the first drug approved by the US Food and Drug Administration (FDA)[Citation84]. It was derived from other TKIs, and showed more potential in blocking CSF1R dependent cells and limited cross-reactivity with other kinases. Besides strong selective activity against CSF1R, it also inhibits KIT and fmls-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD)[Citation85]. In a phase I study by Tap et al., 41 patients were enrolled in various dose-escalation cohorts. 27% had at least one drug-related adverse event of grade 3 or higher, including anemia, increase in aspartate aminotransferase level (AST), and decrease in lymphocytes. The maximum tolerated dose was set at 1000 mg per day taken orally. In the phase II extension study, 23 patients with D-TGCT were enrolled. Frequently experienced AEs were hair color change, fatigue, nausea, dysgeusia, and periorbital edema. Eight patients had grade 3 or higher AEs, including elevated levels of liver enzymes. Two patients discontinued treatment because of AEs. Disease control was observed in 19 (83%) patients, of which 12 had a partial response. Only one patient with metastatic TGCT had disease progression after a stable period of eight months.
The same study group performed a phase III, randomized, mutational, double-blind, then an open-label trial with pexidartinib[Citation86]. One hundred twenty patients were randomly assigned to pexidartinib (n = 61) or placebo (n = 59) treatment. At 25 weeks, the ORR in the pexidartinib group was 39% according to RECIST and 56% by tumor volume score (TVS) compared to 0% in the placebo group for both measurements. In addition, clinical outcomes were significantly improved in the pexidartinib group compared to the placebo group. However, 98% of the patients in the pexidartinib group experienced AEs. Grade 3–4 AEs occurred in 44% and mainly consisted of increased levels of AST, ALT, alkaline phosphatase (AP) and hypertension. Eight patients (13%) discontinued treatment with pexidartinib due to AEs, of which seven were liver-related (including hepatotoxicity and hyperbilirubinaemia requiring liver dialysis procedures). Subsequently, 30 patients from the placebo group were assigned to a crossover group with 800 mg pexidartinib a day. Of this group, 30% had RECIST response, and 57% had TVS response at week 25. Patients in this group experienced fewer liver enzyme elevations and no bilirubin increases or signs of drug-induced cholestatic hepatotoxicity.
Finally, the same study group described the long-term outcomes of pexidartinib by pooling analysis encompassing the three pexidartinib-treated TGCT cohorts described above[Citation87]. One hundred thirty patients received pexidartinib for a median duration of 19 months at data cutoff. With a median follow-up of 39 months, the RECIST ORR was 60%, the TVS ORR was 65%. Tumor response often occurred within six months after treatment but occurred even more after long-term pexidartinib treatment. The median treatment duration was 19 months. A total of 16 (12%) patients progressed on therapy or after treatment discontinuation. 127 (98%) patients experienced one or more treatment-related AEs; 57 patients (44%) had grade 3 or higher treatment-related AEs of which fourteen had treatment-related serious AEs. 119 (92%) patients had aminotransferase elevations, predominantly AST and ALT. Four (3%) patients experienced mixed or cholestatic hepatotoxicity, which started within eight weeks of the first treatment and was reversible.
Because of the risk of hepatotoxicity, frequent monitoring of liver function is needed to help balance the benefit-to-risk. Since long-term safety data did not show late-emerging or cumulative toxicities, monitoring is especially required in the first two months. In the EU the European Medicines Agency (EMA) refused market authorization due to uncertainties on the risk-benefit ratio. In the US, pexidartinib was FDA approved and available through the risk evaluation management system (REMS) program, which ensures appropriate monitoring [Citation88,Citation89].
3.2. Preliminary results of CSF1-CSF1R targeted therapies
3.2.1. Lacnotuzumab
Lacnotuzumab (MCS110) is a monoclonal antibody against CSF1. Preliminary results of lacnotuzumab in TGCT patients in a phase Ib/II study were presented during a congress[Citation90]. Five patients were treated with a single dose of 10 mg/kg intravenously. Lacnotuzumab was well tolerated with no drug-related AEs. Four weeks after dose administration, the tumor volume by MRI was reduced by 40%. Improvement in clinical symptoms and pharmacodynamics effects were also observed. Results of study extension, with multiple-dose administration and a goal of tumor ablation, are awaited.
3.2.2. Cabiralizumab
In 2017, preliminary results of a phase I/II study of cabiralizumab were presented[Citation91]. Cabiralizumab is a monoclonal antibody that inhibits the interaction of CSF1 and IL-34 ligands with their shared receptor CSF1R. Cabiralizumab was intravenously administered in 22 patients with inoperable D-TGCT every two weeks for six months in different dosages. Positive functional status improvements by Ogilvie-Harris scores (combination of pain, synovitis, range of motion and functional capacity on a scale of 0–12) were noted in objective responders (from 2 to 7). Most reported AEs were creatine kinase elevations, rash and other skin disorders, fatigue and edema; 10 grade 3 AEs were reported. Updated results are awaited.
3.2.3. Vimseltinib
Vimseltinib is a selective, orally administered inhibitor of CSF1R. In contrast to small-molecule inhibitors of CSF1R, this drug is designed to be selective not affecting closely related kinases KIT, FLT3, PDGFRA, PDGFRB, and the rest of the kinome[Citation92]. This selectivity potentially leads to a more optimal CSF1R suppression. It is currently being evaluated in a phase I/II clinical study for the treatment of TGCT. The first three TGCT patients, included in the phase I trial, showed rapid, preliminary anti-tumor activity by three cycles with deepening response over time. Vimseltinib was generally well tolerated in these patients, although the authors concluded it was too premature to draw safety conclusions on this limited data set[Citation92].
Results of the first 68 patients included in this phase I/II clinical study were recently presented at the European Society for Medical Oncology congress 2021[Citation93]. The majority of common treatment-emergent AEs were grade 2 or lower; most common grade 3 or 4 AEs were increased levels of creatine phosphokinase in blood. Two serious AEs were reported, consisting of metabolic encephalopathy and vaginal hemorrhage and three (4%) patients discontinued treatment due to treatment-emergent AEs. The median duration of treatment in the phase 1 cohort was 10.1 months and a high ORR of 50% according to RECIST was observed. In the phase II cohort, an ORR of 42% was seen in evaluable patients. The study is still ongoing with continuing follow-up evaluation and a randomized, placebo-controlled, phase III trial evaluating the effect vimseltinib is started (MOTION-study).
3.3. Other therapeutics
3.3.1. Tumor necrosis factor-α blockage
Three studies have reported the effect of TNF-α blockade treatment, with only seven patients in total receiving this treatment [Citation67,Citation94,Citation95]. Off-label intra-articular infliximab injection was administered in one patient and etanercept in six. Improvement in knee function, regression in synovial stromal fibrosis and vasculogenesis, reduction of cellularity and synovial fluid and decreased thickness were observed. On the other hand, no decrement in CSF1 protein of mRNA expression nor synovial tumor shrinkage was seen after infliximab administration. TNF-α blockade treatment affects the reactive component of TGCT but less likely the neoplastic cells. Although Fiocco et al. considered anti-TNF-α antibody injections as a possible neoadjuvant treatment before synovectomy, they concluded that TNF-α blockade alone does not seem to lead to stable remission of D-TGCT and is ineffective in blocking CSF1 secretion[Citation67].
3.3.2. Bevacizumab
Among other activators such as hypoxic stress, CSF1 is also reported to induce angiogenesis via vascular endothelial growth factor (VEGF) expression in monocytes, essential for tumorigenesis [Citation68,Citation96]. Bevacizumab is a humanized monoclonal VEGF antibody and thus inhibits angiogenesis. Nissen et al. investigated the effect of intra-articular injections with bevacizumab as adjuvant therapy after arthroscopic synovectomy in one patient with relapsing D-TGCT located in the knee[Citation97]. During follow-up, complete response was observed, and the patient reported no symptoms nor adverse events (AEs). However, dosage and number of injections and total follow-up duration were not described. Additionally, the effect of synovectomy prior to bevacizumab is unknown. To our knowledge, this is the only report regarding bevacizumab applied for TGCT.
3.4. Preliminary results of other therapeutics
3.4.1. Zaltoprofen
Based on the approaches with zaltoprofen in rheumatoid arthritis and targeted therapy activating PPARγ in other types of cancer, the anti-tumor effect of zaltoprofen was investigated on primary cultured TGCT cells[Citation98]. Zaltoprofen, a non-steroidal anti-inflammatory drug (NSAID), was found to inhibit cell proliferation via activation of PPARγ. Subsequently, a pilot study of zaltoprofen in D-TGCT affecting the knee and ankle joints was conducted, including ten patients. Oral zaltoprofen was given daily for 48 weeks or until disease progression. At 48 weeks, eight patients had stable disease, and one patient showed progressive disease at 72 weeks. Zaltoprofen was well-tolerated. With the results of this pilot study, a study protocol of a double-blind phase II study of zaltoprofen for D-TGCT and unresectable L-TGCT was published. Results are awaited.
4. Conclusion
To date, the preferred choice of treatment for TGCT remains surgical excision. However, there is a need for other therapeutic strategies in patients where local tumor control cannot be achieved and (repeated) surgery is associated with iatrogenic morbidity. Furthermore, the role of (neo)adjuvant radiotherapy is disputed, because it is associated with unacceptable long-term side effects. A better understanding of the pathogenesis of TGCT led to opportunities for the development of medical therapies. In the last decade, new drug targets were discovered, and the efficacy and safety of several drugs have been studied. Currently, pexidartinib is the only drug approved for TGCT by the US FDA. Pexidartinib showed significant radiological and clinical efficacy, but also severe adverse events occurred, including hepatotoxicity. Therefore, active monitoring of liver functions is mandatory. Contrarily, the EMA refused marketing authorization because it was unclear how long treatment effects would last and because of an unbalanced risk/benefit ratio. More recently, new treatments are under research and recent discoveries regarding new therapeutic targets are promising for the development of drugs with even better tolerability and higher efficacy.
5. Expert opinion
Several efforts have been made during the past years to augment the treatment armamentarium for TGCT, especially for patients with relapsing or inoperable D-TGCT. Also, for orphan diseases, such as TGCT, there is a need for tailored therapy. After the discovery of the presence of CSF1 translocations in TGCT, the CSF1-CSF1R axis became the main pathway to target. Imatinib was one of the first TKIs regularly used off-label[Citation57]. Nilotinib was assumed to have a favorable tolerability profile, especially concerning edema. However, the radiological response was inferior to imatinib and a substantial number of patients discontinued treatment[Citation83]. With both drugs, more patients discontinued treatment without tumor progression than usually in oncology trials, which could be attributed to the nonmalignant character of TGCT and thus lower patient-acceptance rate of side-effects. Pexidartinib, another TKI, was designed to have higher selectivity against CSF1R, leading to better response[Citation85]. Nonetheless, rare but serious liver injuries occurred in patients on pexidartinib treatment, resulting in marketing authorization refusal by the EMA. Of the reviewed therapeutic strategies, drugs targeting the CSF1/CSF1R axis showed potential in tumor size reduction and improving quality of life. However, most CSF1R inhibitors also target closely related tyrosine kinases next to CSF1, leading to off-target activity and causing an unfavorable risk-benefit ratio for a subset of patients[Citation92]. Also, the long-term efficacy of most of the CSF1R inhibitors is still unknown to date. The evidence regarding other therapeutic strategies is limited and therefore their efficacy cannot be accurately assessed. Currently, new drugs, such as vimseltinib, are being investigated, aiming to have less interference with other tyrosine kinases than CSF1R, resulting in a more favorable safety profile[Citation92]., 93 Secondly, the effect of intra-articular injections with CSF1R inhibitors is being studied, hoping to cause less systemic adverse events but having at least comparable local efficacy[Citation99]. Finally, blockage of other targets than CSF1 or overlapping pathways may provide new solutions in the future [Citation28,Citation40,Citation62,Citation63,Citation98].
A limitation in all studies is the lack of an appropriate method to monitor the medical treatment effect on TGCT tumor growth. TGCT treatment response is predominantly scored by RECIST, which evaluates target lesions over time by one-dimensional criteria (maximum diameter), although TGCT has an irregular shape and no clear tumor margins. TVS was explicitly developed for TGCT for volumetric quantification of tumor in relation to the maximally distended synovial cavity and is mainly used for tumor assessment in the knee[Citation85]. Still, TVS is an unvalidated, semiquantitative scoring approach. This underlines the need for future approaches for volume measurement in TGCT.
Until now, only patients with inoperable D-TGCT or patients with expected morbidity of operative treatment qualified for TKI therapy, yet inoperable TGCT is not clearly defined. At present, multidisciplinary tumor boards are best capable of determining which patients should receive systemic treatment. Looking at the future, it would be a massive step in the tailored treatment of TGCT to identify predictive and/or prognostic markers to select patients who would benefit from specific drug agents.
Besides not knowing which patients will benefit the most, questions remain about treatment tendencies. Little is known about the optimal treatment duration or length of response. In pexidartinib, tumor response often occurred within six months after the first treatment, but even more tumor responses occurred after long-term treatment[Citation87]. However, in a relatively young population, chronic treatment is undesirable. Emactuzumab showed durable responses despite a relatively short treatment duration, which could also be an advantage for intermittent treatment. A study to evaluate discontinuation and re-treatment with systemic therapy in patients with TGCT is now open to inclusions[Citation100]. Finally, since complete response is not very common, perhaps studies should focus more on partial response, achieving a stable state of disease or improving QoL. Current systemic therapies target the CSF1/CSF1R axis and may not eliminate the neoplastic cells, making it impossible to achieve a true, complete response. Drug cessation is expected to result in a return of the reactive cells and subsequent lesion growth. Therefore, future studies should focus on targeting the neoplastic cell components.
Although systemic therapies are primarily used as a stand-alone treatment in patients with advanced TGCT, their role as (neo)adjuvant therapy and in an earlier stage of the disease is yet to be explored. Starting earlier with such treatment could prevent disease worsening and even secondary joint deterioration. In addition, these therapies may be used for surgical downstaging in inoperable cases or to decrease expected morbidity. However, post-hoc analysis showed no additional effect of surgery performed directly after nilotinib treatment.83 Secondly, the effect of CSF1R inhibition on angiogenesis and the role of macrophages, which are essential in the postoperative course, needs to be further elucidated[Citation60]. Besides the role as (neo)adjuvant therapy, the efficacy of CSF1R inhibitors combined with other therapies, such as anti-VEGF therapy, requires further investigation.
Medical treatment paved the way for those patients not amenable to surgery, despite the uncertainties regarding the optimal treatment. There is an unmet medical need for broader availability of TGCT related drugs, but drug approval can be challenging in rare diseases[Citation101].
Article highlights
TGCT is a monoarticular neoplasm driven by an overexpression of CSF1, leading to an increase in neoplastic TGCT cells and an accumulation of CSF1R presenting cells.
TGCT is mainly treated by surgery, but achieving a cure can be challenging, requiring additional therapies for relapsing or inoperable tumors.
Several molecular pathways have been suggested as drug targets, but the CSF1-CSF1R axis is most targeted to date.
CSF1R inhibitors can have anti-tumor activity, but adverse events often occur.
Only one CSF1R inhibitor is available in the US to date, but new therapies are being investigated.
Future drug development should focus on targeting neoplastic TGCT cells.
List of abbreviations
| TGCT | = | Tenosyonvial giant cell tumor |
| CSF1 | = | Colony-stimulating factor 1 |
| CSF1R | = | Colony-stimulating factor 1 receptor |
| D-TGCT | = | Diffuse-type tenosynovial giant cell tumor |
| L-TGCT | = | Localized-type tenosynovial giant cell tumor |
| PVNS | = | Pigmented villonodular synovitis |
| M-CSF | = | Macrophage colony-stimulating factor 1 |
| COL6A3 | = | Collagen type VI alpha-3 |
| CBL | = | Cas-Br-Murine ecotropic retroviral transforming sequence |
| ALOX5AP | = | Arachidonate 5-lipoxygenase–activating protein |
| SSP1 | = | Osteopontin |
| MMP | = | Matrix metalloproteinase |
| TP53 | = | Tumor suppressor protein p53 |
| FLS | = | Fibroblast-like synoviocytes |
| IL-1β | = | Interleukine-1β |
| TNF-α | = | Tumor necrosis factor-α |
| MRI | = | Magnetic resonance imaging |
| QoL | = | Quality of life |
| RSO | = | Radiosynoviorthesis |
| EBR | = | External beam radiotherapy |
| Abl | = | Abelson proto-oncogene |
| Bcr-Abl | = | Breakpoint cluster region-Abl |
| KIT | = | c-Kit proto-oncogene |
| PDGFR | = | Platelet-derived growth factor receptor |
| TKI | = | Tyrosine kinase inhibitors |
| JAK2 | = | Janus-Kinase-2 |
| PD-L1 | = | Programmed cell death ligand 1 |
| RANKL | = | Receptor-activator of nuclear factor kappa-B ligand |
| cIAP2 | = | Cellular inhibitor of apoptosis 2 |
| ARRB2 | = | β-Arrestin2 |
| VEGF | = | Vascular endothelial growth factor |
| AE | = | Adverse events |
| GIST | = | Gastrointestinal stromal tumors |
| ORR | = | Objective response rate |
| RECIST | = | Response evaluation criteria in solid tumors |
| IgG1 | = | Immunoglobulin 1 |
| OBD | = | Optimal biological dose |
| FDA | = | Food and Druga Administration |
| ALT | = | Alanineaminotransferase |
| FLT3-ITD | = | Fmls-like tyrosine kinase 3 internal tandem duplication |
| AST | = | Aspartate aminotransferase level |
| TVS | = | Tumor volume score |
| REMS | = | Risk evaluation management system |
| PPARγ | = | Proliferator-activated receptor gamma |
| NSAID | = | Non-steroidal anti-inflammatory drugs |
| EMA | = | European Medicines Agency |
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Ottaviani S, Ayral X, Dougados M, et al. Pigmented villonodular synovitis: a retrospective single-center study of 122 cases and review of the literature. Semin Arthritis Rheum. 2011 Jun;40(6):539–546.
- de St. Aubain Somerhausen N, de Rijn M V. Tenosynovial giant cell tumour. In: The WHO Classification of Tumours Editorial Board, editor. 5th World Health Organization Classification Of Tumours Of Soft Tissue And Bone. Lyon:IARC Press.2020.
- Mastboom MJL, Verspoor FGM, Hanff DF, et al. Severity classification of tenosynovial giant cell tumours on MR imaging. Surg Oncol. 2018 Sep;27(3):544–550.
- Rubin BP. Tenosynovial giant cell tumor and pigmented villonodular synovitis: a proposal for unification of these clinically distinct but histologically and genetically identical lesions. Skeletal Radiol. 2007 Apr;36(4):267–268.
- Sansone V, Longhino V. What’s in a name? A call for consistency in the classification of tenosynovial giant cell tumour. Knee. 2018 Dec;25(6):1322–1323.
- Ehrenstein V, Andersen SL, Qazi I, et al. Tenosynovial giant cell tumor: incidence, prevalence, patient characteristics, and recurrence. A registry-based cohort study in denmark. J Rheumatol. 2017 Oct;44(10):1476–1483.
- Mastboom MJL, Verspoor FGM, Verschoor AJ, et al. Higher incidence rates than previously known in tenosynovial giant cell tumors. Acta Orthop. 2017 Dec;88(6):688–694.
- Mastboom MJL, Verspoor FGM, Uittenbogaard D, et al. tenosynovial giant cell tumors in children: a similar entity compared with adults. Clin Orthop Relat Res. 2018 Sep;476(9):1803–1812.
- Gouin F, Noailles T. Localized and diffuse forms of tenosynovial giant cell tumor (formerly giant cell tumor of the tendon sheath and pigmented villonodular synovitis). Orthop Traumatol Surg Res. 2017 Feb;103(1s):S91–s97.
- Gelhorn HL, Tong S, McQuarrie K, et al. Patient-reported symptoms of tenosynovial giant cell tumors. Clin Ther. 2016 Apr;38(4):778–793.
- Bernthal NM, Spierenburg G, Healey JH, et al. The diffuse-type tenosynovial giant cell tumor (dt-TGCT) patient journey: a prospective multicenter study. Orphanet J Rare Dis. 2021 Apr 29;16(1):191.
- van der Heijden L, Mastboom MJ, Dijkstra PD, et al. Functional outcome and quality of life after the surgical treatment for diffuse-type giant-cell tumour around the knee: a retrospective analysis of 30 patients. Bone Joint J. 2014 Aug;96-b(8):1111–1118.
- Verspoor FGM, Mastboom MJL, Hannink G, et al. The effect of surgery in tenosynovial giant cell tumours as measured by patient-reported outcomes on quality of life and joint function. Bone Joint J. 2019 Mar;101-b(3):272–280.
- Lopez-Bastida J, Aranda-Reneo I, Rodríguez-Sánchez B, et al. Economic burden and health-related quality of life in tenosynovial giant-cell tumour patients in Europe: an observational disease registry. Orphanet J Rare Dis. 2021 Jul 2;16(1):294.
- Murphey MD, Rhee JH, Lewis RB, et al. Pigmented villonodular synovitis: radiologic-pathologic correlation. Radiographics. 2008 Sep-Oct;28(5):1493–1518.
- Nishida Y, Tsukushi S, Nakashima H, et al. Osteochondral destruction in pigmented villonodular synovitis during the clinical course. J Rheumatol. 2012 Feb;39(2):345–351.
- Jaffe HL, Lichtenstein L, Sutro CJ. Pigmented villonodular synovitis, bursitis and tenosynovitis. Arch Pathol. 1941;31:731–765.
- Dal Cin P, Sciot R, Samson I, et al. Cytogenetic characterization of tenosynovial giant cell tumors (nodular tenosynovitis). Cancer Res. 1994 Aug 1;54(15):3986–3987.
- Sciot R, Rosai J, Dal Cin P, et al. Analysis of 35 cases of localized and diffuse tenosynovial giant cell tumor: a report from the Chromosomes and Morphology (CHAMP) study group. Mod Pathol. 1999 Jun;12(6):576–579.
- West RB, Rubin BP, Miller MA, et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci U S A. 2006 Jan 17;103(3):690–695.
- Stanley ER, Berg KL, Einstein DB, et al. Biology and action of colony–stimulating factor-1. Mol Reprod Dev. 1997 Jan;46(1):4–10.
- Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Dev Comp Immunol. 2004 May 3;28(5):509–554.
- Nilsson M, Höglund M, Panagopoulos I, et al. Molecular cytogenetic mapping of recurrent chromosomal breakpoints in tenosynovial giant cell tumors. Virchows Arch. 2002 Nov;441(5):475–480.
- Möller E, Mandahl N, Mertens F, et al. Molecular identification of COL6A3‐CSF1 fusion transcripts in tenosynovial giant cell tumors. Genes Chromosomes Cancer. 2008;47(1):21–25.
- Panagopoulos I, Brandal P, Gorunova L, et al. Novel CSF1-S100A10 fusion gene and CSF1 transcript identified by RNA sequencing in tenosynovial giant cell tumors. Int J Oncol. 2014 May;44(5):1425–1432.
- Cupp JS, Miller MA, Montgomery KD, et al. Translocation and expression of CSF1 in pigmented villonodular synovitis, tenosynovial giant cell tumor, rheumatoid arthritis and other reactive synovitides. Am J Surg Pathol. 2007 Jun;31(6):970–976.
- Brahmi M, Alberti L, Tirode F, et al. Complete response to CSF1R inhibitor in a translocation variant of teno-synovial giant cell tumor without genomic alteration of the CSF1 gene. Ann Oncol. 2018 Jun 1;29(6):1488–1489.
- Tsuda Y, Hirata M, Katayama K, et al. Massively parallel sequencing of tenosynovial giant cell tumors reveals novel CSF1 fusion transcripts and novel somatic CBL mutations. Int J Cancer. 2019 Dec 15;145(12):3276–3284.
- Ho J, Peters T, Dickson BC, et al. Detection of CSF1 rearrangements deleting the 3ʹUTR in tenosynovial giant cell tumors. Genes Chromosomes Cancer. 2019 Aug 30; 59(2):96–105.
- Vougiouklakis T, Shen G, Feng X, et al. Molecular profiling of atypical tenosynovial giant cell tumors reveals novel non-CSF1 fusions. Cancers (Basel). 2019 Dec 31;12(1):100.
- Ota T, Urakawa H, Kozawa E, et al. Expression of colony-stimulating factor 1 is associated with occurrence of osteochondral change in pigmented villonodular synovitis. Tumor Biology. 2015 Jul;36(7):5361–5367.
- Mastboom MJL, Hoek DM, Bovee J, et al. Does CSF1 overexpression or rearrangement influence biological behaviour in tenosynovial giant cell tumours of the knee? Histopathology. 2019 Jan;74(2):332–340.
- Finis K, Sültmann H, Ruschhaupt M, et al. Analysis of pigmented villonodular synovitis with genome-wide complementary DNA microarray and tissue array technology reveals insight into potential novel therapeutic approaches. Arthritis Rheum. 2006 Mar;54(3):1009–1019.
- Darling JM, Goldring SR, Harada Y, et al. Multinucleated cells in pigmented villonodular synovitis and giant cell tumor of tendon sheath express features of osteoclasts. Am J Pathol. 1997 Apr;150(4):1383–1393.
- Somerhausen NS, Fletcher CD. Diffuse-type giant cell tumor: clinicopathologic and immunohistochemical analysis of 50 cases with extraarticular disease. Am J Surg Pathol. 2000 Apr;24(4):479–492.
- Yoshida W, Uzuki M, Kurose A, et al. Cell characterization of mononuclear and giant cells constituting pigmented villonodular synovitis. Hum Pathol. 2003 Jan;34(1):65–73.
- Boland JM, Folpe AL, Hornick JL, et al. Clusterin is expressed in normal synoviocytes and in tenosynovial giant cell tumors of localized and diffuse types: diagnostic and histogenetic implications. Am J Surg Pathol. 2009 Aug;33(8):1225–1229.
- O’Connell JX, Fanburg JC, Rosenberg AE. Giant cell tumor of tendon sheath and pigmented villonodular synovitis: immunophenotype suggests a synovial cell origin. Hum Pathol. 1995 Jul;26(7):771–775.
- Ding Y, Griffin JE, Raghavan M, et al. Tenosynovial giant cell tumors lacking giant cells: report of diagnostic pitfalls. Ann Clin Lab Sci Spring. 2014;44(2):222–227.
- Cao C, Wu F, Niu X, et al. Cadherin-11 cooperates with inflammatory factors to promote the migration and invasion of fibroblast-like synoviocytes in pigmented villonodular synovitis. Theranostics. 2020;10(23):10573–10588.
- Li CF, Wang JW, Huang WW, et al. Malignant diffuse-type tenosynovial giant cell tumors: a series of 7 cases comparing with 24 benign lesions with review of the literature. Am J Surg Pathol. 2008 Apr;32(4):587–599.
- Al-Ibraheemi A, Ahrens WA, Fritchie K, et al. Malignant tenosynovial giant cell tumor: the true “synovial sarcoma?” A clinicopathologic, immunohistochemical, and molecular cytogenetic study of 10 cases, supporting origin from synoviocytes. Mod Pathol. 2019 Feb;32(2):242–251.
- Aurégan JC, Klouche S, Bohu Y, et al. Treatment of pigmented villonodular synovitis of the knee. Arthroscopy: J Arthroscopic & Related Surgery: Off publArthroscopy Assoc North Am Int Arthroscopy Assoc. 2014 Oct;30(10):1327–1341.
- Noailles T, Brulefert K, Briand S, et al. Giant cell tumor of tendon sheath: open surgery or arthroscopic synovectomy? A systematic review of the literature. Orthopaedics & Traumatology: Surgery & Research. 2017 Sep;103(5):809–814.
- Mastboom MJL, Staals EL, Verspoor FGM, et al. Surgical treatment of localized-type tenosynovial giant cell tumors of large joints: a study based on a multicenter-pooled database of 31 International Sarcoma Centers. J Bone Joint Surg Am Vol. 2019 Jul 17;101(14):1309–1318.
- Mastboom MJL, Palmerini E, Verspoor FGM, et al. Surgical outcomes of patients with diffuse-type tenosynovial giant-cell tumours: an international, retrospective, cohort study. Lancet Oncol. 2019 Apr 24;20:877–886.
- Palmerini E, Staals EL, Maki RG, et al. Tenosynovial giant cell tumour/pigmented villonodular synovitis: outcome of 294 patients before the era of kinase inhibitors. Eur J Cancer. 2015 Jan;51(2):210–217.
- Healey JH, Bernthal NM, van de Sande M. Management of tenosynovial giant cell tumor: a neoplastic and inflammatory disease. J Am Acad Orthop Surg Glob Res Rev. 2020 Nov;4(11):e2000028.
- Staals EL, Ferrari S, Donati DM, et al. Diffuse-type tenosynovial giant cell tumour: current treatment concepts and future perspectives. Eur J Cancer. 2016 Aug;63:34–40.
- van der Heijden L, Gibbons CL, Hassan AB, et al. A multidisciplinary approach to giant cell tumors of tendon sheath and synovium–a critical appraisal of literature and treatment proposal. J Surg Oncol. 2013 Mar;107(4):433–445.
- Griffin AM, Ferguson PC, Catton CN, et al. Long-term outcome of the treatment of high-risk tenosynovial giant cell tumor/pigmented villonodular synovitis with radiotherapy and surgery. Cancer. 2012 Oct 1;118(19):4901–4909.
- Durr HR, Capellen CF, Klein A, et al. The effects of radiosynoviorthesis in pigmented villonodular synovitis of the knee. Arch Orthop Trauma Surg. 2019 May;139(5):623–627.
- Gortzak Y, Vitenberg M, Frenkel Rutenberg T, et al. Inconclusive benefit of adjuvant (90)Yttrium hydroxyapatite to radiosynovectomy for diffuse-type tenosynovial giant-cell tumour of the knee. Bone Joint J. 2018 Jul;100-b(7):984–988.
- Bickels J, Isaakov J, Kollender Y, et al. Unacceptable complications following intra-articular injection of yttrium 90 in the ankle joint for diffuse pigmented villonodular synovitis. J Bone Joint Surg Am Vol. 2008 Feb;90(2):326–328.
- Mollon B, Lee A, Busse JW, et al. The effect of surgical synovectomy and radiotherapy on the rate of recurrence of pigmented villonodular synovitis of the knee: an individual patient meta-analysis. Bone Joint J. 2015 Apr;97-b(4):550–557.
- Dracham CB, Shankar A, Madan R. Radiation induced secondary malignancies: a review article. Radiat Oncol J. 2018 Jun;36(2):85–94.
- Blay JY, El Sayadi H, Thiesse P, et al. Complete response to imatinib in relapsing pigmented villonodular synovitis/tenosynovial giant cell tumor (PVNS/TGCT). Ann Oncol. 2008 Apr;19(4):821–822.
- Noguchi R, Yoshimatsu Y, Ono T, et al. Establishment and characterization of a novel cell line, NCC-TGCT1-C1, derived from a patient with tenosynovial giant cell tumor. Hum Cell. 2020 Sep 4;34:254–259.
- Tang F, Tie Y, Hong WQ, et al. Patient-derived tumor xenografts plus ex vivo models enable drug validation for tenosynovial giant cell tumors. Ann Surg Oncol. 2021 Mar 21;28:6453–6463.
- Brahmi M, Vinceneux A, Cassier PA. Current systemic treatment options for tenosynovial giant cell tumor/pigmented villonodular synovitis: targeting the CSF1/CSF1R axis. Curr Treat Options Oncol. 2016 Feb;17(2):10.
- Liang J, Zhao W, Tong P, et al. Comprehensive molecular characterization of inhibitors of apoptosis proteins (IAPs) for therapeutic targeting in cancer. BMC Med Genomics. 2020 Jan 21;13(1):7.
- Ding Z, Bai Z, Zhang M, et al. cIAP2 expression and clinical significance in pigmented villonodular synovitis. J Mol Histol. 2021 Feb 18;52:397–406.
- Cao C, Zhang Y, Cheng J, et al. β-Arrestin2 inhibits the apoptosis and facilitates the proliferation of fibroblast-like synoviocytes in diffuse-type tenosynovial giant cell tumor. Cancer Genomics Proteomics. 2021 May-Jun;18(3 Suppl):461–470.
- Rosen ED, Hsu CH, Wang X, et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002 Jan 1;16(1):22–26.
- Takeuchi A, Yamamoto N, Nishida H, et al. Complete necrosis of a giant cell tumor with high expression of PPARγ: a case report. Anticancer Res. 2013 May;33(5):2169–2174.
- O’Keefe RJ, Rosier RN, Teot LA, et al. Cytokine and matrix metalloproteinase expression in pigmented villonodular synovitis may mediate bone and cartilage destruction. Iowa Orthop J. 1998;18:26–34.
- Fiocco U, Sfriso P, Lunardi F, et al. Molecular pathways involved in synovial cell inflammation and tumoral proliferation in diffuse pigmented villonodular synovitis. Autoimmun Rev. 2010 Sep;9(11):780–784.
- Curry JM, Eubank TD, Roberts RD, et al. M-CSF signals through the MAPK/ERK pathway via Sp1 to induce VEGF production and induces angiogenesis in vivo. PloS one. 2008;3(10):e3405.
- Yamagishi T, Kawashima H, Ogose A, et al. Expression profiling of receptor-activator of nuclear factor-kappa b ligand in soft tissue tumors. Tohoku J Exp Med. 2019 Jun;248(2):87–97.
- Palmerini E, Chawla NS, Ferrari S, et al. Denosumab in advanced/unresectable giant-cell tumour of bone (GCTB): for how long? Eur J Cancer. 2017 May;76:118–124.
- Feng W, Guo J, Li M. RANKL-independent modulation of osteoclastogenesis. J Oral Biosci. 2019 Mar;61(1):16–21.
- van der Heijden L, Gibbons CL, Dijkstra PD, et al. The management of diffuse-type giant cell tumour (pigmented villonodular synovitis) and giant cell tumour of tendon sheath (nodular tenosynovitis). J Bone Joint Surg Br. 2012 Jul;94(7):882–888.
- Zheng B, Yu L, Hu J, et al. Expression of PD-L1 in mononuclear cells, multinucleated cells, and foam cells in tenosynovial giant cell tumors. Int J Clin Exp Pathol. 2019;12(3):876–884.
- Hutarew G. PD-L1 testing, fit for routine evaluation? From a pathologist’s point of view. Memo. 2016;9(4):201–206.
- Cassier PA, Gelderblom H, Stacchiotti S, et al. Efficacy of imatinib mesylate for the treatment of locally advanced and/or metastatic tenosynovial giant cell tumor/pigmented villonodular synovitis. Cancer. 2012 Mar 15;118(6):1649–1655.
- Verspoor FGM, Mastboom MJL, Hannink G, et al. Long-term efficacy of imatinib mesylate in patients with advanced Tenosynovial Giant Cell Tumor. Sci Rep. 2019 Oct 10;9(1):14551.
- Mastboom MJL, Lips W, van Langevelde K, et al. The effect of imatinib mesylate in diffuse-type tenosynovial giant cell tumours on MR imaging and PET-CT. Surg Oncol. 2020 Aug 26;35:261–267.
- Stacchiotti S, Crippa F, Messina A, et al. Response to imatinib in villonodular pigmented synovitis (PVNS) resistant to nilotinib. Clin Sarcoma Res. 2013 May 13;3(1):8.
- Ries CH, Cannarile MA, Hoves S, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014 Jun 16;25(6):846–859.
- Cassier PA, Italiano A, Gomez-Roca CA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015 Aug;16(8):949–956.
- Cassier PA, Italiano A, Gomez-Roca C, et al. Long-term clinical activity, safety and patient-reported quality of life for emactuzumab-treated patients with diffuse-type tenosynovial giant-cell tumour. Eur J Cancer. 2020 Nov 5;141:162–170.
- Smart K, Bröske AM, Rüttinger D, et al. PK/PD mediated dose optimization of emactuzumab, a CSF1R inhibitor, in patients with advanced solid tumors and diffuse-type tenosynovial giant cell tumor. Clin Pharmacol Ther. 2020 Jun 23;108:616–624.
- Gelderblom H, Cropet C, Chevreau C, et al. Nilotinib in locally advanced pigmented villonodular synovitis: a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2018 May;19(5):639–648.
- Monestime S, Lazaridis DP. (TURALIO™): the First FDA-Indicated systemic treatment for tenosynovial giant cell tumor. Drugs R D. 2020 Sep;20(3):189–195.
- Tap WD, Wainberg ZA, Anthony SP, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015 Jul 30;373(5):428–437.
- Tap WD, Gelderblom H, Palmerini E, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet. 2019 Jun 19;394:478–487.
- Gelderblom H, Wagner AJ, Tap WD, et al. Long-term outcomes of pexidartinib in tenosynovial giant cell tumors. Cancer. 2020 Nov 16;127(6):884–893.
- Lewis JH, Gelderblom H, van de Sande M, et al. Pexidartinib long-term hepatic safety profile in patients with tenosynovial giant cell tumors. Oncologist. 2021 May;26(5):e863–e73.
- Gelderblom H, de Sande MV. Pexidartinib: first approved systemic therapy for patients with tenosynovial giant cell tumor. Future Oncol. 2020 Jul 23;16:2345–2356.
- Cheng E, Kudlidjian A, Block J MCS110, an anti-CSF-1 antibody, for the treatment of pigmented villonodular synovitis (PVNS). ISOLS/MSTS 2015 Annual Meeting, Orlando, Florida: 2015.
- Sankhala KK, Blay J-Y, Ganjoo KN, et al. A phase I/II dose escalation and expansion study of cabiralizumab (cabira; FPA-008), an anti-CSF1R antibody, in tenosynovial giant cell tumor (TGCT, diffuse pigmented villonodular synovitis D-PVNS). J clin oncol. 2017;35(15_suppl):11078.
- Smith BD, Kaufman MD, Wise SC, et al. Vimseltinib: a precision CSF1R therapy for tenosynovial giant cell tumors and diseases promoted by macrophages. Mol Cancer Ther. 2021 Aug 25;20:2098–2109.
- Gelderblom H, Razak ARA, Sanchez-Gastaldo A, et al. Safety and preliminary efficacy of vimseltinib in tenosynovial giant cell tumor (TGCT). ESMO Congress. 2021 September 16-21;32(supplement 5):S1233–S1234. https://doi.org/https://doi.org/10.1016/j.annonc.2021.08.275
- Kroot EJ, Kraan MC, Smeets TJ, et al. Tumour necrosis factor alpha blockade in treatment resistant pigmented villonodular synovitis. Ann Rheum Dis. 2005 Mar;64(3):497–499.
- Fiocco U, Sfriso P, Oliviero F, et al. Intra-articular treatment with the TNF-alpha antagonist, etanercept, in severe diffuse pigmented villonodular synovitis of the knee. Reumatismo. 2006 Oct-Dec;58(4):268–274.
- Eubank TD, Galloway M, Montague CM, et al. M-CSF induces vascular endothelial growth factor production and angiogenic activity from human monocytes. J Immunol. 2003 Sep 1;171(5):2637–2643.
- Nissen MJ, Boucher A, Brulhart L, et al. Efficacy of intra-articular bevacizumab for relapsing diffuse-type giant cell tumour. Ann Rheum Dis. 2014 May;73(5):947–948.
- Takeuchi A, Nomura A, Yamamoto N, et al. Randomized placebo-controlled double-blind phase II study of zaltoprofen for patients with diffuse-type and unresectable localized tenosynovial giant cell tumors: a study protocol. BMC Musculoskelet Disord. 2019 Feb 9;20(1):68.
- An Open-Label Study of Intra-articular AMB-05X Injections in Subjects With Tenosynovial Giant Cell Tumor of the Knee. [ cited 2021 Nov 01]. Available from: https://ClinicalTrials.gov/show/NCT04731675
- Study to evaluate discontinuation and re-treatment in participants with Tenosynovial Giant Cell Tumor (TGCT). Previously Treated With Pexidartinib. [cited 2021 Nov 01]. Available from: https://ClinicalTrials.gov/show/NCT04526704
- Kasper B. The challenge of drug approval in rare cancers. Cancer. 2020 Nov 16;127(6): 837–839.