
Acute myeloid leukemia (AML) is the most common form of acute leukemia in adults in the United States and with a 5-year overall survival in the contemporary era of only 29.5% it remains a major clinical challenge[Citation1]. The role of leukemia stem cells (LSCs) in disease relapse following response to initial therapy has been increasingly recognized and failure of the immune system to eradicate LSCs has been demonstrated as a major mechanism of disease relapse[Citation2]. A better understanding of the mechanisms of immune escape in AML could therefore offer the potential for better and more effective treatment strategies.
Recent studies have suggested an immunosuppressive tumor microenvironment in the bone marrow niche in patients with AML and myelodysplastic syndromes (MDS) that is characterized by the upregulation of inhibitory immune checkpoint receptors (e.g. programmed death (PD)-1, T-cell immunoglobulin mucin-3 (TIM-3) and lymphocyte activation gene-3 (LAG-3)), changes in T-cell subpopulations (increase in regulatory T-cells), and expansion of immunosuppressive myeloid-derived suppressor cells (MDSC)[Citation3].
The term MDSC was coined in the early 2000’s to describe a heterogenous group of myeloid cells with immune regulatory properties[Citation4]. Chronic inflammatory states including malignancy have been associated with the differentiation of myeloid precursor cells into immunosuppressive MDSCs that are characterized by a reduced phagocytic activity, upregulation of mTOR, MAPK, and IFN-γ regulated pathways, and the increased production of reactive oxygen and nitrogen species[Citation4]. MDSCs are further subdivided based on phenotypic, molecular, and functional properties into granulocytic or polymorphonuclear (PMN-MDSC) and monocytic MDSCs (M-MDSC)[Citation4]. MDSCs can be detected in the peripheral blood or bone marrow by flow cytometry based on the expression of CD11b and Gr-1 with distinct expression profiles for PMN-MDSCs (CD11b+CD14−CD15+, CD11b+CD14−CD66b+, or CD11b+Ly6G+Ly6Clo) and M-MDSCs (CD11b+CD14+HLA-DR−/loCD15− or CD11b+Ly6G−Ly6Chi) having been described in many cases[Citation5]. In various solid and hematologic malignancies the number of peripheral blood MDSCs has been associated with tumor stage and adverse outcomes[Citation6]. Additionally, a higher frequency of MDSCs has been associated with lower response rates to chemotherapy and immune checkpoint blockade in melanoma[Citation7].
MDSCs are also expanded in a subset of AML and MDS patients contributing to the impaired immune surveillance in the bone marrow microenvironment and the number of MDSCs has been shown to inversely correlate with prognosis [Citation8,Citation9]. One study demonstrated that the MDSC population is expanded by up to 60-fold in MDS patients compared to normal donors (6% vs 0.1% of all cells; p < 0.0001) and inversely correlates with the percentage of CD7+ lymphocytes, T-cells, and NK-cells[Citation10]. However, the specific processes that drive MDSC expansion and how they contribute to the immunosuppressive microenvironment in myeloid malignancies are incompletely understood. Preclinical studies have shown that MDSCs in AML cell lines and murine models suppress T-cell responses and T-cell proliferation and that expansion of MDSCs is driven by MUC-1-mediated expression of c-myc[Citation11]. AML-derived extracellular vesicles have been shown to be a key mediator in the initiation of M-MDSC differentiation in AML [Citation11,Citation12]. Interestingly, upregulation of MYC and downregulation of its negative regulator miR-34a has also been associated with the increased expression of PD-L1 in MDS and AML patients with TP53 mutations[Citation13]. This mechanistic association is further supported by the fact that miR-34a transcription is regulated by p53 and the observation that MDSCs are increased in patients with TP53 mutations[Citation13].
The role of mesenchymal stromal cells (MSC) and their interaction with MDSCs in myeloid neoplasms is also becoming an area of increased interest. MSCs have been shown to contribute to chemotherapy resistance, immune regulation and LSC proliferation[Citation14]. The immunomodulatory properties of MSCs are mediated by suppression of T-cell proliferation, induction of T-cell apoptosis via PD-1/PD-L1 interaction, IFN-γ and TNF-α-mediated prostaglandin E2 signaling[Citation14]. Preclinical studies in MDS have also shown that MDS-derived MSCs are able to induce the expansion of MDSCs via an increased expression of the ROS regulator ENC1 and dysregulated TGF-β pathway signaling[Citation15]. This contributes to the suppression of NK cell and T-cell function and the immunosuppressive bone marrow microenvironment observed in MDS patients compared to healthy controls[Citation15].
Targeting MDSCs to re-invigorate the anti-tumor immune response in AML and MDS could be a promising therapeutic strategy. Potential therapeutic strategies include the inhibition of MDSC differentiation, inducing differentiation of MDSCs, inhibition of MDSC activity, or the targeted elimination of MDSCs[Citation16]. illustrates various therapeutic strategies for targeting MDSCs.
Figure 1. Potential therapeutic strategies targeting myeloid-derived suppressor cells.
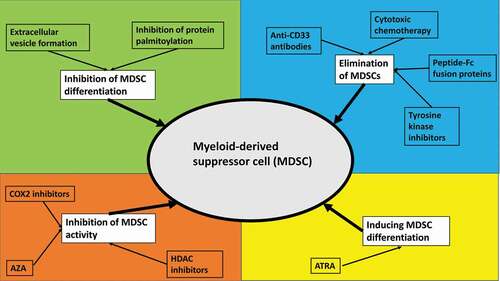
It has been shown that AML-derived extracellular vesicles play a key role in the differentiation of monocytes into M-MDSCs[Citation12]. Preclinical studies suggested that palmitoylated proteins on the surface of these extracellular vesicles are required for MDSC differentiation via activation of Toll-like receptors, which activates downstream Akt/mTOR pathway signaling[Citation12]. Inhibiting protein palmitoylation could therefore be a potential option to interfere with MDSC differentiation.
As M-MDSCs are characterized by the expression of CD33, M-MDSCs could be susceptible to anti-CD33-directed therapies. AMG 330 is a CD3/CD33 bispecific T-cell engager antibody that induces a T-cell response against CD33-expressing cells. In vitro experiments have shown that AMG 330 was able to eradicate CD33+ MDSCs[Citation17]. Similarly, the anti-Cd16/CD33 bispecific killer cell engager antibody construct has been shown to overcome NK cell-mediated immunosuppression and restore NK cell function in MDS[Citation10]. Since CD33 is also expressed on leukemic blasts and LSCs and co-expression with inhibitory immune checkpoints such as TIM-3 has been demonstrated, combination therapies targeting both CD33 and other immune checkpoints could be an effective strategy to eradicate both LSCs and MDSC[Citation18]. Conventional cytotoxic chemotherapy such as gemcitabine, tyrosine kinase inhibitors, or targeting S100A by therapeutic peptide-Fc fusion proteins has been shown to deplete MDSCs in solid tumors. However, their role in AML and MDS is insufficiently characterized to date[Citation16].
As discussed previously, prostaglandin E2 signaling is a key mediator of the immunosuppressive effects of MDSCs and MSCs on T-cell function. Prostaglandin E2 synthesis is regulated by cyclooxygenase-2 (COX-2). COX-2 inhibition has been shown to increase anti-tumor immune responses by cytotoxic T-cells and could have synergistic effects with anti-PD-1 antibodies [Citation16,Citation19]. Along the same line are strategies reducing the production of reactive oxygen and nitrogen species to restore anti-tumor T-cell activity[Citation16].
A final therapeutic strategy to target MDSCs is induction of MDSC differentiation into normal myeloid cell phenotypes. All-trans retinoic acid (ATRA) has been shown to induce the differentiation of MDSCs into mature antigen-presenting cells resulting in a reversal of T-cell suppression in both renal and lung cancer models[Citation16]. Potential mechanisms of action of ATRA in this context include the neutralization of reactive oxygen species and interference with Notch signaling. Finally, epigenetic therapies using the histone deacetylase (HDAC) inhibitor entinostat or the DNA methyltransferase inhibitor azacitidine have been shown to reduce the number of MDSCs in preclinical models of various solid tumors[Citation16]. However, data in AML and MDS are limited although immunomodulatory properties of azacitidine such as the upregulation of inhibitory immune checkpoint receptors have been demonstrated[Citation20]. Combining HDAC inhibitors with anti-PD-1 antibodies could therefore have synergistic effects and is currently being explored in clinical trials in MDS and various solid malignancies (e.g. NCT02936752, NCT02453620).
While substantial advances in our understanding of the role of MDSCs in AML and MDS have been made recently, most data are derived from solid tumor models and patients and how these results apply to AML and MDS is uncertain warranting additional studies. Given the heterogeneity of AML and MDS on a genetic and immunologic level, even more granular studies in genetically defined patient subsets such as those with TP53 mutations are necessary. While immune checkpoint inhibitors had lackluster results when used as monotherapy or in combination with azacitidine, novel combination strategies and alternative immune checkpoint inhibitors such as anti-TIM3 are needed and are currently being tested in clinical trials (STIMULUS program; NCT03946670)[Citation3]. Robust correlative studies evaluating changes in the immune landscape during treatment are important to inform subsequent trials and provide additional insights into MDSC pathobiology in AML and MDS.
Disclosure statement
AM Zeidan has received research institutional funding from Celgene/BMS, Abbvie, Astex, Pfizer, Medimmune/AstraZeneca, Boehringer-Ingelheim, Trovagene/Cardiff oncology, Incyte, Takeda, Novartis, Aprea, and ADC Therapeutics. AM Zeidan has participated in advisory boards, and/or had a consultancy with and received honoraria from AbbVie, Otsuka, Pfizer, Celgene/BMS, Jazz, Incyte, Agios, Boehringer-Ingelheim, Novartis, Acceleron, Astellas, Daiichi Sankyo, Cardinal Health, Taiho, Seattle Genetics, BeyondSpring, Cardiff Oncology, Takeda, Ionis, Amgen, Janssen, Epizyme, Syndax, Gilead, Kura, Chiesi, ALX Oncology, BioCryst, and Tyme. AM Zeidan has served on clinical trial committees for Novartis, Abbvie, Geron and Celgene/BMS, and has received travel support for meetings from Pfizer, Novartis, and Cardiff Oncology. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- Shallis RM, Wang R, Davidoff A, et al. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. 2019;36:70–87.
- Christopher MJ, Petti AA, Rettig MP, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. 2018;379(24):2330–2341.
- Bewersdorf JP, Shallis RM, Zeidan AM. Immune checkpoint inhibition in myeloid malignancies: moving beyond the PD-1/PD-L1 and CTLA-4 pathways. Blood Rev. 2020;45:100709.
- Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–119.
- Bronte V, Brandau S, Chen S-H, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150.
- Zhang S, Ma X, Zhu C, et al. The role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PLoS One. 2016;11(10):e0164514.
- Sade-Feldman M, Kanterman J, Klieger Y, et al. Clinical significance of circulating CD33+CD11b+HLA-DR- myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin Cancer Res. 2016;22(23):5661–5672.
- Kittang AO, Kordasti S, Sand KE, et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. Oncoimmunology. 2016;5(2):e1062208.
- Wang H, Tao Q, Wang Z, et al. Circulating monocytic myeloid-derived suppressor cells are elevated and associated with poor prognosis in acute myeloid leukemia. J Immunol Res. 2020;2020:7363084.
- Gleason MK, Ross JA, Warlick ED, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. 2014;123(19):3016–3026.
- Pyzer AR, Stroopinsky D, Rajabi H, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. 2017;129(13):1791–1801.
- Tohumeken S, Baur R, Böttcher M, et al. Palmitoylated proteins on AML-derived extracellular vesicles promote myeloid-derived suppressor cell differentiation via TLR2/Akt/mTOR signaling. Cancer Res. 2020;80(17):3663–3676.
- Sallman DA, McLemore AF, Aldrich AL, et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood. 136(24): 2812–2823. 2020.
- Kapor S, Santibanez JF. Myeloid-derived suppressor cells and mesenchymal stem/stromal cells in myeloid malignancies. J Clin Med. 2021;10(13):2788.
- Sarhan D, Wang J, Sunil Arvindam U, et al. Mesenchymal stromal cells shape the MDS microenvironment by inducing suppressive monocytes that dampen NK cell function. JCI Insight. 2020;5(5). DOI:https://doi.org/10.1172/jci.insight.130155.
- Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. 2020;9(3):561.
- Jitschin R, Saul D, Braun M, et al. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. 2018;6(1):116.
- Haubner S, Perna F, Kohnke T, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019;33(1):64–74.
- Zelenay S, van der Veen AG, Böttcher JP, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162(6):1257–1270.
- Yang H, Bueso-Ramos C, DiNardo C, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280–1288.