
ABSTRACT
Introduction: Circulating tumor DNA (ctDNA) has become a relevant biomarker in cancer management, allowing tumor assessment through analysis of minimally invasive liquid biopsies. Applications include screening, diagnostics, monitoring of treatment efficacy and detection of minimal residual disease as well as relapse. The potential of ctDNA analysis is significant, but several biological and technical challenges need to be addressed before widespread clinical implementation.
Areas covered: Several clinical applications where ctDNA analysis may be beneficial require detection of individual DNA molecules. Consequently, to acquire accurate and informative data the entire workflow from sampling to final data interpretation needs to be optimized. In this review, we discuss the biological and technical challenges of ctDNA analysis and how preanalytical and analytical approaches affect different cancer applications.
Expert opinion: While numerous studies have demonstrated the potential of using ctDNA in cancer applications, yet few reports about true clinical utility exist. Despite encouraging data, the sensitivity of ctDNA analyses, i.e. the probability to detect presence of cancer in liquid biopsies, is still an issue. Analysis of multiple mutations in combination with simultaneous assessment of other analytes is one solution. Improved standardization and guidelines will also facilitate the introduction of ctDNA analysis into clinical routine.
1. Introduction
With the emergence of technologies capable to detect and quantify few DNA molecules in complex matrices, liquid biopsy analysis has become clinically relevant. In cancer management and research, circulating tumor DNA (ctDNA), released from the tumor mass and sometimes with small contributions from circulating tumor cells [Citation1,Citation2], is used as a clinical biomarker, where detectable levels during therapy are linked to poor patient outcome [Citation3–5]. Besides treatment monitoring and early relapse detection [Citation6–8], ctDNA analysis may also be applied in screening [Citation9–11], prognostics [Citation12,Citation13] and diagnostics [Citation14].
Essentially all mutations and DNA modifications can be assessed using liquid biopsy analysis, including single nucleotide variants, inserts and deletions, and larger chromosomal abnormalities, such as copy number variations and translocations. In addition to mutations, it is also possible to assess ctDNA through analysis of DNA modifications, such as DNA methylation [Citation15] as well as DNA size distribution [Citation16].
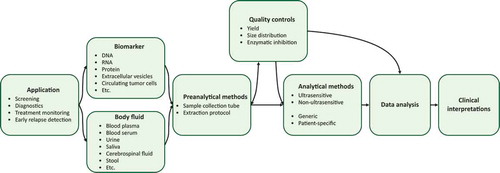
The potential of liquid biopsy analysis is high, but several technical and biological challenges remain to be addressed before widespread clinical implementation. In this review, we discuss the experimental steps using blood plasma and ctDNA analysis with focus on preanalytical parameters (). We also discuss the potential of different technologies in relation to specific cancer applications and how they enable precision medicine.
Figure 1. Overview over the ctDNA analytical workflow. The schematic overview illustrates dependencies between study design and experimental choices
2. Biology of cell-free DNA
Cell-free DNA (cfDNA), which includes ctDNA, is mainly shed from cells through apoptosis, necrosis and active secretion, but also via pyroptosis, autophagy, mitotic catastrophes and NETosis [Citation17], into most body fluids, such as blood, urine, saliva, sputum, cerebrospinal fluid, cyst fluid, stool, sweat, amniotic fluid, bile and bronchoalveolar lavage, among others [Citation18]. Apoptosis is usually believed to be the prime source of cfDNA, where the dominant cfDNA fragment size is around 166 nucleotides [Citation17]. This corresponds to the length of DNA wrapped around a nucleosome and a 20 nucleotide linker bound to histone H1 [Citation19–21]. Multiple nucleosomal repeats are also present [Citation22,Citation23]. Necrosis and active secretion usually generate larger DNA fragments in comparison [Citation17]. Tumor necrosis is associated with aggressive disease and poor prognosis in several cancers [Citation24–26]. Active secretion is poorly understood, but genomic instability and release by extracellular vesicles or alongside protein complexes may be involved [Citation27,Citation28]. The half-life of cfDNA in blood is estimated to be in the range of 15 minutes to 2.5 hours [Citation29]. Clearance occurs through nuclease activity [Citation30,Citation31], renal excretion [Citation32,Citation33] and uptake by spleen and liver [Citation34,Citation35]. Conditions that affect these organs impact on cfDNA clearance [Citation36,Citation37]. However, it is likely that different subpopulations of cells contribute unequally to the total cfDNA pool. For instance, tumor cells with low turnover rate, such as cancer stem cells, probably provide fewer ctDNA molecules compared to dividing cells, just as treatment-resistant cells are expected to release less DNA than treatment-sensitive cells during therapy [Citation38–40]. Location and distance from the liquid analyzed may also affect ctDNA levels and its properties. Hence, even if the ctDNA profile has shown its use as biomarker, it does not necessarily reflect the relative tumor cell compositions [Citation41]. Overall, we know little about release and clearance rates of ctDNA. As a consequence, it is challenging to compare ctDNA levels and profiles between different cancer types and treatments.
In healthy individuals, the total cfDNA concentration is low, generally less than 10 ng per ml blood plasma and rarely higher than 30 ng per ml [Citation42–44], where each ng cfDNA corresponds to approximately 310 haploid human genomes [Citation45]. Most cfDNA is derived from cells of hematopoietic origin and vascular endothelial cells [Citation46]. In cancer patients, cfDNA as well as ctDNA levels vary between individuals and tumor entities [Citation8,Citation47,Citation48], but they have been shown to correlate to tumor burden, defined as either tumor volume [Citation49,Citation50] or tumor stage [Citation51,Citation52], although there are exceptions [Citation53,Citation54]. However, the cfDNA concentration may increase due to various physiological and pathological conditions not necessarily related to cancer, such as physical exercise [Citation55], obesity [Citation56], stroke, sepsis and inflammation, as well as trauma [Citation48], including surgery [Citation57]. Consequently, using only total cfDNA levels to diagnose cancer is not reliable.
3. Preanalytical considerations
To generate accurate and useful ctDNA data, it is essential to consider the entire experimental workflow, including preanalytical factors (). Choice of sample collection tube and extraction protocol may be as critical as the analytical approach. Due to the low amount of cfDNA and often minute ctDNA fractions in most clinical samples, preanalytical strategies preparing high-quality cfDNA in high yields are needed. In addition, to allow straightforward data interpretation the introduction of bias through cellular DNA contamination and substances that inhibit downstream analyses must be minimized [Citation58]. Compared to analytical approaches to assess ctDNA, preanalytical solutions are less evolved, though there is a large and continuously growing number of sample collection tubes and cfDNA extraction kits available. Body fluids have different properties and while some collection tubes are widely used for liquid biopsies, others are optimized for specific body fluids and analytes, such as blood plasma and cfDNA, respectively. The choice of collection tube also determines storage and extraction options. However, it is challenging to compare differences in experimental workflow protocols due to the lack of standardization, with diverse protocols, quality controls, and how downstream analyses are performed. To date, most ctDNA studies use blood plasma [Citation59], which we focus on here.
Figure 2. Overview of preanalytical steps and variable parameters in ctDNA analysis. The use of ctDNA as biomarker is affected by a wide range of preanalytical factors. Experimental steps and common parameters that can vary are shown
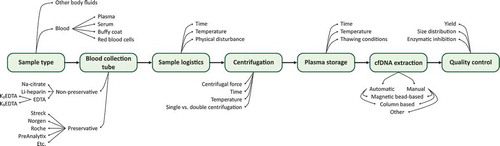
summarizes preanalytical studies, where various blood collection tubes and extraction protocols have been compared and evaluated with different downstream analyses, as well as reports on the effect of time before plasma isolation, time between plasma isolation and DNA extraction, centrifugation speeds and durations, sample storage temperature, inhibition of downstream enzymatic reactions, and the impact of shipping. Most comparative studies evaluating sample collection tubes and extraction protocols use cfDNA yield to determine performance. Both spectrophotometric and fluorometric quantifications are used, the latter being more sensitive and accurate [Citation60]. Sample concentrations can also be deduced from electropherograms generated by capillary gel electrophoresis [Citation61]. To quantify the amount of molecules that can be analyzed with downstream methods, quantitative PCR, or digital PCR may be used [Citation58]. These quantification methods tend to give lower estimates than those based on spectrophotometry and fluorometry. There are several reasons for this difference, including assay accuracy, cfDNA fragmentation, and PCR inhibition, where the latter also can be assessed with quantitative PCR and spike-in controls [Citation58]. Another confounding factor is cellular DNA contamination. Cellular DNA will affect all quantifications of cfDNA yield, except capillary electrophoresis analysis and similar methods where DNA fragment sizes are assessed. Cellular contamination can also be indirectly quantified by quantitative PCR using both a short and a long assay, where the long assay will only detect cellular DNA [Citation58].
Table 1. Overview of comparative preanalytical studies
3.1. Sample collection tubes
In blood, both plasma and serum may be used for cfDNA analysis. Though serum yields more DNA, the cfDNA is often contaminated with larger DNA fragments (150–2000 nucleotides), originating from blood cell lysis during coagulation [Citation88]. Plasma is therefore preferred source of cfDNA [Citation89]. Blood plasma collection tubes are either non-preservative or preservative, where the latter are used when plasma cannot be prepared within a few hours.
Ethylenediaminetetraacetic acid (EDTA) tubes are standard in diagnostic procedures and are thus the most commonly non-preservative collection tube type used for blood [Citation90]. There are two types of EDTA tubes, K2EDTA and K3EDTA, where their difference in relation to cfDNA has to our knowledge not been investigated. EDTA prevents blood clotting by permanently binding Ca2+-ions, which inactivates nucleases that otherwise will degrade nucleic acids [Citation91], but does not prevent blood cell lysis [Citation92]. Other common non-preservative collection tubes are sodium citrate and lithium heparin tubes [Citation93]. Sodium citrate collection tubes can be used for cfDNA analysis, but their properties have been less studied than those of EDTA tubes. Sodium citrate, like EDTA, prevents blot clotting but with lower affinity for calcium and its effect is reversible [Citation94,Citation95]. Heparin-containing tubes should not be used for cfDNA analysis as heparin is a potent enzymatic inhibitor and thus may interfere with downstream analysis [Citation96].
There are numerous reports regarding maximum time from sampling to plasma isolation when using EDTA tubes to minimize the release of cellular DNA from leukocytes. While some studies report an increase of cellular DNA in samples within a few hours of sampling, others observed no increase until 48 hours. However, most studies recommend plasma isolation within either 2 or 6 hours [Citation89,Citation97]. There are also reports demonstrating that the time can be extended to 24 hours [Citation90] or even 48 hours [Citation98] if the collected blood is stored at 4°C. A practical approach is to prepare plasma within 2 hours, but plasma prepared within 6 hours may be considered if the cfDNA is tested for cellular contamination. There are two problems with cellular DNA contamination in ctDNA analysis. First, additional background DNA may affect the sensitivity to detect ctDNA with the applied method. The other potential issue is bias in the estimation of ctDNA levels. For example, if the level of ctDNA is reported as fraction of ctDNA compared to the total amount of cfDNA, contamination of cellular DNA will result in an underestimation of ctDNA. One way to overcome this is to report ctDNA levels as molecules per ml plasma which is independent of cellular DNA contamination. Moreover, if cfDNA is sequenced, capacity will be consumed analyzing basically irrelevant background DNA. When cellular DNA contamination is an issue, sample re-purification to eliminate larger DNA fragments should be considered.
Reports on long-term storage of plasma prepared from non-preservative tubes are contradictory, some claiming that long-term storage at −80°C may lead to substantial loss of DNA [Citation99], while others claim that cfDNA can be extracted from samples stored several years at −20°C with small loss [Citation100]. How different collection tubes affect long-term storage of plasma is poorly understood.
If plasma cannot be prepared within a few hours several preservative blood collection tubes are available, including Cell-Free DNA BCT (Streck), PAXgene Blood ccfDNA tubes (PreAnalytix), Cell-Free DNA Collection Tubes (Roche), and cf-DNA/cf-RNA Preservative Tubes (Norgen BioTek), that allow blood to be stored for longer time (weeks) and transported before plasma isolation. The benefits of these over traditional blood collection tubes have been reported in several studies [Citation90,Citation98,Citation101–103]. Preservative blood collection tubes for cfDNA analysis contain chemical cocktails that both protect cfDNA from degradation by neutralizing nucleases and decrease contamination of cellular DNA by stabilizing blood cells. The effect of temperatures on plasma collected with cfDNA preservative collection tubes has also been investigated with varying results. While one evaluation of the Cell-Free DNA BCT tubes detected an increase in cellular DNA contamination after 24 hours when stored at 23°C–40°C [Citation103], another study reported no cellular contamination after 7 days at 37°C evaluating the same tubes [Citation104]. A limitation of preservative blood collection tubes is that they are optimized for cfDNA analysis and may even prevent analysis of other analytes [Citation105]. Analytes, like metabolites, RNA and proteins, are often technically possible to quantify, but their levels will change over time due to various biological and chemical processes that are not controlled by the preservative reagents. However, most of these effects are not well-characterized.
Regardless the choice of collection tube, the blood sample is generally centrifuged to isolate plasma from blood cells. Standard plasma preparation generates three fractions: red blood cells, an intermediate layer containing white blood cells, referred to as the buffy coat, and plasma. Centrifugation speed, time and temperature may depend on the blood collection tube used. A second high-speed centrifugation step is usually recommended to reduce cellular contamination [Citation89,Citation106]. One important factor that varies across the different blood collection tubes is the final plasma volume as it is affected by the chemical properties, such as osmotic pressure, of respective blood collection tube. This must be considered when reporting the amount of ctDNA as number of molecules per ml plasma. Freeze-thawing of plasma should be avoided as it may cause further DNA fragmentation [Citation107]. While the effects of multiple freeze-thaw cycles are well studied, there are few reports on the importance of plasma thawing conditions in relation to time and temperature [Citation97].
3.2. Cell-free DNA extraction
There is a plethora of kits for cfDNA extraction. Most are based on either silicon membrane-based spin columns or magnetic beads. Both are common as manual extraction kits, while automated methods usually use magnetic beads. Generally, silicon membrane-based spin columns tend to outperform magnetic bead-based kits when comparing cfDNA yield [Citation108]. When choosing extraction kit the blood collection used must be considered for compatibility.
There are several studies comparing manual cfDNA extractions kits, where the commonly used silicon membrane-based spin column, QIAamp Circulating Nucleic Acid Kit (Qiagen), generally performs well with high cfDNA yields [Citation108–110]. However, there are studies showing that other kits, such as the Plasma/Serum Circulating DNA Purification Mini Kit (Norgen Biotek) may be superior. Among manual magnetic bead-based kits no single optimal kit has emerged, although both the QIAamp MinElute ccfDNA Kits (Qiagen) [Citation108] and the MagMAX Cell-Free DNA Isolation Kit (Applied Biosystems) [Citation110] have performed well in comparisons. Regardless if silicon membrane-based spin columns or magnetic beads are used, kits that utilize a lysis step that releases nucleic acids bound to proteins, lipids and contained in vesicles, are preferred [Citation111]. Some extraction kits have the option of adding carrier RNA before extraction to increase cfDNA extraction yield [Citation112]. However, carrier RNA may also negatively affect downstream analysis [Citation113]. There are also various manual cfDNA extraction protocols based on phase separation, e.g. phenol-chloroform protocols [Citation114] and glass-milk-based protocols [Citation115,Citation116], but these are less used today.
Automated cfDNA extraction has been less evaluated than manual approaches. Besides high throughput and low hands-on-time, the use of automated methods reduces variability and the risk of sample-to-sample contamination, though it generates somewhat lower cfDNA yields than manual extraction approaches [Citation117]. There is no automated kit that consistently outperform others, based on the studies in .
4. Circulating tumor DNA analysis
The presence of cfDNA and ctDNA has been known for decades [Citation118,Citation119]. However, it is first with the development of sensitive and specific molecular methods during the last decade that detailed ctDNA analysis has become possible. There are four main obstacles in ctDNA analysis. First, cfDNA is generally present at low concentrations in most body fluids. Secondly, the fraction of ctDNA is usually low, often requiring single molecule detection. Thirdly, cfDNA is highly fragmented. Lastly, handling of naturally occurring background allele variants, including clonal hematopoiesis [Citation120–122] and other age-related accumulations of cancer-associated mutations in healthy tissues [Citation123–125].
Methods like PCR allow analysis of individual mutations, while whole genome sequencing (WGS) targets all mutations. Standard PCR and sequencing only allow detection of variant allele frequencies down to 1–5% [Citation126,Citation127], while digital PCR and ultrasensitive sequencing enable detection of single molecules.
Most ctDNA applications require digital resolution. Ultrasensitive PCR methods, such as digital PCR [Citation128,Citation129] and COLD-PCR [Citation130], are fast, relatively simple, cost-effective and suitable for analysis of single or a few mutations. Another relatively simple method is mass-spectrometry analysis coupled with multiplexed end-point PCR, allowing simultaneous detection of over 70 predefined mutations at allele frequencies down to 0.1% [Citation131]. Ultrasensitive sequencing is enabled by bioinformatical algorithms utilizing unique molecular identifiers that are introduced in library construction [Citation132]. These molecular barcodes are random sequences, usually between four and fourteen nucleotides long, that enable tracking of all sequencing reads back to the original DNA molecule. Subsequently, unique molecular identifiers allow for correction of both DNA polymerase-induced errors [Citation133] and uneven amplification [Citation134] when used in combination with deep sequencing [Citation135,Citation136]. The requirement for deep sequencing limits ultrasensitive sequencing to targeted approaches due to costs. Targeted sequencing, including ultrasensitive sequencing, can be divided into two approaches: amplicon-based and hybridization capture-based. Amplicon-based approaches are simpler and faster than hybridization capture-based strategies, since they are only relying on primers ability to bind target DNA. In comparison, hybridization capture-based approaches are dependent on two steps, i.e. DNA capture and adaptor ligation. In panel development, hybridization capture-based approaches are usually preferred when larger panels are needed. The sensitivity of ctDNA profiling may be improved by selecting for 90–150 nucleotides short DNA fragments that are typical for ctDNA, either experimentally or bioinformatically by enrichment analysis [Citation137]. Similarly, strategies that reduce the amplification of cfDNA but not ctDNA may also improve assay sensitivity [Citation138,Citation139]. Regardless of approach, false positives should be minimized through analysis of non-tumor samples, such as paired peripheral blood cells [Citation140–142].
The wide range of ctDNA applications in cancer management and research has resulted in several technical solutions with their own advantages and disadvantages. Next, we will discuss common cancer applications and their method requirements.
4.1. Circulating tumor DNA analysis – choice of approach
Here, we divide the use of ctDNA analysis in cancer management into four application areas: (i) screening, (ii) diagnostics, (iii) treatment monitoring, and (iv) detection of minimal residual disease and relapse.
4.1.1. Screening
The best chance to survive cancer is detection at an early stage. Several platforms based on ultrasensitive ctDNA analysis have been developed, or are under development, with this aim in mind. To detect and identify cancer in otherwise healthy and non-symptomatic populations at an early stage usually require that mutations can be detected in a large background of cfDNA, corresponding to allele frequencies < 0.1%, i.e. single-molecule resolutions. However, based on theoretical calculations assuming that ctDNA release is proportional to tumor mass, 4 ml of blood plasma is likely to contain less than one cancer genomes if the tumor is smaller than approximately 1 cm3, which corresponds to an early-stage tumor detectable by imaging in a symptomatic patient [Citation143,Citation144]. With few ctDNA molecules expected per ml plasma, there are two options to increase the likelihood of detecting cancer: (i) increase liquid biopsy volume or (ii) increase number of potential mutations that can be detected [Citation58,Citation145], where the last alternative is the most feasible. To enable detection of multiple mutations, an ultrasensitive sequencing panel should be as broad as possible. The drawbacks of large panels, besides cost, are the risk of false positives, especially when patients with conditions associated with increased risk to develop tumor malignancies, including chronic obstructive pulmonary disease [Citation146], Barrett’s esophagus [Citation147], and inflammatory bowel disease [Citation148], are tested due to overlapping mutations. This may result in overdiagnosis, where ctDNA will be detected in some patients where it will never progress to anything clinically relevant, causing additional health-care costs and patient anxiety [Citation143]. In addition, false negative results may also prevent individuals from having any medical examination at the manifestation of symptoms, believing themselves healthy. Another complication is that most mutations are not entity-specific, requiring confirmative imaging and additional sampling. To screen high-risk individuals for a given cancer type is in comparison more straightforward as the mutation landscape often is more limited and well-characterized. Hence, simpler, more focused and cost-effective approaches may be used.
Ultrasensitive sequencing panels are mainly detecting point mutations as well as smaller insertions and deletions. However, many tumor entities are characterized by structural DNA variants, such as copy number variations and translocations. A generic screening approach to identify all these specific mutations requires WGS. Another benefit of WGS compared to most targeted panels is that the tumor entity usually can be determined. However, the limitation of WGS is its sensitivity to find small fractions of ctDNA, minimizing its use in early-stage cancer detection.
Another strategy to increase sensitivity and specificity in early-stage cancer detection is to analyze additional analytes. In addition to mutations, specific methylation- and fragmentation patterns, other biomarkers like circulating tumor cells, proteins, RNA, extracellular vesicles, and tumor-educated platelets, have all shown their use as stand-alone biomarkers in cancer detection [Citation149]. Today, it is technically challenging to analyze different analytes simultaneously in the same sample. Instead, most multi-analyte analyses are performed by aliquoting the samples into parallel analyses. For example, it is usually feasible to combine ctDNA mutational analysis with ctDNA methylation- [Citation150] or protein analysis [Citation9]. Another benefit of multi-analyte analysis is that the cancer entity is easier to determine.
4.1.2. Diagnostics
Today, tissue biopsies are typically used in diagnostics of solid cancers. As tumors are both phenotypically and genetically heterogeneous, it is possible that collected specimens fail to include important subclones essential for comprehensive clinical tumor characterization [Citation151–153]. In addition, some neoplasms are due to their localization or high risk of tumor dissemination not surgically sampled [Citation154]. Instead, diagnosis, treatment monitoring and relapse detection rely on various imaging technologies, lacking in both sensitivity [Citation155] and specificity [Citation156].
Analysis of ctDNA may be used as a complement to tissue biopsies to identify druggable mutations and help stratify patients, reducing over- and undertreatment. Genetic profiling of body fluids, unlike that of tissue biopsies, may also reveal mutations from all locations, i.e. metastases [Citation157–159]. Depending on tumor accessibility and patient health status, liquid biopsy may sometimes even be the only option for initial tumor genomic profiling [Citation160]. For some cancer diagnoses PCR-based methods are enough, whereas others require targeted gene panels, or even WGS, to enable comprehensive diagnosis and identification of potential treatment targets. Whole genome analysis is suited for applications where cancer is already confirmed and needs a broader characterization. The fraction of tumor-derived DNA in liquid biopsies is minute in comparison to tissue samples. However, at diagnosis the ctDNA fraction is often high, allowing standard PCR-based or non-ultrasensitive sequencing approaches to be used. For instance, in patients with non-small cell lung cancer where tissue sampling is not always possible, the quantitative PCR-based cobas EGFR Mutation Test v2 from Roche Diagnostics is available [Citation161]. This was the first ctDNA-based companion diagnostic test to be approved by FDA and is clinically used today to help guide the use of EGFR tyrosine kinase inhibitors. Recently, Guardant360 CDx from Guardant Health using a hybridization-based capture technology became an FDA approved liquid biopsy companion diagnostic that uses sequencing to assess EGFR status in non-small cell lung cancer [Citation162]. Besides EGFR, the Guardant360 assay covers alterations in more than 70 genes, which opens up for detection of more treatment-predictive mutations. Foundation Medicine’s FoundationOne Liquid CDx assay, targeting over 300 genes is another hybridization-based capture approach that is approved as a companion diagnostic test to identify prostate cancer and non-small lung cancer patients that may benefit from PARP inhibition treatment and tyrosine kinase inhibitor therapy, respectively [Citation163].
In addition to specific mutations, the tumor mutational burden is also an emerging biomarker in cancer management. For example, tumor mutational burden is used as a predictive biomarker in immunotherapy where high tumor mutational burden is associated with increased response rate [Citation164], such as to the tumor-agnostic programmed death 1 (PD-1) protein inhibitor pembrolizumab [Citation165]. As a surrogate to tissue-based assessments, it is possible to measure tumor mutational burden through ctDNA analysis [Citation166]. However, blood-based tumor mutational burden assessments require approaches that target larger sequence regions with representative number of mutations [Citation167] and is dependent on sufficient ctDNA levels [Citation8,Citation166].
4.1.3. Monitoring of treatment efficacy
The use of ctDNA analysis has the potential to complement or sometimes even replace imaging during monitoring of treatment efficacy. In cases with high tumor load, standard sequencing, such as WGS and whole exome sequencing, have been used for longitudinal assessment of treatment efficacy. However, reliable treatment monitoring requires ultrasensitive technologies that are able to detect individual ctDNA molecules. Thus, there are mainly two ultrasensitive approaches for ctDNA analysis: (i) generic cancer panels or (ii) patient-specific solutions. The main benefit of broad generic panels is that they always include the same targets with no additional assay development. The major limitation is that some tumors only have few or no hotspot mutations interrogated by these panels. Furthermore, cost may be an issue if each patient is sampled continuously over a long time period. An alternative is to use patient-specific solutions, targeting only patient-specific mutations and sequences related to treatment resistance. The foremost benefit of this strategy is that only sequences of interests are analyzed, simplifying analysis and reducing cost per sample. The drawbacks are the time and costs associated with developing patient-specific panels. Furthermore, patient-specific panels will not identify any mutations that may occur during treatment that are not directly related to known resistance mechanisms. If individual mutations are monitored, methods like digital PCR are often preferable over targeted sequencing, due to cost and simplicity. However, when multiple mutations and sequences are monitored, ultrasensitive targeted sequencing is preferred. Even if multiplexing is feasible with digital PCR, the benefits of sequencing increase with increasing number of targets.
To generate patient-specific assays relevant mutations first need to be identified. These mutations can be identified by WGS, whole exome sequencing, or targeted hotspot panels using tumor tissue biopsies. An alternative is to use liquid biopsies as described above. One disadvantage of applying smaller sequencing panels is that the number of identified mutations may be relatively low, resulting in small patient-specific panels with reduced sensitivity. The number of assays to be included in panels is a tradeoff between increased probability of cancer detection and the efforts in assay development [Citation58]. Here, simple and flexible sequencing methods, such as SiMSen-Seq [Citation168,Citation169], allowing rapid design and validation of individual assays are preferred. Though the initial effort may be deterring, the time to generate patient-specific panels will decrease with increasing number of assays that are developed, since many mutations are recurrent.
4.1.4. Detection of minimal residual disease and relapse
Following tumor resection, liquid biopsy analysis can be used to identify patients at high risk of recurrence and help guide adjuvant treatment decisions [Citation3,Citation170]. Several studies have demonstrated that ctDNA may predict relapse months or even years before regular clinical follow-ups [Citation171,Citation172]. Though conceptually different from monitoring of treatment efficacy, similar analytical possibilities and limitations apply in the detection of minimal residual disease and relapse.
5. Conclusions
The possibility to detect and diagnose cancer, monitor treatment efficacy, and detect relapse through analysis of liquid biopsies has long been expected to overcome many of the current challenges within cancer management. Furthermore, simple, minimally invasive, less traumatic and decentralized sampling will permit higher numbers of patients to access advanced health care and precision medicine. Assessment of ctDNA has received considerable attention during the last decade with the emergence of new and ultrasensitive technologies that facilitate single molecule detection. Alongside the development of more sensitive tools for ctDNA analysis, the preanalytical aspects of ctDNA analysis have also evolved. In recent years, there have been numerous studies demonstrating the potential clinical use of ctDNA analysis in all areas of cancer management. Several reliable analytical approaches and workflows exist, but no single strategy is optimal for all applications, complicating their use.
6. Expert opinion
The field of ctDNA analysis is rapidly evolving, but a substantial part of all reports are still performed as proof-of-principle studies examining the feasibility of ctDNA analysis in various body fluids and/or for specific patient cohorts. Overall, most reported data are encouraging, demonstrating that ctDNA can reliably be detected in several areas of cancer management, providing novel information. However, there is still a limited number of reports demonstrating true clinical utility of ctDNA analysis.
Despite technological advances, most applications are limited by DNA yield and inability to analyze replicates of clinical samples. To fully realize the potential of ctDNA analysis, we also need to improve our biological understanding of both ctDNA release and its clearance as well as their dependency on cancer type, relation to clinical parameters and choice of treatment. Thus far, most ctDNA studies have assessed the presence of single somatic mutations. Lately, simultaneous analysis of multiple mutations using large generic- or patient-specific panels has become more frequently applied and we expect this development to continue, increasing the likelihood to detect the presence of tumors in liquid biopsies. Another strategy that is emerging is analysis of additional biomarkers alongside ctDNA, including DNA-fragmentation and methylation patterns, but also of other analytes, such as proteins, RNAs, circulating tumor cells, extracellular vesicles, immune cell repertoires, as well as imaging. Multi-omics approaches do not only increase the sensitivity but also specificity, allowing for direct identification of specific tumor type, which is essential in screening efforts in otherwise healthy individuals. This was recently demonstrated in a prospective study, where women with no prior history of cancer were tested for multiple cancers. If a woman’s blood sample was positive, she subsequently underwent positron emission tomography-computed tomography imaging to both confirm and localize site and extent of cancer if present [Citation10]. Further technology development will likely also allow for simultaneous detection of multiple analytes in the same reaction tube, without dividing samples into parallel workflows, which today is the common strategy.
Another obstacle that limits a more widespread clinical implementation of ctDNA analysis is the lack of consensus and standardization in the field. Today, numerous experimental workflows exist, especially on the preanalytical side. This includes a large variety in sample collection tubes and extraction protocols used, but also in how experimental steps are performed and how data are reported, rendering comparisons difficult [Citation126]. An additional challenge is the number of possible ctDNA applications in cancer management, each with different technical requirements. For example, while the use of generic cancer screening platforms is practical and allows the detection of hotspot mutations in common cancer types, the same approach is less suited for cancer types with mainly private mutations and may become too costly for extensive testing during treatment monitoring and relapse detection. For longitudinal studies, the use of patient-specific panels is instead a more suitable approach. We expect that both generic and patient-specific platforms will continue to be developed but for different applications.
Article highlights
There are numerous studies in the field of circulating tumor DNA analysis that demonstrate its clinical potential in precision medicine.
Cell-free DNA is highly fragmented and present at low concentrations. Suitable technologies need to handle these parameters and enable detection of individual circulating tumor DNA molecules.
Accurate circulating tumor DNA analysis remains technically challenging. Hence, the entire experimental workflow: from sampling, via preanalytical steps and circulating tumor DNA analysis, to clinical data interpretation, needs to be optimized and standardized.
There are several applications in cancer management that use circulating tumor DNA analysis, including screening, primary diagnostics, prognostics, monitoring of treatment efficacy, as well as early detection of minimal residual disease and relapse.
Each cancer application has its own technical and clinical constraints, resulting in that no single and generic method exists.
We expect that generic as well as patient-specific approaches to analyze circulating tumor DNA will continue to be developed, most likely in combination with analyses of other analytes and independent readouts, such as imaging. In addition, we also predict further improvements and standardization of the preanalytical steps.
Declaration of interest
M Kubista declares stock ownership in TATAA Biocenter. M Kubista is also a board member of TATAA Biocenter and SiMSen Diagnostics. A Ståhlberg declares stock ownership in TATAA Biocenter, Iscaff Pharma and SiMSen Diagnostics. A Ståhlberg is also a board member of Iscaff Pharma and SiMSen Diagnostics. A Ståhlberg is inventor of SiMSen-Seq (United States Patent No. 10,557,134) and has a pending patent application (Swedish patent application No. 2,050,673-9). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Thierry AR, El Messaoudi S, Gahan PB, et al. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35(3):347–376.
- Crowley E, Di Nicolantonio F, Loupakis F, et al. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013 Aug;10(8):472–484.
- Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8(346):346ra92.
- Reinert T, Henriksen TV, Christensen E, et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019;5(8):1124–1131.
- Wang Y, Li L, Cohen JD, et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019;5(8):1118–1123.
- Wan JCM, Heider K, Gale D, et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci Transl Med. 2020;12(548):eaaz8084.
- Mok T, Wu YL, Lee JS, et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin Cancer Res. 2015;21(14):3196–3203.
- Zill OA, Banks KC, Fairclough SR, et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin Cancer Res. 2018;24(15):3528–3538.
- Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–930.
- Lennon AM, Buchanan AH, Kinde I, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science. 2020;369(6499):eabb9601.
- Chen X, Gole J, Gore A, et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun. 2020;11(1):3475.
- Cavallone L, Aguilar-Mahecha A, Lafleur J, et al. Prognostic and predictive value of circulating tumor DNA during neoadjuvant chemotherapy for triple negative breast cancer. Sci Rep. 2020;10(1):14704.
- Pietrasz D, Pecuchet N, Garlan F, et al. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clin Cancer Res. 2017;23(1):116–123.
- Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;611:112–123.
- Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563(7732):579–583.
- Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–389.
- Bronkhorst AJ, Ungerer V, Holdenrieder S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol Detect Quantif. 2019;17:100087.
- Andersson D, Kubista M, Stahlberg A. Liquid biopsy analysis in cancer diagnostics. Mol Aspects Med. 2020;72:100839.
- Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2(61):61ra91.
- Zheng YW, Chan KC, Sun H, et al. Nonhematopoietically derived DNA is shorter than hematopoietically derived DNA in plasma: a transplantation model. Clin Chem. 2012;58(3):549–558.
- Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–238.
- Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61(4):1659–1665.
- Ivanov M, Baranova A, Butler T, et al. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics. 2015;16(Suppl 13):S1.
- Swinson DEB, Jones JL, Richardson D, et al. Tumour necrosis is an independent prognostic marker in non-small cell lung cancer: correlation with biological variables. Lung Cancer. 2002 Sep;37(3):235–240.
- Langner C, Hutterer G, Chromecki T, et al. Tumor necrosis as prognostic indicator in transitional cell carcinoma of the upper urinary tract. J Urol. 2006;176(3):910–913.
- Hiraoka N, Ino Y, Sekine S, et al. Tumour necrosis is a postoperative prognostic marker for pancreatic cancer patients with a high interobserver reproducibility in histological evaluation. Br J Cancer. 2010;103(7):1057–1065.
- Bronkhorst AJ, Wentzel JF, Aucamp J, et al. Characterization of the cell-free DNA released by cultured cancer cells. Biochim Biophys Acta. 2016;1863(1):157–165.
- Bronkhorst AJ, Wentzel JF, Ungerer V, et al. Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumour Biol. 2018;40(9):1010428318801190.
- Stewart CM, Tsui DWY. Circulating cell-free DNA for non-invasive cancer management. Cancer Genet. 2018;228-229:169–179.
- Tamkovich SN, Cherepanova AV, Kolesnikova EV, et al. Circulating DNA and DNase activity in human blood. Ann N Y Acad Sci. 2006;1075:191–196.
- Cherepanova AV, Tamkovich SN, Bryzgunova OE, et al. Deoxyribonuclease activity and circulating DNA concentration in blood plasma of patients with prostate tumors. Ann Ny Acad Sci. 2008;1137:218–221.
- Yu SC, Lee SW, Jiang P, et al. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin Chem. 2013;59(8):1228–1237.
- Reckamp KL, Melnikova VO, Karlovich C, et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J Thorac Oncol. 2016;11(10):1690–1700.
- Chused TM, Steinberg AD, Talal N. The clearance and localization of nucleic acids by New Zealand and normal mice. Clin Exp Immunol. 1972 Dec;12(4):465–476.
- Minchin RF, Carpenter D, Orr RJ. Polyinosinic acid and polycationic liposomes attenuate the hepatic clearance of circulating plasmid DNA. J Pharmacol Exp Ther. 2001;296(3):1006–1012.
- Lau TW, Leung TN, Chan LY, et al. Fetal DNA clearance from maternal plasma is impaired in preeclampsia. Clin Chem. 2002 Dec;48(12):2141–2146.
- Nelson M, Eagle C, Langshaw M, et al. Genotyping fetal DNA by non-invasive means: extraction from maternal plasma. Vox Sang. 2001 Feb;80(2):112–116.
- Larzabal L, El-Nikhely N, Redrado M, et al. Differential effects of drugs targeting cancer stem cell (CSC) and non-CSC populations on lung primary tumors and metastasis. PLoS One. 2013;8(11):e79798.
- Uchida J, Imamura F, Kukita Y, et al. Dynamics of circulating tumor DNA represented by the activating and resistant mutations in epidermal growth factor receptor tyrosine kinase inhibitor treatment. Cancer Sci. 2016;107(3):353–358.
- Hur W, Yoon SK. Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells. Int J Mol Sci. 2017;18(12):2749.
- Andersson D, Fagman H, Dalin MG, et al. Circulating cell-free tumor DNA analysis in pediatric cancers. Mol Aspects Med. 2020;72:100819.
- Mead R, Duku M, Bhandari P, et al. Circulating tumour markers can define patients with normal colons, benign polyps, and cancers. Br J Cancer. 2011;105(2):239–245.
- Mouliere F, El Messaoudi S, Pang DL, et al. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol Oncol. 2014;8(5):927–941.
- Haller N, Helmig S, Taenny P, et al. Circulating, cell-free DNA as a marker for exercise load in intermittent sports. PLoS One. 2018;13(1):e0191915.
- Piovesan A, Pelleri MC, Antonaros F, et al. On the length, weight and GC content of the human genome. BMC Res Notes. 2019;12(1):106.
- Moss J, Magenheim J, Neiman D, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9(1):5068.
- Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
- Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–437.
- Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):552–558.
- Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446–451.
- Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102(45):16368–16373.
- Xu RH, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16(11):1155.
- Garcia-Saenz JA, Ayllon P, Laig M, et al. Tumor burden monitoring using cell-free tumor DNA could be limited by tumor heterogeneity in advanced breast cancer and should be evaluated together with radiographic imaging. BMC Cancer. 2017;17(1):210.
- Nygaard AD, Holdgaard PC, Spindler KL, et al. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br J Cancer. 2014;110(2):363–368.
- Atamaniuk J, Vidotto C, Tschan H, et al. Increased concentrations of cell-free plasma DNA after exhaustive exercise. Clin Chem. 2004;50(9):1668–1670.
- Haghiac M, Vora NL, Basu S, et al. Increased death of adipose cells, a path to release cell-free DNA into systemic circulation of obese women. Obesity (Silver Spring). 2012;20(11):2213–2219.
- Paunel-Gorgulu A, Wacker M, El Aita M, et al. cfDNA correlates with endothelial damage after cardiac surgery with prolonged cardiopulmonary bypass and amplifies NETosis in an intracellular TLR9-independent manner. Sci Rep. 2017;7(1):17421.
- Johansson G, Andersson D, Filges S, et al. Considerations and quality controls when analyzing cell-free tumor DNA. Biomol Detect Quantif. 2019;17:100078.
- Keller L, Belloum Y, Wikman H, et al. Clinical relevance of blood-based ctDNA analysis: mutation detection and beyond. Br J Cancer. 2021;124(2):345–358.
- Ponti G, Maccaferri M, Manfredini M, et al. The value of fluorimetry (Qubit) and spectrophotometry (NanoDrop) in the quantification of cell-free DNA (cfDNA) in malignant melanoma and prostate cancer patients. Clin Chim Acta. 2018;479:14–19.
- Kumar M, Choudhury Y, Ghosh SK, et al. Application and optimization of minimally invasive cell-free DNA techniques in oncogenomics. Tumour Biol. 2018;40(2):1010428318760342.
- Alidousty C, Brandes D, Heydt C, et al. Comparison of Blood Collection Tubes from Three Different Manufacturers for the Collection of Cell-Free DNA for Liquid Biopsy Mutation Testing. J Mol Diagn. 2017;19(5):801–804.
- Barrett AN, Thadani HA, Laureano-Asibal C, et al. Stability of cell-free DNA from maternal plasma isolated following a single centrifugation step. Prenat Diagn. 2014;34(13):1283–1288.
- Bartak BK, Kalmar A, Galamb O, et al. Blood Collection and Cell-Free DNA Isolation Methods Influence the Sensitivity of Liquid Biopsy Analysis for Colorectal Cancer Detection. Pathol Oncol Res. 2019;25(3):915–923.
- Board RE, Williams VS, Knight L, et al. Isolation and extraction of circulating tumor DNA from patients with small cell lung cancer. Ann N Y Acad Sci. 2008;1137:98–107.
- Breitbach S, Tug S, Helmig S, et al. Direct quantification of cell-free, circulating DNA from unpurified plasma. PLoS One. 2014;9(3):e87838.
- Denis MG, Knol AC, Theoleyre S, et al. Efficient Detection of BRAF Mutation in Plasma of Patients after Long-term Storage of Blood in Cell-Free DNA Blood Collection Tubes. Clin Chem. 2015;61(6):886–888.
- Fleischhacker M, Schmidt B, Weickmann S, et al. Methods for isolation of cell-free plasma DNA strongly affect DNA yield. Clin Chim Acta. 2011;412(23–24):2085–2088.
- Henao Diaz E, Yachnin J, Gronberg H, et al. The In Vitro Stability of Circulating Tumour DNA. PLoS One. 2016;11(12):e0168153.
- Hidestrand M, Stokowski R, Song K, et al. Influence of temperature during transportation on cell-free DNA analysis. Fetal Diagn Ther. 2012;31(2):122–128.
- Kim K, Shin DG, Park MK, et al. Circulating cell-free DNA as a promising biomarker in patients with gastric cancer: diagnostic validity and significant reduction of cfDNA after surgical resection. Ann Surg Treat Res. 2014;86(3):136–142.
- Kloten V, Ruchel N, Bruchle NO, et al. Liquid biopsy in colon cancer: comparison of different circulating DNA extraction systems following absolute quantification of KRAS mutations using Intplex allele-specific PCR. Oncotarget. 2017;8(49):86253–86263.
- Mazurek AM, Fiszer-Kierzkowska A, Rutkowski T, et al. Optimization of circulating cell-free DNA recovery for KRAS mutation and HPV detection in plasma. Cancer Biomark. 2013;13(5):385–394.
- Medina Diaz I, Nocon A, Mehnert DH, et al. Performance of Streck cfDNA Blood Collection Tubes for Liquid Biopsy Testing. PLoS One. 2016;11(11):e0166354.
- Mehrotra M, Singh RR, Chen W, et al. Study of Preanalytic and Analytic Variables for Clinical Next-Generation Sequencing of Circulating Cell-Free Nucleic Acid. J Mol Diagn. 2017;19(4):514–524.
- Page K, Guttery DS, Zahra N, et al. Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS One. 2013;8(10):e77963.
- Perez-Barrios C, Nieto-Alcolado I, Torrente M, et al. Comparison of methods for circulating cell-free DNA isolation using blood from cancer patients: impact on biomarker testing. Transl Lung Cancer Res. 2016;5(6):665–672.
- Rothwell DG, Smith N, Morris D, et al. Genetic profiling of tumours using both circulating free DNA and circulating tumour cells isolated from the same preserved whole blood sample. Mol Oncol. 2016;10(4):566–574.
- Schmidt B, Reinicke D, Reindl I, et al. Liquid biopsy - Performance of the PAXgene(R) Blood ccfDNA Tubes for the isolation and characterization of cell-free plasma DNA from tumor patients. Clin Chim Acta. 2017;469:94–98.
- Sherwood JL, Corcoran C, Brown H, et al. Optimised Pre-Analytical Methods Improve KRAS Mutation Detection in Circulating Tumour DNA (ctDNA) from Patients with Non-Small Cell Lung Cancer (NSCLC). PLoS One. 2016;11(2):e0150197.
- Swinkels DW, Wiegerinck E, Steegers EA, et al. Effects of blood-processing protocols on cell-free DNA quantification in plasma. Clin Chem. 2003;49(3):525–526.
- Toro PV, Erlanger B, Beaver JA, et al. Comparison of cell stabilizing blood collection tubes for circulating plasma tumor DNA. Clin Biochem. 2015;48(15):993–998.
- van Dessel LF, Beije N, Helmijr JC, et al. Application of circulating tumor DNA in prospective clinical oncology trials - standardization of preanalytical conditions. Mol Oncol. 2017;11(3):295–304.
- van Ginkel JH, van den Broek DA, van Kuik J, et al. Preanalytical blood sample workup for cell-free DNA analysis using Droplet Digital PCR for future molecular cancer diagnostics. Cancer Med. 2017;6(10):2297–2307.
- Warton K, Yuwono NL, Cowley MJ, et al. Evaluation of Streck BCT and PAXgene Stabilised Blood Collection Tubes for Cell-Free Circulating DNA Studies in Plasma. Mol Diagn Ther. 2017;21(5):563–570.
- Wolf A, Beller K, Groemminger S, et al. Purification of Circulating Cell-Free DNA from Plasma and Urine Using the Automated Large-Volume Extraction on the QIAsymphony(R) SP Instrument. Adv Exp Med Biol. 2016;924:179–185.
- Zhao Y, Li Y, Chen P, et al. Performance comparison of blood collection tubes as liquid biopsy storage system for minimizing cfDNA contamination from genomic DNA. J Clin Lab Anal. 2019;33(2):e22670.
- Parpart-Li S, Bartlett B, Popoli M, et al. The Effect of Preservative and Temperature on the Analysis of Circulating Tumor DNA. Clin Cancer Res. 2017;23(10):2471–2477.
- Trigg RM, Martinson LJ, Parpart-Li S, et al. Factors that influence quality and yield of circulating-free DNA: a systematic review of the methodology literature. Heliyon. 2018;4(7):e00699.
- Gerber T, Taschner-Mandl S, Saloberger-Sindhoringer L, et al. Assessment of Pre-Analytical Sample Handling Conditions for Comprehensive Liquid Biopsy Analysis. J Mol Diagn. 2020;22(8):1070–1086.
- Barra GB, Santa Rita TH, de Almeida Vasques J, et al. EDTA-mediated inhibition of DNases protects circulating cell-free DNA from ex vivo degradation in blood samples. Clin Biochem. 2015;48(15):976–981.
- Risberg B, Tsui DWY, Biggs H, et al. Effects of Collection and Processing Procedures on Plasma Circulating Cell-Free DNA from Cancer Patients. J Mol Diagn. 2018;20(6):883–892.
- Lam NY, Rainer TH, Chiu RW, et al. EDTA is a better anticoagulant than heparin or citrate for delayed blood processing for plasma DNA analysis. Clin Chem. 2004;50(1):256–257.
- Helgadottir H, Olafsson I, Andersen K, et al. Stability of thromboxane in blood samples. Vasc Health Risk Manag. 2019;15:143–147.
- Keowmaneechai E, McClements DJ. Influence of EDTA and citrate on physicochemical properties of whey protein-stabilized oil-in-water emulsions containing CaCl2. J Agric Food Chem. 2002;50(24):7145–7153.
- Beutler E, Gelbart T, Kuhl W. Interference of heparin with the polymerase chain reaction. Biotechniques. 1990;9(2):166.
- Ungerer V, Bronkhorst AJ, Holdenrieder S. Preanalytical variables that affect the outcome of cell-free DNA measurements. Crit Rev Clin Lab Sci. 2020;57(7):484–507.
- Nikolaev S, Lemmens L, Koessler T, et al. Circulating tumoral DNA: preanalytical validation and quality control in a diagnostic laboratory. Anal Biochem. 2018;542:34–39.
- Sozzi G, Roz L, Conte D, et al. Effects of prolonged storage of whole plasma or isolated plasma DNA on the results of circulating DNA quantification assays. J Natl Cancer Inst. 2005;97(24):1848–1850.
- Sato A, Nakashima C, Abe T, et al. Investigation of appropriate pre-analytical procedure for circulating free DNA from liquid biopsy. Oncotarget. 2018;9(61):31904–31914.
- Norton SE, Lechner JM, Williams T, et al. A stabilizing reagent prevents cell-free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR. Clin Biochem. 2013;46(15):1561–1565.
- Ward Gahlawat A, Lenhardt J, Witte T, et al. Evaluation of Storage Tubes for Combined Analysis of Circulating Nucleic Acids in Liquid Biopsies. Int J Mol Sci. 2019;20(3):704.
- Wong D, Moturi S, Angkachatchai V, et al. Optimizing blood collection, transport and storage conditions for cell free DNA increases access to prenatal testing. Clin Biochem. 2013;46(12):1099–1104.
- Norton SE, Luna KK, Lechner JM, et al. A new blood collection device minimizes cellular DNA release during sample storage and shipping when compared to a standard device. J Clin Lab Anal. 2013;27(4):305–311.
- Sorber L, Zwaenepoel K, Jacobs J, et al. Circulating Cell-Free DNA and RNA Analysis as Liquid Biopsy: optimal Centrifugation Protocol. Cancers (Basel). 2019;11(4):458.
- Raymond CK, Hernandez J, Karr R, et al. Collection of cell-free DNA for genomic analysis of solid tumors in a clinical laboratory setting. PLoS One. 2017;12(4):e0176241.
- Chan KC, Yeung SW, Lui WB, et al. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem. 2005;51(4):781–784.
- Diefenbach RJ, Lee JH, Kefford RF, et al. Evaluation of commercial kits for purification of circulating free DNA. Cancer Genet. 2018;228-229:21–27.
- Devonshire AS, Whale AS, Gutteridge A, et al. Towards standardisation of cell-free DNA measurement in plasma: controls for extraction efficiency, fragment size bias and quantification. Anal Bioanal Chem. 2014;406(26):6499–6512.
- Markus H, Contente-Cuomo T, Farooq M, et al. Evaluation of pre-analytical factors affecting plasma DNA analysis. Sci Rep. 2018;8(1):7375.
- Sorber L, Zwaenepoel K, Deschoolmeester V, et al. A Comparison of Cell-Free DNA Isolation Kits: isolation and Quantification of Cell-Free DNA in Plasma. J Mol Diagn. 2017;19(1):162–168.
- Warton K, Graham LJ, Yuwono N, et al. Comparison of 4 commercial kits for the extraction of circulating DNA from plasma. Cancer Genet. 2018;228-229:143–150.
- Gyanchandani R, Kvam E, Heller R, et al. Whole genome amplification of cell-free DNA enables detection of circulating tumor DNA mutations from fingerstick capillary blood. Sci Rep. 2018;8(1):17313.
- Mauger F, Dulary C, Daviaud C, et al. Comprehensive evaluation of methods to isolate, quantify, and characterize circulating cell-free DNA from small volumes of plasma. Anal Bioanal Chem. 2015;407(22):6873–6878.
- Rykova EY, Laktionov PP, Skvortsova TE, et al. Extracellular DNA in breast cancer: cell-surface-bound, tumor-derived extracellular DNA in blood of patients with breast cancer and nonmalignant tumors. Ann N Y Acad Sci. 2004;1022:217–220.
- Skvortsova TE, Rykova EY, Tamkovich SN, et al. Cell-free and cell-bound circulating DNA in breast tumours: DNA quantification and analysis of tumour-related gene methylation. Br J Cancer. 2006;94(10):1492–1495.
- Ordonez E, Rueda L, Canadas MP, et al. Evaluation of sample stability and automated DNA extraction for fetal sex determination using cell-free fetal DNA in maternal plasma. Biomed Res Int. 2013;2013:195363.
- Stroun M, Anker P, Maurice P, et al. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology. 1989;46(5):318–322.
- Mandel P, Metais P. Les acides du plasma sanguin chez l'homme. C R Seances Soc Biol Fil. 1948 Feb;142(3–4):241–243.
- Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014 Dec 25;371(26):2488–2498.
- Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478.
- Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487.
- Salk JJ, Loubet-Senear K, Maritschnegg E, et al. Ultra-Sensitive TP53 Sequencing for Cancer Detection Reveals Progressive Clonal Selection in Normal Tissue over a Century of Human Lifespan. Cell Rep. 2019;28(1):132–144e3.
- Yizhak K, Aguet F, Kim J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364(6444):eaaw0726.
- Alexandrov LB, Jones PH, Wedge DC, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47(12):1402–1407.
- Elazezy M, Joosse SA. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput Struct Biotechnol J. 2018;16:370–378.
- Fox EJ, Reid-Bayliss KS, Emond MJ, et al. Accuracy of Next Generation Sequencing Platforms. Next Gener Seq Appl. 2014;1:1000106.
- Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96(16):9236–9241.
- Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–8610.
- Li J, Wang L, Mamon H, et al. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14(5):579–584.
- Belloum Y, Janning M, Mohme M, et al. Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay. Cells. 2020;9(11):2337.
- Huang CC, Du M, Wang L. Bioinformatics Analysis for Circulating Cell-Free DNA in Cancer. Cancers (Basel). 2019;11(6):805.
- Filges S, Yamada E, Stahlberg A, et al. Impact of Polymerase Fidelity on Background Error Rates in Next-Generation Sequencing with Unique Molecular Identifiers/Barcodes. Sci Rep. 2019;9(1):3503.
- Johansson G, Kaltak M, Rimniceanu C, et al. Ultrasensitive DNA Immune Repertoire Sequencing Using Unique Molecular Identifiers. Clin Chem. 2020;66(9):1228–1237.
- Kinde I, Wu J, Papadopoulos N, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(23):9530–9535.
- Jabara CB, Jones CD, Roach J, et al. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A. 2011;108(50):20166–20171.
- Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10(466):eaat4921.
- Scimia M, Du J, Pepe F, et al. Evaluation of a novel liquid biopsy-based ColoScape assay for mutational analysis of colorectal neoplasia and triage of FIT+ patients: a pilot study. J Clin Pathol. 2018;71(12):1123–1126.
- Gan Q, Cui Z, Tang S, et al. A novel XNA-based NGS panel for cancer diagnostics. J Clin Oncol. 2020;38(15_suppl):e16142.
- Chan HT, Nagayama S, Chin YM, et al. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol Oncol. 2020;14(8):1719–1730.
- Hu Y, Ulrich BC, Supplee J, et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res. 2018;24(18):4437–4443.
- Barbany G, Arthur C, Lieden A, et al. Cell-free tumour DNA testing for early detection of cancer - a potential future tool. J Intern Med. 2019;286(2):118–136.
- Fiala C, Diamandis EP. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018;16(1):166.
- Bronkhorst AJ, Ungerer V, Holdenrieder S. Early detection of cancer using circulating tumor DNA: biological, physiological and analytical considerations. Crit Rev Clin Lab Sci. 2019:1–17.
- Siravegna G, Mussolin B, Venesio T, et al. How liquid biopsies can change clinical practice in oncology. Ann Oncol. 2019;30(10):1580–1590.
- Caramori G, Ruggeri P, Mumby S, et al. Molecular links between COPD and lung cancer: new targets for drug discovery? Expert Opin Ther Targets. 2019;23(6):539–553.
- Weaver JMJ, Ross-Innes CS, Shannon N, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46(8):837–843.
- Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: the role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol. 2016;22(20):4794–4801.
- De Rubis G, Rajeev Krishnan S, Bebawy M. Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol Sci. 2019;40(3):172–186.
- Xia S, Ye J, Chen Y, et al. Parallel serial assessment of somatic mutation and methylation profile from circulating tumor DNA predicts treatment response and impending disease progression in osimertinib-treated lung adenocarcinoma patients. Transl Lung Cancer Res. 2019;8(6):1016–1028.
- Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892.
- Bedard PL, Hansen AR, Ratain MJ, et al. Tumour heterogeneity in the clinic. Nature. 2013;501(7467):355–364.
- Baslan T, Kendall J, Ward B, et al. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 2015;25(5):714–724.
- Berry JL, Xu L, Murphree AL, et al. Potential of Aqueous Humor as a Surrogate Tumor Biopsy for Retinoblastoma. JAMA Ophthalmol. 2017;135(11):1221–1230.
- Pantel K, Alix-Panabieres C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol. 2019;16(7):409–424.
- Lee JH, Long GV, Menzies AM, et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients With Metastatic Melanoma Treated With Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018;4(5):717–721.
- Murtaza M, Dawson SJ, Tsui DW, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–112.
- Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8(1):1324.
- Chicard M, Colmet-Daage L, Clement N, et al. Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin Cancer Res. 2018;24(4):939–949.
- Seoane J, De Mattos-Arruda L, Le Rhun E, et al. Cerebrospinal fluid cell-free tumour DNA as a liquid biopsy for primary brain tumours and central nervous system metastases. Ann Oncol. 2019;30(2):211–218.
- Jenkins S, Yang JC, Ramalingam SS, et al. Plasma ctDNA Analysis for Detection of the EGFR T790M Mutation in Patients with Advanced Non-Small Cell Lung Cancer. J Thorac Oncol. 2017;12(7):1061–1070.
- Guardant Health Guardant360 CDx First FDA-Approved Liquid Biopsy for Comprehensive Tumor Mutation Profiling Across All Solid Cancers [Internet]. Redwood City (CA): Guardant Health; [cited 2021 Jan 18]. Available from: https://investors.guardanthealth.com/press-releases/press-releases/2020/Guardant-Health-Guardant360-CDx-First-FDA-Approved-Liquid-Biopsy-for-Comprehensive-Tumor-Mutation-Profiling-Across-All-Solid-Cancers/default.aspx
- FDA Approves Foundation Medicine’s FoundationOne Liquid CDx, a Comprehensive Pan-Tumor Liquid Biopsy Test with Multiple Companion Diagnostic Indications for Patients with Advanced Cancer [Internet]. Cambridge (MA): Foundation Medicine; [cited 2021 Jan 18]. Available from: https://www.foundationmedicine.com/press-releases/445c1f9e-6cbb-488b-84ad-5f133612b721
- Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med. 2017;377(25):2500–2501.
- Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–1365.
- Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441–1448.
- Cescon DW, Bratman SV, Chan SM, et al. Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer. 2020;1(3):276–290.
- Ståhlberg A, Krzyzanowski PM, Egyud M, et al. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat Protoc. 2017;12(4):664–682.
- Ståhlberg A, Krzyzanowski PM, Jackson JB, et al. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res . 2016;44(11):e105.
- Tan L, Sandhu S, Lee RJ, et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann Oncol. 2019;30(5):804–814.
- Sausen M, Phallen J, Adleff V, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686.
- Olsson E, Winter C, George A, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol Med. 2015;7(8):1034–1047.