
1. Introduction
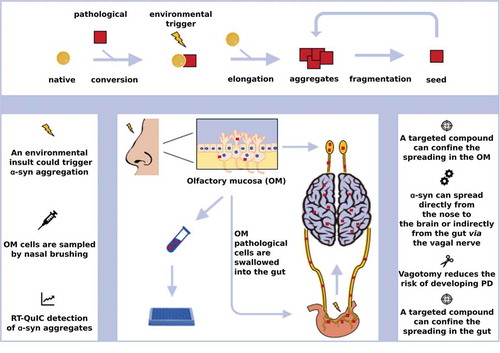
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the progressive accumulation of aberrant aggregates of α-synuclein, resulting in a clinical syndrome defined by motor and non-motor manifestations. Alpha-synuclein inclusions are known as Lewy-bodies (LBs) and Lewy neurites (LNs), whether they accumulate in neuronal perikarya or in neuronal processes, respectively [Citation1]. Braak seminal studies on the pattern of distribution of pathological α-synuclein in PD postmortem human brains suggested a stereotypical and disease-course related accumulation of α-synuclein in different areas of the central and also the enteric and autonomous nervous system [Citation2]. In 2008, two independent research teams identified classical α-synuclein pathology in the embryonic neurons which had been transplanted into the brains of PD patients as a potential stem cell treatment [Citation3,Citation4]. In the next year, those findings were provided a possible explanation with the evidence that α-synuclein aggregation demonstrates a capability of transmitting from neurons to neural stem cells in vitro and in vivo [Citation5]. In light of all these findings, it was suggested that α-synuclein could behave like a prion, being able to recruit its endogenous physiological counterpart and template onto it its aberrant conformation, leading, ultimately, to neurodegeneration [Citation6] (). This hypothesis was subsequently supported by several studies performed using in vitro produced recombinant α-synuclein aggregates, which efficiently induced pathology both in vitro and in vivo [Citation7,Citation8]. Notably, despite aggregated α-synuclein shares some features with prions, to date prion diseases are the only neurodegenerative disorders showing infectious transmissible protein species capable of recapitulating clinical disease [Citation9,Citation10]. No clear evidence exists to naturally occurring human-to-human transmission of other neurodegenerative conditions [Citation11,Citation12]. Similar to prions, α-synuclein aggregates may present in different conformers (i.e. strains) [Citation13,Citation14] with relevant pathological and clinical implications [Citation15]. The prion hypothesis for α-synuclein also accounts for Braak observations, which suggest that α-synuclein accumulation could begin either in the olfactory bulb or in the gastrointestinal tract and subsequently spread to the ventral midbrain, where it selectively affects dopamine neurons of the substantia nigra pars compacta (SNpc). According to the ‘dual hit’ hypothesis, proposed to explain PD pathological progression, an unknown pathogenic assault could reach the brain through two routes: the nose and the gut [Citation16]. These two compartments are, indeed, both in close and constant contact with the external environment, and so, with potential detrimental effectors. If the pathological process starts in the olfactory mucosa (OM) it can reach the brain directly through the olfactory system or retrogradely, via the enteric plexus and then the vagal nerve. In fact, affected olfactory cells containing α-synuclein aggregates can detach from OM and be subsequently swallowed within saliva and mucus reaching the stomach and then the enteric plexus (). Accordingly, clinical signs of an early involvement of gut and olfactory compartments, such as hyposmia and gastrointestinal dysfunction (e.g. reduced bowel movements) are frequently reported in PD early phases [Citation16]. This gut to brain spread has been recently reproduced in vivo with α-synuclein in vitro preformed fibrils (PFF) injected in the stomach and duodenum of wild-type mice [Citation17]. Interestingly, the absence of endogenous α-synuclein and truncal vagotomy completely prevented this spreading in the injected animals.
Figure 1. The prion-like replication process and α-synuclein spreading in PD. The upper panel describes the prion-like replication process. A misfolded protein (red square) interacts with its native isoform (yellow circle) and templates on it its aberrant conformation. This phenomenon can be triggered by environmental stressors facilitating the pathological conversion. The rescue of other molecules of the native protein leads to elongation and the formation of aggregates, which can be fragmented generating the so-called seed (i.e. the smallest amount of the pathological protein able to replicate the cycle). The lower image describes the ‘dual hit’ hypothesis for PD and some of its diagnostic and therapeutic implications.
2. Therapeutic implications
The prion concept leads to several direct and indirect consequences and implications. First, considering the spreading of the pathology, the starting point and an endpoint for α-synuclein deposition are postulated by the Braak hypothesis. Sites identified as the starting points of α-synuclein aggregation could be sampled and checked for the presence of aberrant α-synuclein deposition, thus, performing the earliest possible diagnosis before the disease has spread to the brain. Similar strategies have already been explored with promising results, exploiting the high detection power of new generation ultrasensitive prion amplification techniques, such as the Real Time Quaking Induced Conversion reaction (RT-QuIC) in the analysis of OM from patients with PD and other α-synucleinopathies [Citation18]. The association between non-motor symptoms and the risk of developing PD has been postulated by several large cohort prospective studies reporting that non-motor symptoms could precede motor manifestations of many years [Citation19]. Hyposmia might manifest within 4 years prior to PD development [Citation20,Citation21] while in the case of constipation this timeframe could be even longer, from 12 to 20 years before motor disease onset [Citation22,Citation23]. Once adapted to OM α-synuclein detection, RT-QuIC could help to select subjects with a high likelihood of developing PD and eventually to treat them in the earliest disease phase, sparing the pathological involvement of their central nervous system (CNS). Due to its anatomical connections, vagal nerve is likely to be the primary route of α-synuclein spreading from the enteric plexus to the CNS. This hypothesis is supported by the observation that subjects which underwent a truncal vagotomy displayed a lower risk of developing PD during their life span [Citation24] and by results of the aforementioned study conducted by Kim and colleagues, in which vagotomy prevented the spreading to CNS of α-synuclein fibrils injected in the stomach of the animal [Citation17]. These data suggest the intriguing possibility that, at least in some selected cases, the disease could be identified in its early phases and confined in the enteric system, blocking its spreading via the vagal nerve. The RT-QuIC serves not only as a diagnostic tool since it also provides a ‘clean’ environment for prion replication, suitable for high-throughput screening of a large number of molecules possibly interfering with the replication process. Masuda and colleagues screened a large number of compounds by means of a Thioflavin T-based assay, identifying several molecules able to interfere with in vitro α-synuclein fibrillization [Citation25]. Starting from the observation that dopamine (DA) may be a direct interactor with α-synuclein, Latawiec et al., performed α-synuclein in vitro fibrillization assays in the presence of five dopamine analogs which, indeed, affected the aggregation propensity of the protein [Citation26]. The prion-like properties of in vitro produced α-synuclein PFF were also instrumental for the development of cellular and animal models crucial to mimic and study the α-synuclein pathophysiological transmission and replication process. Such models are essential for the identification of pathways and molecular partners involved in the uptake and propagation of α-synuclein fibrils. The cellular prion protein, PrPC, is known to be a receptor for amyloid-β (Aβ) oligomers and, intriguingly, it showed also a complex interplay with α-synuclein fibrils [Citation27]. It was observed that a more consistent uptake of fibrillar forms of α-synuclein takes place in the immortalized murine neuroblastoma cell line (N2a) constitutively expressing PrPC compared to the same cell line ablated for PrPC, with the same effect in mouse primary cortical neurons [Citation28]. These findings have a direct impact on therapeutic approaches to PD, suggesting that antibodies can be used to target PrPC and thus reduce the uptake of α-synuclein fibrils and the associated neurodegenerative cascade. Another consequence deriving from the prion hypothesis is that, as it is for the prion protein, we now know that pathological α-synuclein should be considered as a population of aberrant conformers or strains with different properties and specific selectivity for the most vulnerable environment of replication (e.g. dopaminergic neurons, glial cells, cortical neurons). In fact, recently it has been elegantly demonstrated that α-synuclein pathological species originating from recombinant fibrils or derived from several types of synucleinopathy affected brains exhibited features reminiscent of prion strains, when injected in transgenic mice [Citation14]. In the near future scenario, clinicians should be able to identify a specific α-synuclein strain at a single patient level and so to administer the targeted treatment designed against that exact conformer. Also in this case, the RT-QuIC technique could be exploited as both an early diagnostic tool and as an instrument to amplify the specific conformer in order to better characterize its peculiar biochemical features. Recently, Shahnawaz and colleagues exploited the RT-QuIC amplification power to demonstrate that α-synuclein seeds present in the CSF of PD and Multiple System Atrophy (MSA) patients are representative of those that accumulate in diseased brains. Moreover, they showed that PD and MSA derived aggregates displayed distinct features that, intriguingly, were maintained after several rounds of RT-QuIC amplification [Citation29]. These results suggest that pathological α-synuclein exists in different strains, and confirm the great potential of the RT-QuIC technique for future applications in clinical trials and targeted medicine development. In fact, as for Alzheimer’s Disease, active and passive immunotherapy is a promising strategy to treat PD and evidence from preclinical trials in animal models suggests that immunizing against α-synuclein aggregates could be an effective and disease-modifying approach [Citation30,Citation31]. Since α-synuclein aggregates are the target of immunotherapy trials in PD, the identification of specific conformers (i.e. strains) is key to develop the most appropriate and efficient immune response.
3. Conclusions
The prion-like paradigm has emerged as a revolutionary hypothesis and a unifying pathological process for all neurodegenerative disorders [Citation32]. For what concerns PD, it is now recognized that there is a stereotypical spread of pathology which starts from sites particularly exposed to environmental insults, namely the gut and the OM, with a subsequent progression to the CNS via the vagal nerve. Moreover, we now possess a series of tools for exploring the prion-like phenomenon ranging from in vitro fibrillization assays, cellular models replicating the transmission of pathogenic α-synuclein oligomers and fibrils and animal models. Recently, in vivo models replicated two key aspects of α-synuclein pathological progression: the gut to brain spreading and the strain propagation phenomenon. Lastly, α-synuclein prion-like features are exploited to set up ultrasensitive diagnostic tools, such as the RT-QuIC technology, enabling early diagnosis and strain-specific characterization of the pathological entity involved in the disease.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345(1):27–32.
- Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
- Kordower JH, Chu Y, Hauser RA, et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506.
- Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503.
- Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015.
- Olanow CW, Prusiner SB. Is Parkinson’s disease a prion disorder? Proc Natl Acad Sci U S A. 2009;106:12571–12572.
- Luk KC, Song C, O’Brien P, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056.
- Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–1138.
- Moore RA, Taubner LM, Priola SA. Prion protein misfolding and disease. Curr Opin Struct Biol. 2009;19(1):14–22.
- Smethurst P, Sidle KC, Hardy J. Review: prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol Appl Neurobiol. 2015;41:578–597.
- Holmes BB, Diamond MI. Amyotrophic lateral sclerosis and organ donation: is there risk of disease transmission? Ann Neurol. 2012;72:832–836.
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–540.
- Peelaerts W, Bousset L, Van der Perren A, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–344.
- Lau A, So JC, Lau HH, et al. α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci. 2020;23(1):21–31. .
- Scialo C, De Cecco E, Manganotti P, et al. Prion and prion-like protein strains: deciphering the molecular basis of heterogeneity in neurodegeneration. Viruses. 2019;11(3):261.
- Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci. 2009;1170:615–622.
- Kim S, Kwon SH, Kam TI, et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103:627–641 e7.
- De Luca CMG, Elia AE, Portaleone SM, et al. Efficient RT-QuIC seeding activity for alpha-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl Neurodegener. 2019;8:24.
- Chen H, Burton EA, Ross GW, et al. Research on the premotor symptoms of Parkinson’s disease: clinical and etiological implications. Environ Health Perspect. 2013;121:1245–1252.
- Ross GW, Abbott RD, Petrovitch H, et al. Association of olfactory dysfunction with incidental Lewy bodies. Mov Disord. 2006;21:2062–2067.
- Ross GW, Petrovitch H, Abbott RD, et al. Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann Neurol. 2008;63:167–173.
- Abbott RD, Ross GW, Petrovitch H, et al. Bowel movement frequency in late-life and incidental Lewy bodies. Mov Disord. 2007;22:1581–1586.
- Gao X, Chen H, Schwarzschild MA, et al. A prospective study of bowel movement frequency and risk of Parkinson’s disease. Am J Epidemiol. 2011;174:546–551.
- Svensson E, Horvath-Puho E, Thomsen RW, et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78:522–529.
- Masuda M, Suzuki N, Taniguchi S, et al. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006;45:6085–6094.
- Latawiec D, Herrera F, Bek A, et al. Modulation of alpha-synuclein aggregation by dopamine analogs. PLoS One. 2010;5:e9234.
- De Cecco E, Legname G. The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion. 2018;12:23–27.
- Aulic S, Masperone L, Narkiewicz J, et al. Alpha-synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci Rep. 2017;7:10050.
- Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 2020;578:273–277.
- George S, Brundin P. Immunotherapy in Parkinson’s Disease: micromanaging alpha-synuclein aggregation. J Parkinsons Dis. 2015;5:413–424.
- Wang Z, Gao G, Duan C, et al. Progress of immunotherapy of anti-alpha-synuclein in Parkinson’s disease. Biomed Pharmacother. 2019;115:108843.
- Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci. 2015;38:87–103.