
ABSTRACT
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disease and the most common cause of dementia. It has a complex pathophysiology that is not yet completely understood, where multiple central, systemic, and environmental factors play a key role in disease progression. Understanding the multifactorial nature of AD is paramount to formulate new therapies.
Areas covered
The authors reviewed the role of the amyloid-β-binding, antioxidant, and immunomodulatory properties of albumin in AD and the use of therapeutic plasma exchange (PE) in neurology. The results from the Alzheimer Management By Albumin Replacement (AMBAR) trial that combined the use of PE with albumin replacement in patients with mild-to-moderate AD, are also analyzed.
Expert opinion
Findings from the AMBAR study provide encouraging results in the treatment of AD with PE and albumin replacement, especially in patients at the moderate stage of the disease, who showed less cognitive decline from baseline compared with placebo in most of the variables analyzed. Further research is warranted to ascertain the possible mechanisms of action underlying these results. Different cohorts of patients that may also benefit from this treatment, such as those with mild cognitive impairment or other types of dementia, could also be the target of additional studies.
1. Introduction: a multifactorial approach to Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disease and the most common cause of dementia. It affects more than 47 million people worldwide, and it is characterized by cognitive and functional deficits [Citation1]. The exact cause of AD is still poorly understood, and new knowledge is continuously emerging that proves the multifactorial nature of its pathophysiology [Citation2].
At a cellular level, the two major molecules involved in AD are amyloid-β peptide (Aβ), and tau protein. Aβ is a small peptide derived from proteolysis of the amyloid precursor protein [Citation1]. Among the different Aβ isoforms, Aβ1-42 is the most neurotoxic and prone to aggregation at a central level [Citation3]. Aβ self-aggregates into oligomers that can be soluble and affect synaptic function, or arrange into sheets to form the amyloid plaques that are the hallmark of AD [Citation1,Citation4]. Maintaining a balance between Aβ production and clearance is essential to preserve brain health as Aβ accumulation will lead to increased oligomer production and neurotoxicity [Citation3,Citation5].
In recent years, the role of inflammation in AD has gained relevance. Aβ oligomers interact with microglia as part of their regular elimination process; this triggers microglia activation and the secretion of proinflammatory cytokines and chemokines that recruit further microglia and astrocytes to the inflammatory site. Under normal circumstances, this process is well controlled; however, Aβ excess dysregulates this response and causes a disproportionate release of pro-inflammatory mediators that contribute to neurodegeneration [Citation6,Citation7].
The other key molecule implicated in AD pathophysiology is tau. In pathological conditions, tau may undergo post-translational modifications (PTMs) that lead to microtubule disassembly in axons, synaptic dysfunction, fibrillary tangle formation, and deterioration of neuronal function [Citation1,Citation8]. Aβ and tau interact in many neuronal compartments, with Aβ contributing to increased spreading of tau pathology [Citation1,Citation9,Citation10]. Aβ pathology is therefore predominant in the earlier stages of AD and it contributes to the progression of the tau-based neurofibrillary pathology that is associated with the clinical manifestations of the later stages of AD [Citation9].
The ε4 allele of apolipoprotein E (APOEε4) is also known to be a major risk factor of late-onset AD as it reduces clearance and exacerbates accumulation of Aβ [Citation7,Citation11]. However, the risk factor with the highest impact on neurodegeneration is, by far, the aging process itself. Cells with high levels of age-associated DNA damage, become senescent, stop proliferating, and acquire proinflammatory properties that contribute to AD progression [Citation12].
Systemic conditions such as diabetes, obesity, or metabolic syndrome can contribute to creating an inflammatory environment that promotes harmful effects on neuronal function [Citation5]; dyslipidemia, infections and sleep impairment may also prompt the advancement of underlying AD pathology [Citation13,Citation14]. Recent research has highlighted that the disruption of metal homeostasis causes neuronal dysfunction and death, playing a key role in the development and progression of AD [Citation15]. Other factors such as gut microbiota metabolites, plasma proteins, exercise-induced metabolites and peripheral immune cells directly cross or indirectly transmit signals across the blood-brain barrier (BBB) to modulate or interfere with brain function [Citation16]. Evidence also suggests that protective factors, such as cognitive reserve, may help to prevent or delay the onset of AD [Citation17].
The pathophysiological process of AD is thought to start many years, if not decades, before clinical disease onset. However, the natural history of asymptomatic biomarker positivity in the preclinical phase toward the subsequent appearance of clinical manifestations is yet to be better understood. Even though strong evidence supports the role of Aβ aggregation in initiating AD pathogenesis, to date, a number of Aβ-based therapies appear to be ineffective in modifying the course of the disease once it is already symptomatic. It is only now that anti-Aβ antibodies such as aducanumab, gantenerumab, lecanemab or donanemab are contributing to the understanding of the Aβ pathophysiology [Citation18–21]. Furthermore, the accumulation of Aβ might not be enough on its own to precipitate the clinical symptomatology of AD [Citation14,Citation17,Citation22].
2. Therapeutic plasma exchange in neurology
Therapeutic plasma exchange (TPE) is a procedure in which blood is passed through a device that separates and removes the plasma from the cellular components. The removed plasma is discarded, and replaced with either a colloid solution (like albumin) or a combination of crystalloid and colloid solutions [Citation23]. TPE has been used in neurology for many years, and not all the indications for which it is performed are mediated by autoantibodies. Some of them, such as multiple sclerosis, are mediated by neuroinflammation. Therefore, the aim of TPE is to remove damaging cytokines, chemokines and other damaging substances from the blood [Citation24,Citation25].
As previously discussed, together with Aβ and tau pathology, microglial activation and neuroinflammation play an important role in AD pathology. When activated, microglia release cytokines and neurotoxic agents that contribute to synaptic and neuronal damage [Citation26].
Microglial NLR family pyrin domain containing 3 (NLRP3) inflammasome activation has been extensively studied in acute and chronic central nervous system (CNS) disorders, including AD. Inflammasomes are intracellular multiprotein complexes that mediate innate immune responses. The NLRP3 inflammasome is formed by a sensor protein (NLRP3), an adaptor molecule (apoptosis-associated speck-like protein [ASC]), and an effector enzyme (caspase-1). Different stimuli activate the formation of the NLRP3 inflammasome complex, thereby initiating signaling cascades that promote the secretion of the proinflammatory mediators interleukin (IL)-1b and IL-18. This process is essential for the clearance of pathogens or injured cells. However, when dysregulated, it contributes to neuronal damage and neurodegeneration [Citation27].
Several studies have shown that Aβ aggregates activate the NLRP3 inflammasome [Citation28] and, in turn, the activated inflammasome exacerbates amyloid pathology [Citation29]. Aggregated tau has also been shown to activate the NLRP3 inflammasome and the activated inflammasome promotes a prion-like seeding and spreading of tau pathology in mice in vivo [Citation30].
Microglial activation and neuroinflammatory processes are locked in a vicious cycle with amyloidosis [Citation31]. TPE could potentially have a beneficial effect to break up both arms of this cycle by removing excess Aβ and proinflammatory mediators. In addition, the pleiotropic properties of albumin could play a key role if used as the replacement fluid in the PE-based therapeutic strategy for AD treatment.
3. The role of albumin in Alzheimer’s disease
Human serum albumin is a single polypeptide that is synthesized primarily in the liver. It is composed of 585 amino acids and presents three structurally similar domains (I, II, and III) with each domain further subdivided into A and B subdomains [Citation32,Citation33]. Albumin is the most abundant protein in plasma, and it binds and transports both endogenous and exogenous molecules throughout the body, from fatty acids, drugs, metals and metabolites, to proteins and peptides such as Aβ. Albumin is also the main extracellular antioxidant and has immunomodulatory properties [Citation34,Citation35].
Approximately 90% of plasma Aβ is bound to albumin, and only a small fraction is free [Citation36]. This capacity to bind Aβ has been assessed in several studies that proved that albumin inhibited Aβ self-association by selectively binding Aβ aggregates rather than monomers and by preventing further growth of the Aβ assemblies [Citation37,Citation38]. Even though dissociation constant values for Aβ monomers are in the submillimolar range as opposed to the micromolar range ones of aggregated Aβ, they are still physiologically relevant because of the ~0.6–0.7 mM plasma albumin concentration [Citation39].
Aβ can shift from the brain into the blood in the direction of the decreasing Aβ concentration gradient [Citation40]. Albumin’s capacity to prevent most plasma and cerebrospinal fluid (CSF) Aβ from forming aggregates [Citation41–43] may help to maintain a constant free-Aβ concentration in the blood and a balance with its shift from the brain. Albumin-bound Aβ plasma levels decrease with the progression of AD, which possibly indicates that this equilibrium gradient is altered in AD patients [Citation40].
Aβ can cause a series of downstream changes that result in nitration, glycation and oxidation of proteins such as albumin, thereby dramatically affecting their function [Citation34,Citation44]. Reduced albumin-bound Aβ levels observed in AD patients could be due to a decreased capacity of albumin to bind Aβ due to these PTMs that occur in disease states which cause cellular stress [Citation34]. Changes in this apparently protective role of albumin in the clearance and transportation of Aβ could therefore contribute to the pathophysiology of AD [Citation44]. Furthermore, disturbed binding/transport properties of post translationally modified albumin may impair traffic of endogenous substances, and also the delivery of drugs that are critical in the treatment of human disease [Citation34], and this may impact the clinical management of not only AD but also other concomitant pathologies in these patients.
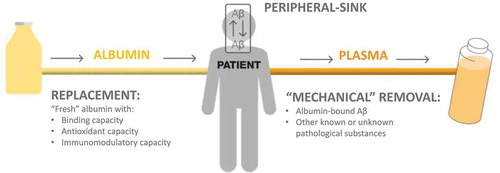
The pathophysiological events discussed above provide support to the rationale of the Alzheimer Management By Albumin Replacement (AMBAR) program that uses different modalities of PE with albumin replacement. According to the AMBAR rationale, PE would remove albumin-bound Aβ from plasma and subsequently change the equilibrium between CSF and plasma Aβ concentrations. PE with albumin replacement may induce an Aβ shift from CSF to plasma through the BBB, thereby reducing Aβ brain levels. This is known as the ‘peripheral sink’ hypothesis. The volume of plasma removed would be replaced by ‘fresh’ albumin with binding, antioxidant and immunomodulatory capacities. During PE, other plasma components might also be removed, including possible pro-aging systemic factors () [Citation45–47].
Figure 1. The AMBAR program rationale and the ‘peripheral sink’ that would cause an Aβ shift from cerebrospinal fluid to plasma through the blood-brain barrier
To date, the AMBAR program has completed phase 1 (EudraCT#: 2005–001616-45), phase 2 (EudraCT#: 2007–000414-36; ClinicalTrials.gov ID: NCT00742417) and phase 2b/3 (EudraCT#: 2011–001598-25; ClinicalTrials.gov ID: NCT01561053) clinical trials in patients with mild-to-moderate AD.
A series of basic research studies have been carried out in plasma and CSF samples from patients in the phase 2 trial [Citation48], to characterize albumin PTMs such as oxidation and early glycation occurring in AD patients as compared to healthy age-matched controls [Citation49,Citation50].
According to the oxidation status of the thiol group (-SH) at the Cys34 residue, three forms of albumin with decreasing antioxidant capacity can be distinguished: reduced, reversibly oxidized and irreversibly oxidized [Citation49]. Albumin oxidation in plasma and CSF was assessed using anionic exchange high performance liquid chromatography coupled to a fluorescent detector. Albumin was shown to be mainly reversibly oxidized in plasma and irreversibly oxidized in CSF of AD patients whereas, in healthy controls, reduced albumin forms were predominant [Citation49].
Using mass spectrometry analysis in albumin-enriched CSF and plasma samples, 17 different albumin PTMs were identified (several oxidized forms, glycated and truncated isoforms). Principal components analysis (PCA) of these albumin PTMs showed good separation between CSF and plasma albumin from AD patients. PCA also showed good separation with CSF albumin from AD patients and controls while plasma albumin from AD patients and controls showed some similarities. PCA only based on four albumin oxidation-related PTMs showed similar separation. This analysis showed that albumin was more oxidized in AD patients than in healthy controls and this effect was much more prominent in CSF than in plasma [Citation49].
Mass spectrometry and a quantitative enzymatic assay (Lucica GA-L) were used to analyze albumin glycation. By mass spectrometry analysis, noticeable increases in the cysteinylated+glycated and the oxidized+glycated albumin forms were observed in both plasma and CSF samples from AD patients. Furthermore, AD patients showed higher glycation in plasma than in CSF (50).
By means of Lucica GA-L analysis, characterization of early glycated albumin levels in plasma from the same cohorts after PE with albumin showed a temporary reduction of glycated albumin levels [Citation50,Citation51].
After characterizing the albumin PTMs from samples taken during the phase 2 study, the next step will be to characterize the albumin PTMs from larger cohorts in the AMBAR study, a Phase 2b/3 clinical trial. In these cohorts, an initial analysis confirmed that different modalities of PE with albumin replacement produce a temporary reduction in glycated albumin levels in plasma, especially during the intensive treatment phase () [Citation52].
Figure 2. Changes from baseline in early glycated albumin (eGA) after TPE (conventional therapeutic PE for 6 weeks) and LVPE (low-volume PE for 12 months) periods. Reproduced from poster (however the Figure itself has not been published) [Citation52]
![Figure 2. Changes from baseline in early glycated albumin (eGA) after TPE (conventional therapeutic PE for 6 weeks) and LVPE (low-volume PE for 12 months) periods. Reproduced from poster (however the Figure itself has not been published) [Citation52]](/cms/asset/1bc9456f-7914-4495-82c6-0ea6377df7ed/iern_a_1960823_f0002_oc.jpg)
4. Plasma exchange with albumin replacement in patients with Alzheimer’s disease
The phase 2 clinical trial showed that mobilization of Aβ in plasma and CSF of patients with AD who underwent PE with therapeutic albumin (Albutein®, Grifols) replacement, was associated with a positive trend in cognition outcomes which persisted after treatment was discontinued [Citation48]. The AMBAR study was then designed as a phase 2b/3 multicenter, randomized, blinded and placebo-controlled, parallel-group trial in mild-to-moderate AD patients to further evaluate these trends by carrying out PE with different replacement volumes of Albutein®, alternated with or without intravenous immunoglobulin (IVIG) 5% (Flebogamma® DIF, Grifols) to correct any potential immunological deficit [Citation53].
The AMBAR study consisted of an initial 6-week treatment period with weekly sessions of TPE with Albutein® 5% for all the active groups through peripheral or central access, followed by a 12-month period of monthly sessions with low-volume plasma exchange (LVPE) through a peripheral line with three different regimens of Albutein® 20% alternated with or without IVIG administration every 4 months. The control group underwent a simulated PE through a noninvasive procedure that mimicked both PE procedures but without any actual fluid replacement [Citation54].
There were two co-primary clinical efficacy variables: the Alzheimer’s Disease Cooperative Study–Activities of Daily Living (ADCS-ADL), as a functional scale, and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog), as a cognitive scale, and changes were assessed from baseline to 14 months. Two global assessment scales were used as secondary efficacy variables in the AMBAR study: the Clinical Dementia Rating Sum of Boxes (CDR-sb), and the ADCS-Clinical Global Impression of Change (ADCS-CGIC). Changes in CSF biomarkers (Aβ1-40, Aβ1-42, total tau and phosphorylated tau) were also assessed [Citation53,Citation54]. Results for the moderate (baseline Mini Mental State Examination [MMSE]: 18–21) and mild (baseline MMSE: 22–26) AD cohorts were analyzed separately. The main safety variable was the percentage of PE procedures (TPE and LVPE) associated with at least one adverse event (AE) or serious adverse event (SAE) [Citation53,Citation54]. Further details of study design, methods and global primary results have already been provided elsewhere [Citation53,Citation54].
In summary, results at the final study visit showed a beneficial shift of cognitive, functional, and global assessment decline in PE-treated patients compared to the placebo group. That is, 52% less decline in ADCS-ADL (p = 0.03); 66% less decline in ADAS-cog (p = 0.06); 71% less decline in CDR-Sb (p = 0.002); and 100% less decline in ADCS-CGIC (p < 0.0001). For the two co-primary outcomes, the beneficial effect of PE treatment was more prominent in moderate than in mild AD patients, whereas for the secondary efficacy outcomes both cohorts performed better than placebo. Regarding biomarkers, at the end of the treatment period, CSF Aβ1-42 levels in moderate AD patients remained stable in the PE-treated group while the placebo group showed a longitudinal decrease (p = 0.05) (). Occurrence of AEs was similar to that expected for patients undergoing PE [Citation53,Citation54].
Figure 3. Least square (LS) mean change from baseline in cerebrospinal fluid levels ± standard error of the mean (SEM) of Aβ1-42 between the finalization and beginning of each of the two PE with albumin replacement periods (TPE: conventional therapeutic PE up to month 2; LVPE: low-volume PE up to month 14) performed on moderate AD patients. Reproduced from [Citation54] under CC BY-NC-ND 4.0 license
![Figure 3. Least square (LS) mean change from baseline in cerebrospinal fluid levels ± standard error of the mean (SEM) of Aβ1-42 between the finalization and beginning of each of the two PE with albumin replacement periods (TPE: conventional therapeutic PE up to month 2; LVPE: low-volume PE up to month 14) performed on moderate AD patients. Reproduced from [Citation54] under CC BY-NC-ND 4.0 license](/cms/asset/af95d0c4-3b3c-4db1-9048-52fe54271841/iern_a_1960823_f0003_oc.jpg)
5. Conclusions
Results from the series of basic research studies support the role of glycation and oxidative stress in AD and deserve further investigation. Furthermore, they suggest that albumin PTMs, especially those relating to oxidation and glycation, occur in people with AD.
The AMBAR study provides encouraging results for the treatment of AD with different modalities of PE and albumin replacement, especially in the moderate AD patient cohort at the beginning of the study who showed a slowdown from baseline in the progression of cognitive and functional decline.
6. Expert opinion
AD is a complex process where clinical manifestations result from the confluence of multiple brain and systemic pathophysiological mechanisms together with a number of risk and protective factors. In this regard, the amyloidosis-neuroinflammation feedback loop can be a relevant therapeutic target in AD. There is a need to design clinical trials that use combination therapies based on approved, safe, and efficacious anti-neuroinflammatory agents such as anti-IL-1 signaling agents in combination with anti-Aβ antibodies that have demonstrated to be safe in multiyear trials [Citation31]. Importantly, as a possible new approach to treating AD, TPE exerts both effects simultaneously as it eliminates both the pathological deposit of Aβ, and the proinflammatory elements involved in AD pathogenesis.
The AMBAR study was designed to assess the efficacy and safety of PE with albumin replacement, and showed promising results in patients with mild-to-moderate AD. Given the diversity of factors involved in the pathophysiology of AD, further studies are needed to ascertain any other mechanisms of action, beyond Aβ binding, that could have contributed to the slowing and stabilizing of disease progression observed in the AMBAR study. More in-depth exploration of neuroinflammation in AD, and the use of TPE to remove damaging cytokines and chemokines or any other detrimental substances, is also necessary.
The AMBAR program may very well represent an ideal setting for the progress of this type of research. On one hand, investigations may focus on mechanisms of action beyond amyloid clearance and investigate other factors related to neuroinflammation, metabolism, or pro- and anti-aging factors. On the other hand, the promising results of TPE in patients with mild-to-moderate AD demand confirmation in further clinical trials in larger cohorts of patients in the early and intermediate stages of AD.
Furthermore, we argue that the findings from the AMBAR study warrant further investigation of the effects of PE with albumin, not only in mild-to-moderate AD, but also in cohorts of patients with mild cognitive impairment and other types of dementia, as PE could potentially remove a whole array of damaging substances from the body that are products of the normal aging process and cellular senescence. Additionally, we believe the role of PE with albumin in removing harmful substances in other neurodegenerative diseases should also be further explored. For instance, albumin is also known to bind to extracellular alpha-synuclein, a protein that aggregates into the Lewy bodies that are the hallmark of Parkinson’s disease (PD) and which contributes to PD pathogenesis through a prion-like transmission mechanism [Citation55].
Another aspect of the basic research studies worth mentioning is that they focused primarily on the effect of albumin oxidation and glycation, as these are the PTMs most commonly related to aging. However, the binding of long-chain non-esterified fatty acids to albumin is also a known modulator of albumin- Aβ interactions as fatty acids compete with Aβ oligomers for binding to albumin’s domain 3 [Citation56,Citation57]. The effect this interaction could have on the results of the AMBAR study may also be worth examining.
As described throughout this review, extremely encouraging advances have been made in the understanding of the pathophysiology of AD. We must keep the momentum going to turn those advances into successful treatments that will lead to better outcomes for AD patients.
Article highlights
Given the complex nature of the pathophysiology of Alzheimer’s disease (AD), a comprehensive strategy for treatment and prevention targeting brain and systemic factors simultaneously is required.
Post-translational modifications of albumin, such as those related to oxidation and glycation, are different between healthy subjects and AD patients.
Further research is required around the involvement of neuroinflammation in AD and the role of therapeutic plasma exchange in removing cytokines, chemokines and other factors from blood.
Findings from the Alzheimer Management By Albumin Replacement (AMBAR) study provide encouraging results regarding the treatment of patients with mild-to-moderate AD with plasma exchange and albumin replacement.
Declaration of interest
P Martínez-Lage has received fees for lectures from Grifols. M Boada has been a consultant for Araclon, Avid, Bayer, Elan, Grifols, Janssen/Pfizer, Lilly, Neuroptix, Nutricia, Roche, Sanofi, Biogen, and Servier; and received fees for lectures and funds for research from Araclon, Esteve, Grifols, Janssen, Novartis, Nutricia, Piramal, Pfizer-Wyeth, Roche and Servier. P Serrano-Castro received fees for lectures from Grifols; M Costa and A Páez are full-time employees of Grifols, a manufacturer of plasmatic therapeutic albumin and intravenous immunoglobulin. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Polanco JC, Li C, Bodea LG, et al. Amyloid-β and tau complexity - Towards improved biomarkers and targeted therapies. Nat Rev Neurol. 2018;14:22–40.
- Loeffler DA. AMBAR, an encouraging Alzheimer’s trial that raises questions. Front Neurol. 2020;11:1–7.
- Wang J, Gu BJ, Masters CL, et al. A systemic view of Alzheimer disease - Insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612–623.
- Querfurth HW, LaFerla FM. Alzheimer’s disease: mechanism of disease. N Engl J Med. 2010;362(4):329–344.
- Forloni G, Alzheimer’s Disease BC. Oligomers, and Inflammation. J Alzheimer’s Dis. 2018;62(3):1261–1276.
- Minter MR, Taylor JM, Crack PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem. 2016;136(3):457–474.
- Montoliu-Gaya L, Mulder SD, Herrebout MAC, et al. Aβ-oligomer uptake and the resulting inflammatory response in adult human astrocytes are precluded by an anti-Aβ single chain variable fragment in combination with an apoE mimetic peptide. Mol Cell Neurosci. 2018;89:49–59.
- Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5–21.
- Pontecorvo MJ, Devous MD, Navitsky M, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140:748–763.
- Jacobs HIL, Hedden T, Schultz AP, et al. Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat Neurosci. 2018;21(3):424–431.
- Yamazaki Y, Zhao N, Caulfield TR, et al. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–518.
- Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–581.
- Iriondo A, García-Sebastian M, Arrospide A, et al. Plasma lipids are associated with white matter microstructural changes and axonal degeneration. Brain Imaging Behav. 2020;15(2):1043–1057.
- Long JM, Holtzman DM. Alzheimer Disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–339.
- Shamsi A, Shahwan M, Khan MS, et al. Elucidating the interaction of human ferritin with quercetin and naringenin: implication of natural products in neurodegenerative diseases: molecular docking and dynamics simulation insight. ACS Omega. 2021;6(11):7922–7930.
- Pluvinage JV, Wyss-Coray T. Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat Rev Neurosci. 2020;21(2):93–102.
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7(3):280–292.
- Cummings J, Aisen P, Lemere C, et al. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimer’s Res Ther. 2021;13(1):98.
- Klein G, Delmar P, Voyle N, et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimer’s Res Ther. 2019;11(1):101.
- Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res Ther. 2021;13(1):80.
- Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384(18):1691–1704.
- Hansson O, Zetterberg H, Buchhave P, et al. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5(3):228–234.
- Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice - Evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145–284.
- Jamshidian A, Abd-Nikfarjam B, Khademi Z, et al. Therapeutic plasma exchange may adjust IL-6 and TGF-β signals in relapsed MS patients peripheral blood. J Clin Apher. 2020;35(2):72–78.
- Padmanabhan A, Connelly-Smith L, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice - evidence-based approach from the Writing Committee of the American Society for Apheresis: the eighth special issue. J Clin Apher. 2019;34:171–354.
- Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimer’s Dement. 2016;12(6):719–732.
- Olcum M, Tastan B, Kiser C, et al. Microglial NLRP3 inflammasome activation in multiple sclerosis. Adv Protein Chem Struct Biol. 2020;119:247–308.
- Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol. 2008;9(8):857–865.
- Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678.
- Stancu IC, Cremers N, Vanrusselt H, et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137(4):599–617.
- Bartfai T, Lees GV. Alzheimer drug trials: combination of safe and efficacious biologicals to break the amyloidosis-neuroinflammation vicious cycle. ASN Neuro. 2020;12:1–7.
- Shamsi A, Ahmed A, Khan MS, et al. Understanding the binding between Rosmarinic acid and serum albumin: in vitro and in silico insight. J Mol Liq. 2020;311:113348.
- Shamsi A, Ahmed A, Khan MS, et al. Rosmarinic acid restrains protein glycation and aggregation in human serum albumin: multi spectroscopic and microscopic insight - possible therapeutics targeting diseases. Int J Biol Macromol. 2020;161:187–193.
- Colombo G, Clerici M, Giustarini D, et al. Redox albuminomics: oxidized albumin in human diseases. Antioxid Redox Signal. 2012;17(11):1515–1527.
- Casulleras M, Flores-Costa R, Duran-Güell M, et al. Albumin internalizes and inhibits endosomal TLR signaling in leukocytes from patients with decompensated cirrhosis. Sci Transl Med. 2020;12(566):eaax5135.
- Biere AL, Ostaszewski B, Stimson ER, et al. Amyloid β-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271(51):32916–32922.
- Costa M, Ortiz AM, Jorquera JI. Therapeutic albumin binding to remove amyloid-β. J Alzheimer’s Dis. 2012;29(1):159–170.
- Milojevic J, Costa M, Ortiz AM, et al. In vitro amyloid-β binding and inhibition of amyloid-β self-association by therapeutic albumin. J Alzheimer’s Dis. 2014;38(4):753–765.
- Algamal M, Ahmed R, Jafari N, et al. Atomic-resolution map of the interactions between an amyloid inhibitor protein and amyloid β (Aβ) peptides in the monomer and protofibril states. J Biol Chem. 2017 Oct;20(292):17158–17168.
- Yamamoto K, Shimada H, Koh H, et al. Serum levels of albumin-amyloid beta complexes are decreased in Alzheimer’s disease. Geriatr Gerontol Int. 2014;14(3):716–723.
- Kuo YM, Kokjohn TA, Kalback W, et al. Amyloid-β peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem Biophys Res Commun. 2000;268(3):750–756.
- Bohrmann B, Tjernberg L, Kuner P, et al. Endogenous proteins controlling amyloid β-peptide polymerization. J Biol Chem. 1999;274(23):15990–15995.
- Rózga M, Kłoniecki M, Jabłonowska A, et al. The binding constant for amyloid Aβ40 peptide interaction with human serum albumin. Biochem Biophys Res Commun. 2007;364(3):714–718.
- Ramos-Fernández E, Tajes M, Palomer E, et al. Posttranslational nitro-glycative modifications of albumin in alzheimer’s disease: implications in cytotoxicity and amyloid-β peptide aggregation. J Alzheimer’s Dis. 2014;40(3):643–657.
- Katsimpardi L, Litterman NK, Schein PA, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344(6184):630–634.
- Loffredo FS, Steinhauser ML, Jay SM, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153(4):828–839.
- Villeda SA, Plambeck KE, Middeldorp J, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014;20(6):659–663.
- Boada M, Anaya F, Ortiz P, et al. Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-β concentrations and cognition outcomes in Alzheimer’s disease patients: a multicenter, randomized, controlled clinical trial. J Alzheimer’s Dis. 56(1): 129–143. 2017.
- Costa M, Horrillo R, Ortiz AM, et al. Increased albumin oxidation in cerebrospinal fluid and plasma from Alzheimer’s disease patients. J Alzheimer’s Dis. 63(4): 1395–1404. 2018.
- Costa M, Mestre A, Horrillo R, et al. Cross-sectional characterization of albumin glycation state in cerebrospinal fluid and plasma from Alzheimer’s disease patients. J Prev Alzheimer’s Dis. 2019;6:139–143.
- Costa M. Albumin as a pharmaceutical active ingredient for Alzheimer’s disease. In J.L. Cummings (chair), AMBAR (Alzheimer management by albumin replacement) phase iib/iii results: clinical and biomarker update. Symposium at the 14th Int Conference on AD and PD. Lisbon (Portugal); 2019
- Ortiz AM, Minguet C, Mestre A, et al. Albumin glycation in Alzheimer’s disease patients: results from the AMBAR trial. Alzheimer’s Dement. 2020;16(S3):32916–32922.
- Boada M, López O, Núñez L, et al. Plasma exchange for Alzheimer’s disease management by albumin replacement (AMBAR) trial: study design and progress. Alzheimer’s Dement Transl Res Clin Interv. 5(1): 61–69. 2019.
- Boada M, López OL, Olazarán J, et al. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: primary results of the AMBAR Study. Alzheimer’s Dement. 16(10): 1412–1425. 2020.
- Ahmed R, Huang J, Weber DK, et al. Molecular mechanism for the suppression of alpha synuclein membrane toxicity by an unconventional extracellular chaperone. J Am Chem Soc. 2020;142:9686–9699.
- Algamal M, Milojevic J, Jafari N, et al. Mapping the Interactions between the Alzheimer’s Aβ-Peptide and human serum albumin beyond domain resolution. Biophys J. 2013;105(7):1700–1709.
- Jafari N, Ahmed R, Gloyd M, et al. Allosteric sensing of fatty acid binding by NMR: application to human serum albumin. J Med Chem. 2016;59(16):7457–7465.