Abstract
The discovery of new topoisomerase I inhibitors is necessary since most of the antitumor drugs are targeted against type II and only a very few can specifically affect type I. Topoisomerase poisons generate toxic DNA damage by stabilization of the covalent DNA-topoisomerase cleavage complex and some have therapeutic efficacy in human cancer. Two iridoids, aucubin and geniposide, have shown antitumoral activities, but their activity against topoisomerase enzymes has not been tested. Here it was found that both compounds are able to stabilize covalent attachments of the topoisomerase I subunits to DNA at sites of DNA strand breaks, generating cleavage complexes intermediates so being active as poisons of topoisomerase I, but not topoisomerase II. This result points to DNA damage induced by topoisomerase I poisoning as one of the possible mechanisms by which these two iridoids have shown antitumoral activity, increasing interest in their possible use in cancer chemoprevention and therapy.
1 Introduction
Iridoids represent a wide group of cyclopentane[c]pyran monoterpenoids which are known to be associated with diverse biological properties, including analgesic, anti-inflammatory, liver protective, choleretic, purgative, antimicrobial, sedative and antitumor activities [Citation1].
The iridoids which are the object of our study are aucubin and geniposide (). Previous studies have shown that these compounds have some chemoprevention and cancer therapy potential. It has been reported that this anticancer activity is mediated by different mechanism of action involved in cancer initiation, promotion and progression Citation2-14.
Figure 1 Chemical structure of aucubin and geniposide.
DNA topoisomerases (topos) are essential enzymes that govern DNA topology through transient DNA cleavage, strand passing and religation during fundamental nuclear metabolic processes, such as replication, transcription or recombination. Topo I acts by forming a transient single strand break through which the other DNA strand passes to achieve relaxation and topo II is able to do so with the two strands that make up duplex DNA, creating a DNA-linked protein gate through which another intact duplex passes [Citation15]. Poisons of topoisomerases allow the enzyme to cut and covalently bind to DNA, but prevent the subsequent rejoining of the molecule after relieving the torsional stress causing stabilization of the covalent topo-DNA cleavage complex. Stabilization of the cleavage complexes may not be directly cytotoxic. One attractive model that has experimental support holds that collision of DNA replication forks with cleavage complexes causes the complex to fall apart without rejoining DNA, thereby generating lethal double strands breaks [Citation16,Citation17].
With these precedents, and as part of our research on natural products as topoisomerase poisons Citation18-22, we have evaluated the topoisomerase poison activity of aucubin and geniposide, two iridoids present in Plantago species. These plants have been employed in traditional medicine to treat digestive and respiratory problems, wounds, ulcers and cancer [Citation23].
2 Materials and methods
2.1 Enzymes, nucleic acids and chemicals
Purified enzyme human topoisomerase I and II, pRYG DNA and the positive inhibitory controls camptothecin and etoposide were purchased from TopoGen, Inc (Columbus, OH, USA). Proteinase K was from Sigma Chemical Co. The iridoid aucubin was obtained from Extrasynthese SA (Genay, France). The iridoid geniposide was isolated from a methanol extract from Plantago bellardii [Citation24]. Stock solutions of these compounds were dissolved in dimethyl sulfoxide (DMSO) at 40 mM and were diluted in water containing 2.5% DMSO before use.
2.2 DNA cleavage reactions with topoisomerase I and II
Cleavage topo I buffer contained 10 mM Tris–HCl (pH 7.9), 1 mM EDTA, 0.15 M NaCl, 0.1% BSA, 0.1 mM spermidine and 5% glycerol (pH 8). Cleavage topo II buffer contained 30 mM Tris–HCl (pH 7.6), 60 mM NaCl, 15 mM mercaptoethanol, 8 mM MgCl2 and 3 mM ATP. The cleavage reaction (20 μL) contained cleavage buffer (2 μL), the tested drugs dissolved in 2 μL DMSO/H2O (2.5%), pRYG DNA (0.25 μg in 1 μL of buffer), and 2.5 μL (5 units) of human topoisomerase I, or 2 μL (4 units) of human topoisomerase II, and water up to 20 μL. Reactions were incubated at 37°C for 30 min, terminated by the addition of 2 μL SDS 10% and 1 μL proteinase K 2 mg/ml and followed by an additional 30 min incubation at 37°C for topoisomerase I, or 15 min incubation for topoisomerase II. Subsequently, the samples were extracted with chloroform:isoamyl alcohol, and 2 μL bromophenol blue was added. For topoisomerase I assays, samples were loaded on 1% agarose gels and electrofocussed at 3 V/cm for 6 h in Tris-acetate-EDTA buffer with ethidium bromide to a final concentration of 0.5 μg/mL. Gels were washed in a large amount of water. In topoisomerase II tests, samples were loaded on 1% agarose gels and electroforesed at 6 V/cm for 3 h in Tris-acetate-EDTA buffer. Gels were stained with ethidium bromide and washed in water. For the quantitative determination of topo I, and II, videoimpression was densitometrically measured using Image J software. After integration of the bands, the nicked open circle DNA (OC) form for topo I, and the linear and nicked open circle DNA (OC) forms for topo II, induced by the tested drugs was expressed as the percentage of total DNA in relation to these DNA forms obtained in the absence of drug.
3 Results
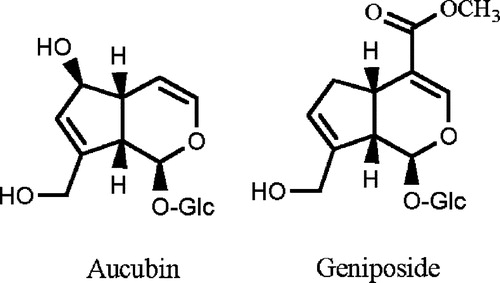
Despite preliminary assays having shown that neither aucubin nor geniposide were cytotoxic on the human cancer cell lines TK-10, MCF-7 and UACC-62 (data not shown), both iridoids, were studied to determined their capacity to stabilise covalent attachments of the topoisomerase I and II subunits to DNA at sites of DNA strand breaks, so generating cleavage complexes intermediates that could be detected in agarose gels. Both compounds were assayed at concentrations of 50 and 100 μM. Camptothecin and etoposide were used as positive control at a concentration of 100 μM for both topo I and topo II assays. The Gel showed that aucubin and geniposide were able to induce open circle (OC) DNA. Then, after staining the gels with ethidium bromide, the nicked open circle (OC) DNA band was densitometrically measured using Image J software. Nicked OC DNA form was expressed as a percentage of total DNA () and it was be observed that aucubin and, in a higher percentage, geniposide induced the formation of DNA cleavage complex at the two tested concentrations, acting as DNA topoisomerase I poisons. It seems that there was not a dose-response relationship, since the lower dose showed higher activity.
Figure 2 DNA-topoisomerase I mediated DNA cleavage: 1. Topo I+ pRYG DNA+50 μM aucubin. 2. Topo I+ pRYG DNA+100 μM aucubin. 3. Topo I+ pRYG DNA+50 μM geniposide. 4. Topo I+ pRYG DNA+100 μM geniposide. 5. Topo I+ pRYG DNA+100 μM camptothecin. 6. Topo I+ pRYG DNA 7. pRYG DNA.
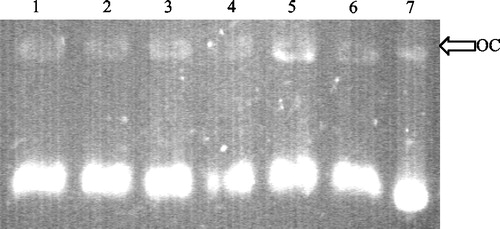
Figure 3 Topoisomerase I-mediated DNA cleavage, expressed as percentage of OC (open circular) DNA, induced by (1) camptothecin 100 μM, (2) aucubin 50 μM, (3) aucubin 100 μM, (4) geniposide 50 μM and (5) geniposide 100 μM. The percentage of DNA cleavage of topoisomerase I in presence of these compounds was determined by gel scanning.
However, neither aucubin nor geniposide were able to induce linear and nicked open circle (OC) DNA at both tested concentrations, thereby not showing any activity as topoisomerase II poisons when the assay was performed against the positive control, etoposide at 100 μM (not shown).
4 Discussion
Studies on aucubin and geniposide have previously shown that these compounds have some chemoprevention and cancer therapy potential. Aucubin inhibited phase I enzymes [Citation2], was proapoptotic through the inhibition of IkB degradation [Citation3], and inhibited the production of TNF-α or IL-6 induced by antigens [Citation3]. Moreover, it has been shown that its aglycon is able to inhibit DNA and RNA polymerases, and both, glycoside and aglycon, can interact with DNA [Citation4]. Aucubin cytotoxicy has been tested in different cell lines, such as leukemic or human lymphoid cells, but showed a very weak activity in all of them Citation4-6. These results corroborate our preliminary cytotoxic assays, using other cell lines such as TK-10 (renal adenocarcinoma), MCF-7 (breast adenocarcinoma) or UACC-62 (melanoma).
Geniposide like aucubin, has shown antitumoral activity. Geniposide is able to inhibit tumorigenesis induced by different carcinogens [Citation7,Citation8], can induce phase II enzymes [Citation9] and inhibiting, at the same time, phase I enzymes [Citation10] and other enzymes involved in the tumoral processes [Citation8]. It has shown antimutagenic [Citation11] and anti-angiogenic properties [Citation12].
Therefore, although have been mentioned different antitumoral activities related to these compounds this is the first report of an iridoid acting as a topoisomerase poison.
The results obtained here lead to three important aspects for comment.
First, the higher activity showed by geniposide, could be due to two situations. First, the absence of the hydroxyl moiety at C-6 and second, the presence of the methylester moiety in C-4, able to interact with a free amino group so increasing possible covalent binding in relation to aucubin, which does do not possess this group.
Second, the reduction in topo I cleavable complexes at 100 μM for both compounds seemed paradoxical, since less activity is shown than at 50 μM. One of the possibilities for the lack of a dose-response relationship could be explained based on literature data, where it has been shown that there are two kinds of topoisomerase inhibitors; poisons, that stabilize the cleavable complexes and stimulate single or double-stranded DNA cleavage, and catalytic inhibitors, that prevent the catalytic cycle of the enzymes at steps other than cleavage intermediates [Citation25]. The stabilization of topoisomerase cleavable complexes can decrease if the catalytic activity of the enzyme is inhibited. Pre-treatment with a catalytic inhibitor of topoisomerase can prevent the stabilization of cleavable complexes in cells exposed to topo poisons. Using this approach the topo II poison idarubicin has been suggested to act as a topo II catalytic inhibitor at high concentrations [Citation26]. The reduction in the topo I poison activity of aucubin and geniposide observed at high concentrations () may indicate that they are acting as topo I catalytic inhibitors.
Finally, with regard to the lack of activity shown by these compounds in vivo, the iridoid metabolites genipin and aucubigenin, aglycones of geniposide and aucubin respectively, might be the agents responsible for the effect in cells [Citation27]. Even more, interesting is the fact that the open chain aglycone of these iridoids can form an imine bond with a nucleophilic site on protein, through a Schiff's reaction [Citation28,Citation29]. This irreversible binding may partially contribute to their biological effect. Therefore, further assays with these compounds and in vivo topoisomerase activity are suggested.
Interest concerning topoisomerase I poisons has increased since 1997, when the camptothecin derivatives topotecan and irinotecan were introduced into the clinic for the treatment of refractory ovarian and colorectal cancer, respectively [Citation30], constituting topoisomerase I poisons as a novel family of antitumor agents.
As mentioned above, these iridoids can be considered as non-toxic anticancer compounds, since they are weak or non-cytotoxic against different cell lines. Therefore, they could be useful in combination with toxic drugs currently being used in cancer therapy in order to reduce their toxic effects. Also the possible usefulness of the their combination with other topo II poisons is apparent since there is a synergistic effect between them [Citation31]. Moreover, different authors have shown that aucubin and geniposide metabolites are also active as antitumoral agents, even more so than the parent molecules.
In conclusion, the present results suggest that DNA damage induced by topoisomerase I poisoining is one of the possible mechanism by which aucubin and geniposide exert their antitumoral activity. This fact integrated with the literature available concerning aucubin and geniposide as safe anticancer compounds, suggests that this group of compounds should be borne in mind as possible candidates not only for cancer prevention but also for cancer therapy, with the further testing of other iridoids to determine a structure-activity relationship as well as the influence of the sugar moieties. As far as cancer therapy is concerned, it is suggested that a combination of aucubin or geniposide with specific currently used anticancer drugs should be examined.
Acknowledgements
We are most grateful to Dr Cortés research group (Biología Celular, Facultad de Biología, Universidad de Sevilla). We thank the National Cancer Institute (NCI), USA, for the generous gift of the human cancer cell lines used in the present investigation. This work has been supported by the Spanish Ministry of Science and Technology (SAF 2000-0167).
Related Research Data
References
- Ghisalberti EL. Phytomedicine 1998; 5: 147–163
- Bartholomaeus A, Ahokas J. Toxicol Lett 1995; 80: 75–83
- Jeong HJ, Koo HN, Na HJ, Kim MS, Hong SH, Eom JW, Kim KS, Shin TY, Him HM. Cytokine 2002; 18: 252–259
- Lee DH, Cho IG, Park MS, Kim KN, Chang IM, Mar WC. Arch Pharmacol Res 2001; 24: 55–63
- Chiang LC, Chiang W, Chang MY, Ng LT, Lin CC. Am J Chinese Med 2003; 31: 37–46
- Zong YY, Che CT. Planta Med 1995; 61: 585–586
- Ueda S, Iwashashi Y. J Nat Prod 1991; 54: 1677–1680
- Lee MJ, Hsu JD, Wang CJ. Anticancer Res 1995; 15: 411–416
- Wang CJ, Wang SW, Lin JK. Cancer Lett 1991; 60: 95–102
- Kang JJ, Wang HW, Liu TY, Chen YC, Ueng TH. Food Chem Toxicol 1997; 35: 957–965
- Nakamura T, Nakazawa Y, Onizuka S, Satoh S, Chiba A, Sekihashi K, Miura A, Yasagahira N, Sasaki YF. Mut Res 1997; 388: 7–20
- Koo HJ, Lee S, Shin KH, Kim BC, Lim CJ, Park EH. Planta Med 2004; 70: 467–469
- Ishiguro K, Yamaki M, Takayi S, Ikeda Y, Kawakani K, Ito K, Nose T. J Nat Prod 1983; 46: 532–536
- Wang SW, Lai CY, Wang CJ. Cancer Lett 1992; 65: 133–137
- Wang JC. Ann Rev Biochem 1985; 54: 665–697
- Hsiang YH, Lihou MG, Liu LF. Cancer Res 1989; 49: 5077–5082
- Kauffman WK. Proc Soc Exp Biol Med 1998; 217: 327–334
- Martín-Cordero C, López-Lázaro M, Piñero J, Cortés F, Ayuso MJ. J Enz Inhib 2000; 15: 455–460
- López-Lázaro M, Martín-Cordero C, Ayuso MJ. Z Naturforch 2000; 55: 898–902
- López-Lázaro M, Martín-Cordero C, Bermejo A, Cortes D, Ayuso MJ. Anticancer Res 2001; 21: 3493–3498
- López-Lázaro M, Martín-Cordero C, Toro MV, Ayuso MJ. J Enz Inhib Med Chem 2002; 17: 25–29
- Martín-Cordero C, López-Lázaro M, Gálvez M, Ayuso MJ. J Enz Inhib Med Chem 2003; 18: 505–509
- Samuelsen AB. J Ethnopharmacol 2000; 71: 1–21
- Gálvez M. PhD Doctoral Thesis. University of Seville, Spain 2004
- Umemura K, Yanase K, Suzuki M, Okutani K, Yamori T, Andoh T. Biochem Pharmacol 2003; 66: 481–487
- Willmore E, Errington F, Tilby MJ, Austin CA. Biochem Pharmacol 2002; 63: 1807–1815
- Akao T, Kobashi K, Aburada M. Biol Pharm Bull 1994; 17: 1573–1676
- Koo HJ, Song YS, Kim HJ, Lee HY, Hong SM, Kim SJ, Kim BC, Jin C, Lim CJ, Park EH. Eur J Pharmacol 2004; 495: 201–208
- Kim DH, Kim BR, Kim JY, Jeong YC. Toxicol Lett 2000; 114: 181–188
- Bailly C. Curr Med Chem 2000; 7: 39–58
- Cortés F, Piñero J. Cancer Chemother Pharmacol 1994; 34: 411–415