Abstract
The synthesis of new xanthanolide derivatives is reported starting from xanthatin, a sesquiterpenic lactone isolated from Xanthium macrocarpum (Asteraceae). In vitro evaluation of their antifungal activity has been investigated.
Introduction
The frequency of severe fungal infections has escalated over the past two decades [Citation1]. This increase is linked with the growing number of immunocompromised patients as the result of a primary infection (AIDS) or therapies used in case of cancer and transplants. Contrary to antibiotics, the antifungal armamentorium is essentialy limited to two classes, namely the polyenes and the azole derivatives. Their use for the treatment of invasive fungal infection is sometimes limited by their secondary effects such as the dose-dependent polyene nephrotoxicity [Citation2], or as the azole hepatotoxicity [Citation3]. The closeness of the azole target, the fungal lanosterol-14-α-demethylase, to human cytochrome P450 also resulted in many drug-interactions [Citation4]. Moreover, azole resistance is a well established cause of treatment failure, contributing to mortality rates as high as 80% in patients with leukemia [Citation5].
Taken into account the limited number of effective agents, the search for new antifungal drugs is still an urgent task and natural products could represent a source of structural diversity towards this goal [Citation6]. Indeed, caspofungin has been recently approved for use against invasive aspergillosis resistant to other therapies. This compound is representative of the echinocandins group, originating from the fermentation of the fungus Glarea lozoyensis and its antifungal activity is related to inhibition of the synthesis of the β-1,3-D-glucan, an essential component of the fungal cell wall [Citation7]. In our group, the exploration of the natural kingdom as a source of new compounds for the treatment of fungal diseases led us to report the antifungal activities of several phenolic natural products like phenylpropanoids [Citation8], and xanthones [Citation9,Citation10]. These activities had legitimised our efforts towards the study of their total synthesis, that culminated with the discovery of a new pathway to ortho-(2-hydroxy-3-methylbut-3-enyl)phenol derivatives [Citation11,Citation12]. More recently, we have also reported on the antifungal activities of xanthatin 1 [Citation13], a xanthanolide previously isolated from Xanthium macrocarpum [Citation14]. This type of sesquiterpenic lactones is already known for their antimicrobial [Citation15], antimalarial [Citation16], cytotoxic [Citation17] and farnesyl transferase inhibitory activities [Citation18]. The above mentioned biological properties of these unsaturated lactones as well as their known allergenic properties and toxicity are associated with their ability to react in vivo, as a Michael acceptor, with endogenous nucleophiles [Citation19]. In the case of xanthatin, both the α-methylene-γ-lactone and the unsaturated ketonic side chain could show such reactivity. Therefore, in order to clarify the role of each part of the molecule on the antifungal activity, we decided to synthesize several reduced xanthatin derivatives and their in vitro antifungal activity was evaluated against three human pathogenic fungi: Candida albicans, C. glabrata and Aspergillus fumigatus. However, the total synthesis of such derivatives seems a hard task to achieve. The access to the 5,7-fused system has only been reported for compounds without the α-methylene-γ-lactone moiety [Citation20,Citation21]. We therefore thought that semisynthesis of xanthatin derivatives would be more fruitful, starting from xanthatin 1 and xanthinin 4 isolated from Xanthium macrocarpum as already described [Citation14].
Materials and methods
Chemistry
Compounds 1 and 4 were extracted and isolated from the leaves of Xanthium macrocarpum [Citation14]. All reagents and solvents were general purpose grade. Column chromatography was performed on Si gel 60 (Marcherey-Nagel, 230–400 mesh) and preparative TLC with Si gel plates (Marcherey-Nagel, SIL G/UV254, 0.25 mm). The spots were detected under UV light at 250 nm. Infrared (IR) spectra were determined on a BRUKER FT IR Vector 22 using neat liquid films. 1H-NMR spectra and 13C-NMR spectra were recorded in CDCl3 with HCCl3 at 7.26 ppm as the internal standard on a BRUKER AVANCE DRX 500 or on a JEOL GSX 270 WB spectrometer.
Synthesis of the α-methyl-γ-lactone series
11,13-Dihydro-4-hydroxyxanthatin 2
Sodium borohydride (38 mg, 1 mmol) was added in small portions to a solution of xanthatin 1 (75 mg, 0.3 mmol) in methanol (8 mL) at 0°C. After 3 h, the mixture was acidified to pH 4 with 10% HCl. Methanol was removed under reduced pressure and the aqueous solution was extracted with dichloromethane (3 × 10 mL). The organic extract was successively washed with saturated aqueous NaHCO3 (2 × 10 mL), 20% aqueous NH4Cl (2 × 10 mL), brine (2 × 10 mL) and water (3 × 20 mL). The dichloromethane extract was dried with Na2SO4, concentrated under reduced pressure and purified by column chromatography over silica gel with a mixture of dichloromethane and ethyl acetate (9:1) as the eluent to afford the alcohol 2 (liquid) as an inseparable diastereoisomeric mixture (72 mg, 0.29 mmol) in 95% yield. IR νmax 3436, 1765, 1455, 1380 cm− 1; 1H-NMR (270 MHz, CDCl3) δH ppm, 6.07 (1H, d, J = 15.6 Hz, 2-H), 5.77 (1H, dd, J = 9.2 Hz and J = 3.4 Hz, 5-H), 5.67 (1H, dd, J = 15.6 Hz and J = 6.4 Hz, 3-H), 4.35 (1H, m, 4-H), 4.27 (1H, td, J = 12.4 Hz, 10.2 Hz, and 2.8 Hz, 8-H), 3.01 (1H, m, 10-H), 2.42 (1H, m, 7-H), 2.32 (1H, m, 11-H), 2.29 (1H, m, 6-Hα), 2.07 (1H, m, 6-Hβ), 1.67 (1H, m, 9-Hα), 1.61 (1H, m, 9-Hβ), 1.29 (3H, d, J = 6.2 Hz, 15-H), 1.20 (3H, d, J = 6.8 Hz, 13-H), 1.13 (3H, d, J = 7.5 Hz, 14-H), 13C-NMR (67.5 MHz; CDCl3), δC ppm, 178.5 (C-12), 144.6 (C-1), 133.3 and 133.2 (C-2), 130.1 (C-2), 129.9 (C-5), 81.8 (C-8), 68.9 (C-4), 51.2 (C-7), 41.8 (C-11), 36.6 and 36.1 (C-9), 29.1 and 29.0 (C-10), 27.6 and 27.4 (C-6), 23.6 (C-15), 18.4 (C-14), 12.5 (C13).
11,13-Dihydroxanthatin 3
2,3-Dichloro-5,6-dicyanobenzoquinone (65 mg, 0.29 mmol) was added to a solution of the alcohol 2 (72 mg, 0.29 mmol) in toluene (10 mL). The mixture was heated under reflux for 1 h and the black precipitate was filtered. Toluene was removed under reduced pressure and the residue was purified on preparative TLC, 10% ethyl acetate in dichloromethane as eluent, yielding the unsaturated ketone 3 as a liquid (23 mg, 0.09 mmol) in 32% yield. IR νmax 1765, 1661, 1589 cm− 1, m/z (%) 248.1400 (100.0), 1H-NMR (270 MHz, CDCl3) δH ppm, 7.04 (1H, d, J = 15.9 Hz, 2-H), 6.24 (1H, dd, J = 9.2 Hz and J = 3.2 Hz, 5-H), 6.18 (1H, d, J = 15.9 Hz, 3-H), 4.31 (1H, td, J = 12.0 Hz, J = 10.2 Hz and 2.5 Hz, 8-H), 3.05 (1H, m, 10-H), 2.58 (1H, qd, J = 9.2 Hz, 7.1 and J = 2.1 Hz, 11-H), 2.37 (1H, m, 6-Hα and 6-Hβ), 2.33 (1H, m, 7-H), 2.30 (3H, s, 15-H), 2.17 (1H, m, 7-H), 1.69 (2H, m, 9-Hα and 9-Hβ), 1.25 (3H, d, J = 7.1 Hz, 13-H), 1.17 (3H, d, J = 7.5 Hz, 14-H), 13C-NMR (67.5 MHz; CDCl3), δC ppm, 198.5 (C-4), 178.1 (C-12), 148.3 (C-2), 144.6 (C-1), 138.8 (C-5), 124.4 (C-3), 81.3 (C-8), 50.7 (C-7), 41.9 (C-11), 36.1 (C-9), 29.0 (C-10), 28.3 (C-6), 27.9 (C-15), 18.5 (C-14), 12.6 (C-13).
Synthesis of the α-methylene-γ-lactone series
11-Hydro-13-phenylselanylxanthatin 6
A yellow solution of diphenyldiselenide (173 mg, 0.53 mmol) in 10 mL of dry ethanol was reduced with NaBH4 (38 mg, 1.06 mmol) under nitrogen before xanthinin 4 (166 mg, 0.53 mmol) was added. The mixture was stirred at room temperature for a few minutes. The resulting black precipitate was filtered and washed with cold ethanol. The filtrate was acidified to pH 4 with 10% HCl and ethanol was removed under reduced pressure. The acidified solution was extracted with dichloromethane (3 × 10 mL) and the extract was successively washed with saturated aqueous NaHCO3 (2 × 10 mL), 20% aqueous NH4Cl (2 × 10 mL), brine (2 × 10 mL) and water (3 × 20 mL). The dichloromethane extract was dried with Na2SO4 and concentrated under reduced pressure. The residue was adsorbed on silica gel (5 g) for 48 h at room temperature to give 6 which was purified on silica gel with 1% to 3% of methanol in chloroform to give the liquid 6 (175 mg, 0.43 mmol) in 80% yield. IR νmax 2930, 1769, 1662, 1589 cm− 1, 1H-NMR (270 MHz, CDCl3) δH ppm, 7.51 (2H, m, 3′-H and 5′-H), 7.25 (3H, m, 2′-H, 4′-H and 6′-H), 6.98 (1H, d, J = 16.2 Hz, 2-H), 6.14 (1H, d, J = 16.2 Hz, 3-H), 6.00 (1H, dd, J = 9.2 Hz and J = 2.9 Hz, 5-H), 4.27 (1H, td, J = 9.5 Hz, J = 2.5 Hz and J = 2.2 Hz, 8-H), 3.32 (1H, dd, J = 13.3 Hz and J = 4.5 Hz, 13-Hα), 3.13 (1H, dd, J = 13.0 Hz and J = 6.0 Hz, 13-Hβ), 3.00 (1H, m, 10-H), 2.72 (1H, m, 11-H), 2.58 (1H, qd, J = 10.6 Hz, J = 9.2 Hz and J = 1.6 Hz, 6-Hα), 2.32 (1H, m, 9-Hα), 2.27 (3H, s, 15-H), 2.06 (1H, m, 6-Hβ), 2.01 (1H, m, 7-H), 1.70 (1H, m, 9-Hβ), 1.10 (3H, d, J = 7.3 Hz, 14-H), 13C-NMR (67.5 MHz; CDCl3), δC ppm, 198.4 (C-4), 175.9 (C-12), 148.2 (C-3), 144.4 (C-1), 138.7 (C-5), 132.9 (C-3′ and C-5′), 129.3 (C-2′ and C-6′), 127.4 (C-4′), 124.3 (C-2), 106.3 (C-1′), 81.2 (C-8), 48.6 (C-7), 47.3 (C-11), 36.1 (C-9), 28.9 (C-10), 28.4 (C-6), 27.9 (C-15), 25.5 (C-13), 18.3 (C-14).
11-Hydro-4-hydroxy-13-phenylselanylxanthatin 7
Preparation: Following the method for 2. (from 6 (114 mg, 0.28 mmol) with NaBH4 (17 mg, 0.42 mmol)). The residue was purified on preparative TLC with 5% ethyl acetate in dichloromethane as the eluent to give the liquid 7 as an inseparable diastereoisomeric mixture (37 mg, 0.09 mmol) in 32% yield. IR νmax 3469, 1770 cm− 1, 1H-NMR (270 MHz, CDCl3) δH ppm, 7.53 (2H, m, 3′-H and 5′-H), 7.25 (3H, m, 2′-H, 4′-H and 6′-H), 6.01 (1H, d, J = 15.9 Hz, 2-H), 5.62 (1H, dd, J = 15.9 Hz and J = 6.3 Hz, 3-H), 5.50 (1H, dd, J = 8.8 Hz and J = 1.8 Hz, 5-H), 4.37 (1H, m, 4-H), 4.27 (1H, td, J = 11.3 Hz, J = 8.8 Hz and J = 2.1 Hz, 8-H), 3.33 (1H, dd, J = 12.4 Hz and J = 4.6 Hz, 13-Hα), 3.13 (1H, dd, J = 13.0 Hz and J = 6.0 Hz, 13-Hβ), 3.01 (1H, m, 10-H), 2.71 (1H, m, 11-H), 2.50 (1H, m, 6-Hα), 2.29 (1H, m, 6-Hβ), 1.99 (1H, m, 9-Hα), 2.01 (1H, m, 7-H), 1.73 (1H, m, 9-Hβ), 1.32 (3H, d, J = 6.7 Hz, 15-H), 1.12 (3H, d, J = 7.4 Hz, 14-H), 13C-NMR (67.5 MHz; CDCl3), δC ppm, 176.3 (C-12), 144.5 (C-1), 134.3 (C-2), 134.2 (C-3), 132.9 (C-2′ and C-6′), 130.0 (C-5), 129.9 (C-1′), 129.2 (C-3′ and C-5′), 127.3 (C-4′), 81.7 (C-8), 69.0 and 68.9 (C-4), 49.2 (C-7), 47.4 (C-11), 36.2 (C-9), 29.0 (C-10), 27.8 (C-6), 25.6 (C-13), 23.7 and 23.6 (C-15), 18.4 (C-14).
4-Hydroxyxanthatin 8
10% H2O2 (75 μL, 0.23 mmol) was added to a solution of the alcohol 7 (37 mg, 0.09 mmol) in dichloromethane (5 mL). The mixture was heated under reflux for 2 h and was then washed with water (3 × 5 mL), dried with Na2SO4 and concentrated under reduced pressure. The residue was purified on preparative TLC with 3% methanol in chloroform to give the liquid 8 (6 mg, 0.02 mmol) in 25% yield. IR νmax 3367, 1769, 1713 cm− 1, m/z (%) 247.1314 (28.4), 246.0511 (33.5), 230.1288 (100.0), 1H-NMR (270 MHz, CDCl3) δH ppm, 6.18 (1H, d, J = 3.2 Hz, 13-Hα), 6.09 (1H, d, J = 16.6 Hz, 2-H), 5.84 (1H, dd, J = 9.2 Hz and J = 3.2 Hz, 5-H), 5.71 (1H, dd, J = 16.6 Hz and J = 6.4 Hz, 3-H), 5.46 (1H, dd, J = 3.2 Hz, 13-Hβ), 4.39 (1H, m, 4-H), 4.27 (1H, td, J = 12.0 Hz, J = 9.9 Hz and J = 2.5 Hz, 8-H), 3.08 (1H, m, 7H), 2.66 (1H, qd, J = 9.2 Hz, J = 7.5 Hz and J = 2.5 Hz, 10-H), 2.53 (1H, m, 6-Hα), 2.35 (1H, m, 6-Hβ), 2.18 (1H, m, 9-Hα), 1.84 (1H, m, 9-Hβ), 1.33 (3H, d, J = 6.4 Hz, 15-H), 1.15 (3H, d, J = 7.5 Hz, 14-H), 13C-NMR (67.5 MHz; CDCl3), δC ppm, 173.0 (C-12), 144.9 (C-1), 139.7 (C-11), 130.5 (C-2), 130.4 (C-3), 128.5 (C-15), 118.2 (C-13), 81.9 (C-8), 69.1 (C-4), 47.9 (C-7), 36.7 (C-9), 29.7 (C-10), 26.6 (C-6), 23.6 (C-15), 18.8 (C-14).
Antifungal activity
Antifungal susceptibilities of compounds 1–3 and 8 were evaluated on the following fungi: Candida albicans (ATCC 66–390), Candida glabrata (LMA 9061085) and Aspergillus fumigatus (CBS 113–26). They were previously cultured on yeast peptone dextrose agar at 37°C during 48 h for yeasts and 72 h for Aspergillus. For all the compounds, a modified disk diffusion method was used [Citation22]. Briefly, compounds were dissolved in DMSO and 250 μg aliquots were applied on 12 mm diameter paper disks (ref 06234304, Prolabo 33173 Gradigan). After evaporating the solvent, disks were placed in the center of 90 mm-diameter casitone Petri dishes previously flooded with 10 ml of spore suspensions. Aspergillus suspension was prepared by fragmenting the culture in sterile distilled water with a ground-glass grinder and the fungal suspensions were finally adjusted spectrophotometrically to an A450 of 0.6 [Citation23]. A positive control was made with amphotericin B paper disk and a negative control with drug-free DMSO.
Results and discussion
Chemistry
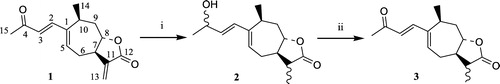
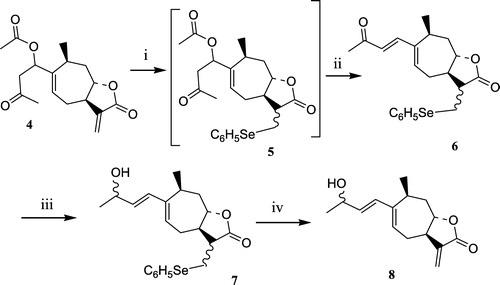
The reduction of the α-methylene-γ-lactone function leading to the α-methyl-γ-lactone series could be expected by the use of a soft reducing agent like NaBH4 [Citation24]. In the case of xanthatin, such conditions would also lead to the reduction of the ketonic carbonyl group. Indeed, low temperature reduction (NaBH4, MeOH, − 78°C) of 1 yielded the alcohol 2, however in an unseparable mixture with the mono reduced alcohol 8, in a 55/45 ratio. By an increase of the temperature (NaBH4, MeOH, 0°C), 2 was obtained as the sole product (Scheme ). Obtaining the desired Michael acceptor 3 in the α-methyl-γ-lactone series then necessitated reoxidation of the allylic alcohol 2 which was realized under Ganem conditions (DDQ) [Citation25]. Keeping in mind the prone reduction of the α-methylene-γ-lactone function, we reasoned that the synthesis of derivatives only reduced on the lateral side chain would necessitate protection transiently of the methylenic double bond. Therefore, the preparation of a phenylselenide adduct 5 was envisaged (Scheme ) [Citation26]. Its synthesis was realized in an unacceptable low yield (23%) when the sodium phenylselenoate addition was directly performed on 1. The synthesis of 5 was optimized when 1 was replaced by xanthinin 4, a natural product isolated from X. macrocarpum, but bearing only one electrodeficient double bond. The phenylselenide intermediate 5 thus obtained was unstable, yielding the elimination product 6 during its chromatography [Citation27]. Therefore, 5 was not purified but treated with SiO2 to yield 6 in a good 80% overall yield. Reduction of the ketonic side chain was then realized leading to the allylic alcohol 7. Finally, a gentle elimination of the corresponding selenoxide (H2O2, 0°C) gave the desired Michael acceptor 8.
Scheme 1 Synthesis in the α-methyl-γ-lactone series: compound 3. Reagents: (i) NaBH4, MeOH; (ii) DDQ, Toluene.
Scheme 2 Synthesis in the α-methylene-γ-lactone series: compound 8. Reagents: (i) Ph2Se2, NaBH4, EtOH; (ii) SiO2; (iii) NaBH4, MeOH; (iv) H2O2, CH2Cl2.
Pharmacology
The antifungal activity of xanthatin 1 and its reduced derivatives 2, 3 and 8 was evaluated in vitro against three strains of fungal species, Candida glabrata, C. albicans and Aspergillus fumigatus. 1 and 8 revealed an antifungal activity similar to the positive control (amphotericin B). Comparison of the antifungal activity of xanthatin 1, (active against the 3 strains of fungi), with its α-methyl-γ-lactone derivative 3, lacking of any antifungal activity at a dose of 250 μg, clearly indicated out the essential role of the α-methylene-γ-lactone function as the support of the biological response. The same conclusion also arose when the antifungal activity of the allylic alcohol 8 and its dihydro derivative 2 were compared. Therefore, the influence of the lateral side chain on the antifungal activity was obviously determined in the α-methylene-γ-lactone series, when the efficacy of xanthatin 1 and of the allylic alcohol 8 were evaluated. Both compounds inhibited the development of the three strains, however the antifungal activity of the allylic alcohol 8 was always lower compared to xanthatin 1. The highest antifungal activity of 1 may rely on the alkylating properties of the unsaturated ketonic side chain, but the slight difference in antifungal activity may also be explained by other parameters such as a difference in the ability to cross the fungal cell wall.
Table I. In vitro antifungal activity of the tested compounds expressed by measurement of the inhibition diameter of the growth of yeasts (in mm) after inoculums of 250 μg.
In conclusion, an efficient chemical synthesis of xanthatin derivatives was realized for the first time and represented an access to the natural product 3 [Citation28]. Moreover, the evaluation of the antifungal activity of xanthatin 1 and its reduced derivatives in the α-methyl-γ-lactone series clearly indicated the prime role of the α-methylene-γ-lactone in the biological activity, probably due to alkylating properties [Citation19]. The superior in vitro antifungal activity of the unsaturated ketone 1 compared to the allylic alcohol 8, could be explained by their difference of lipophilicity. In fact, a low polarity seems to be required for sesquiterpenes lactones to pass through the fungal cell wall [Citation29]. More biological investigations would therefore be needed in order to determine the target of these sesquiterpenic lactones and to explain the contribution of the unsaturated ketonic side chain to the antifungal activity of xanthatin.
Related Research Data
References
- Groll AH, Shah PM, Mentzel C, Schneider M, Just-Nuebling G, Huebner K. J Infect 1996; 33: 23–32
- Harbarth S, Pestotnik SL, Lloyd JF, Burke JP, Samore MH. Am J Med 2001; 111: 528–534
- Gearhart MO. Ann Pharmacother 1994; 28: 1177–1181
- Pea F, Furlanut M. Clin Pharmacokinet 2001; 40: 833–868
- Lin SJ, Schranz J, Teutsch SM. Clin Inf Dis 2001; 32: 358–366
- Vicente MF, Basilio A, Cabello A, Pelaez F. Clin Microbiol Infect 2003; 9: 15–32
- Kartsonis NA, Nielsen J, Douglas CM. Drug Resist Update 2003; 6: 197–218
- Oger JM, Richomme P, Guinaudeau H, Bouchara JP, Fournet A. J Essent Oil Res 1994; 6: 493–497
- Morel C, Séraphin D, Teyrouz A, Larcher G, Bouchara JP, Litaudon M, Richomme P, Bruneton J. Planta Med 2002; 68: 41–44
- Larcher G, Morel C, Tronchin G, Landreau A, Séraphin D, Richomme P, Bouchara JP. Planta Med 2004; 70: 569–571
- Oger JM, Morel C, Helesbeux JJ, Litaudon M, Séraphin D, Dartiguelongue C, Larcher G, Richomme P, Duval O. Nat Prod Res 2003; 17: 195–199
- Helesbeux JJ, Guilet D, Séraphin D, Duval O, Richomme P, Bruneton J. Tetrahedron Lett 2000; 41: 4559–4562
- Lavault M, Landreau A, Larcher G, Bouchara JP, Pagniez F, Le Pape P, Richomme P. Fitoterapia 2005, in press
- Lavault M, Bruneton J. Ann Pharm Fr 1979; 37: 59–63
- Sato Y, Oketani H, Yamada T, Singyouchi KI, Ohtsubo T, Kihara M, Shibata H, Higuti T. J Pharm Pharmacol 1997; 49: 1042–1044
- Joshi SP, Rojatkar SR, Nagasampagi BA. J Med Aromat Plant Sci 1997; 19: 366–368
- Cui B, Lee YH, Chai H, Tucker JC, Fairchild CR, Raventos-Suarez C, Long B, Lane KE, Menendez AT, Beecher CWW, Cordell GA, Pezzuto JM, Kinghorn AD. J Nat Prod 1999; 62: 1545–1550
- Kim YS, Kim JS, Park SH, Choi SU, Lee CO, Kim SK, Kim YK, Kim SH, Ryu SY. Planta Med 2003; 69: 375–377
- Bruneton J. Sesquiterpenoid lactones. Pharmacognosy: Phytochemistry medicinal plants2nd ed., J Bruneton. Intercept Ltd., London, Paris, New York 1999; 619–636
- Nosse B, Chhor RB, Jeong WB, Böhm C, Reiser O. Org Lett 2003; 5: 941–943
- Rudler H, Parlier A, Certal V, Lastennet G, Audouin M, Vaissermann J. Tetrahedron Lett 2004; 45: 2409–2411
- Barry AL, Brown SD. J Clin Microbiol 1996; 34: 2154–2157
- Korting HC, Ollert M, Abeck D. Antimicrob Agents Chemother 1995; 39: 1206–1208
- Biswanath D, Venkataiah B, Kashinatham A. Tetrahedron 1999; 55: 6585–6594
- McKittrick BA, Ganem B. J Org Chem 1985; 50: 5897–5898
- Barbetti P, Fardella G, Chiappini I, Scarcia V, Furlani-Candiani A. Eur J Med Chem 1989; 24: 299–305
- Majewski M, Lasny R. J Org Chem 1995; 60: 5825–5830
- Mahmoud AA. Planta Med 1998; 64: 724–727
- Barrero AF, Oltra JE, Alvarez M, Raslan DS, Saude DA, Akssira M. Fitoterapia 2000; 71: 60–64