Abstract
Thymidine phosphorylase (EC 2.4.2.4) catalyses the reversible phosphorolysis of pyrimidine 2′-deoxynucleosides, forming 2-deoxyribose-1-phosphate and pyrimidine. 5-Chloro-6-(2-imino-pyrrolidin-1-yl)methyl-uracil hydrochloride (TPI, 1) and its 5-bromo analogue (2), 6-(2-amino-imidazol-1-yl)methyl-5-bromo-uracil (3) and its 5-chloro analogue (4) act as tight-binding stoichiometric inhibitors of recombinant E. coli thymidine phosphorylase, and thus can be used as the first active-site titrants for it using either thymidine or 5-nitro-2′-deoxyuridine as substrate.
Introduction
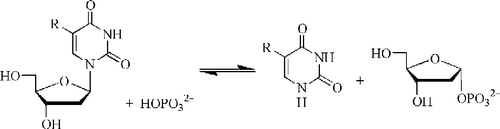
Thymidine phosphorylase (TP, EC 2.4.2.4) catalyses the reversible phosphorolysis of (substituted)uracil 2′-deoxynucleosides () forming 2-deoxyribose-1-phosphate and (substituted)uracil, thus providing both catabolic and salvage routes for pyrimidines. It also provides such metabolic routes for anti-pyrimidine therapeutic agents, such as anticancer drugs (e.g. 5-fluorouracil) and antivirals. The 2-deoxyribose-1-phosphate is rapidly dephosphorylated in the cell to produce the angiogenic factor 2-D-deoxyribose (also known as endothelial cell chemoattractant), which is then exported from the cell [Citation1].
Figure 1 The reaction catalysed by thymidine phosphorylase.
TP is attracting attention as a cancer target as it plays a role in tumour angiogenesis Citation2-5, and is expressed at higher levels in the plasma of patients with cancer and in solid tumours relative to normal tissues. Inhibitors of TP inhibit angiogenesis and metastasis as well as promoting apoptosis [Citation1,Citation2,Citation4,Citation6,Citation7]. Inhibitors of TP also potentiate the actions of anticancer and antiviral pyrimidine-based drugs; for example, 5-fluoro-2′-deoxyuridine, (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU) and 5-trifluoromethyl-2′-deoxyuridine are metabolised and deactivated by TP: therefore coadministration with a TP inhibitor enhances their cytotoxicity or antiviral activity [Citation2,Citation8]. TP inhibition, however, can reduce the therapeutic effects of some prodrugs: 5-fluorouracil has to be converted to its deoxynucleotide in order to inhibit thymidylate synthase and TP-catalysed deoxyribosylation provides one route towards this entity. TPI (1) (), the most potent inhibitor of TP widely investigated to date, is reported to inhibit the growth of human KB epidermoid carcinoma cells and decrease the growth rate of experimental tumours in mice [Citation9].
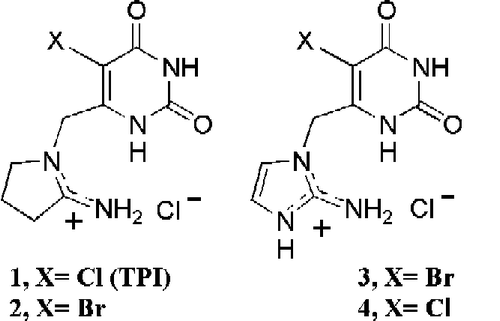
Figure 2 Active-site titrant inhibitors of E.coli thymidine phosphorylase.
Inhibition studies in this area were boosted by the solution of the X-ray crystal structures for TP from both Escherichia coli [Citation10] and Bacillus stearothermophilus [Citation11]. Knowledge of the three-dimensional structure has led to the report of novel multisubstrate analogue inhibitors of the E. coli enzyme [Citation12,Citation13], as well as the rationalisation of the strong inhibition by TPI (1) of human TP [Citation14]. However, the problem is complicated by the conformational dynamism of TP [Citation15,Citation16]. Recently an X-ray diffraction study of human TP has led to the determination of the structure of TPI bound to human TP in a closed conformation [Citation17]. The structure does not have phosphate ion (none was present in the crystallization solution) or any diffusible anion in the expected location, somewhat restricting its interpretation in terms of possible active site geometry. TPI is reported as a competitive inhibitor [Citation18] of human TP, with a Ki value of 17 nM.
During our studies [Citation14,Citation19] of TP inhibitor design we determined the inhibition kinetics of TPI (1) and a novel aromatic analogue 3 towards TP from E. coli in detail, as well as those for analogues 2 and 4 (). For these inhibitors we found extremely tight, stoichiometric inhibition of E. coli TP, effectively irreversible under the assay conditions used. Thus, this study provides evidence for the first active-site titrants for the E. coli enzyme under suitable concentration conditions.
Materials and methods
Reagents were of highest quality available. Thymidine phosphorylase (recombinant E. coli (T 2807)) and thymidine were from Sigma-Aldrich Ltd. (Poole, U.K). 2′-Deoxyuridine was from Lancaster Research Chemicals (Morecambe, U.K). All water used was distilled and purified by ion exchange and charcoal using a MilliQ system (Millipore Ltd). Melting points (corrected) were determined on a Köfler melting point apparatus microscope (Reichert, Austria).
Current assay methods for TP are single point, and typically involve measuring thymidine and thymine levels through HPLC [Citation20], radioisotopes [Citation21] or single-point spectrophotometric [Citation22] methods in alkaline solution. Therefore, we adapted a published spectrophotometric [Citation23,Citation24] method of the conversion of 5-nitro-2′-deoxyuridine to 5-nitrouracil at 355 nm (, R = NO2) to multi-well monitoring. Diacetylation of 2′-deoxyuridine to 3′,5′-diacetyl-2′-deoxyuridine, m.p.107–110°C (Lit. [Citation25] m.p. 108–109 °C), followed by transfer nitration of this via N-nitropyrazole, and deacetylation by sodium methoxide, gave 5-nitro-2′-deoxyuridine [Citation26] as a pale-yellow solid: m.p. 149–151 °C (lit.[Citation26] m.p. 154–155 °C). TPI (1) and (2) were synthesized from 5-halo-6-(chloromethyl)uracil and 2-iminopyrrolidine [Citation8]. Compounds (3) and (4) were prepared by reaction of 5-halo-6-(chloromethyl)uracil with the potassium salt of 2-nitroimidazole followed by reduction of the nitro group [Citation19,Citation24].
Enzyme rate spectral scans, studies of stoichiometric inhibition and absorbance readings at fixed wavelengths were conducted using a Peltier-thermostatted cuvette holder in a Cary 4000 UV-visible spectrophotometer and the Cary Enzyme Kinetics Software. Assays used either thymidine or 5-nitro-2′-deoxyuridine as substrate. The assay mixture contained (in 1 ml) 20 μM thymidine, 0.1 M potassium phosphate (pH 7.4), and limiting amounts of TP (ca.0.00177 units per assay). The reaction was initiated by addition of enzyme and the change in absorbance was monitored at 265 nm at 25 °C [Citation27]. For some substrate and inhibition studies a thermostatted 96-well spectrophotometer (Molecular Devices, Spectramax®) instrument with pathlength calibration was used. In this case assays were conducted at 355 nm, 25 °C in 0.2 mL total volume containing 0.13 mM 5-nitro-2′-deoxyuridine, 0.1 M potassium phosphate (pH 7.4), and limiting amounts of TP (ca.0.00883 units per assay). Reaction was initiated by addition of TP. Enzyme kinetics data were analysed using Grafit version 3 software (Erithacus software). Protein concentrations were determined initially by the Coomassie Blue binding method [Citation28], but once the discovery had been made that the active site concentration of TP could be determined by direct titration with certain tight binding inhibitors, stock TP concentrations (E. coli) were routinely determined by such titrations.
Results
Repetitive spectral scanning of reacting mixtures of thymidine or 5-nitro-2′-deoxyuridine in 0.1 M phosphate (pH 7.4) in the presence of recombinant E.coli TP at 25 °C showed that convenient wavelengths to follow the enzyme-catalyzed reactions were 265 nm (Δε 9300 M− 1 cm− 1) and 355 nm (Δε 7482 M− 1 cm− 1), respectively (changes in extinction coefficient for the respective reactions are given in parentheses). The reactions followed Michaelis–Menten kinetics for both substrates. Values [Citation24] of kcat, Km and kcat/Km were 1775 ± 68 s− 1, 0.24 ± 0.02 mM and 7.4 × 106 M− 1 s− 1, respectively, for thymidine and 20 ± 0.6 s− 1, 0.23 ± 0.02 mM and 8.7 × 104 M− 1 s− 1, respectively, for 5-nitro-2′-deoxyuridine. The nitro group decreases the value of kcat by 89-fold compared to the methyl group, but has little effect on Km. The value of Km for thymidine measured for enzyme isolated from E.coli is 0.38 mM [Citation29], and a value of 0.6 mM has been quoted for recombinant E. coli TP [Citation12]. We used both substrates, the nitro substrate sometimes for its spectral convenience (it is usable in 96-well polystyrene plates and when stabilizing BSA was present at high concentration) and at other times because its lower kcat allows one to avoid using fast reaction methodologies that would be needed if thymidine was substrate and the free-enzyme concentration was greater than ∼20 nM.
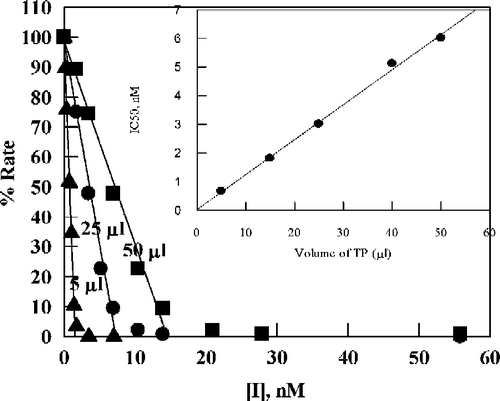
When percent activity using thymidine as substrate was plotted against TPI concentration, the activity went to zero at high inhibitor concentrations (). The apparent IC50 values were found to depend on the concentration of enzyme, indicating that a process other than simple inhibition is occurring. Straight lines plotted through the data in the activity range 10–100% give intercepts on the [TPI] axis that are proportional to the amount of enzyme added (shown as the inset to : the experimental results are shown as they were performed, using volumes, as only later were we able to relate volume to active enzyme concentration).
Figure 3 Plot of percent activity for E. coli TP versus concentration of TPI for various added enzyme volumes (indicated by each line in μL), using 20 μM thymidine as substrate in 0.1 M potassium phosphate (pH 7.4) at 25 °C. The straight lines are by linear regression analysis of data corresponding to the region 10–100% activity: the lines were used to determine apparent IC50 values. The inset is a plot of the apparent IC50 values versus the amount of E. coli TP added (the line is by linear regression analysis).
The simplest explanation is very tight binding. To assess this, a mixture of enzyme and TPI (1:1.4 molar ratio) was gel-filtered over a HiPrep 26/10 column (2.6 × 10 cm, void volume 15 mL, bed volume 53 mL, Sephadex G-25) eluted with 0.1 M (pH 7.4) phosphate buffer at room temperature. The activity of the eluted protein was 2.5 ± 0.5% of control similarly treated in the absence of TPI. Thus, the inhibition is sufficiently tight not to be reversed by gel filtration under these conditions. A separate calibration experiment was performed using the competitive inhibitor 6-amino-5-bromouracil (for which we determined the linear competitive Ki value to be 2.0 ± 0.2 μM under these conditions) and 93.3 ± 7.8% of enzyme activity was recovered after gel filtration, consistent with the rapid-equilibrium reversible inhibition kinetics of this compound and confirming the validity of the separation procedure. However, when elution of the TP/TPI complex was performed with 0.1M Tris/HCl buffer at pH 7.4 (enzyme/inhibitor mixture applied in phosphate-buffer), the outcome depended on eluant flow rate: at a flow rate of 0.5 mL/min and a 1.4-fold excess of TPI over TP, the TP activity after gel filtration was 40.0 ± 3.0% of control; under identical conditions, but at a flow rate of 0.1 mL/min, the TP activity recovered from the column was identical to control. Thus in Tris buffer the inhibition by TPI is fully reversible if an adequate time is allowed for the column step, but in phosphate buffer the inhibition was not reversed under the conditions used. In molecular modeling studies, phosphate, modeled as the dianion, makes a major contribution to the binding energy of TPI through direct hydrogen bonding with the cationic NH2 group of the protonated iminopyrrolidine, and through the very strong electrostatic attraction of the contact dianion-cation pair. During gel filtration, phosphate ion can diffuse out of the enzyme active site and become separated from the protein. This allows easier release of TPI since chloride anion, from the Tris/HCl buffer, is unlikely to substitute for phosphate: the TP/TPI supernatant of the crystals used in the X-ray diffraction study of human TP [Citation17] contained chloride, but neither chloride, nor any other diffusible anion, was present in the protein-enclosed TPI binding site. The inhibition by the imidazole analogue 3 was also shown to be irreversible by this gel filtration criterion: the TP activity recovered was 0.52 ± 0.13% from a mixture of 60 μM TP and 78 μM 3 using 0.1 M phosphate buffer for elution).
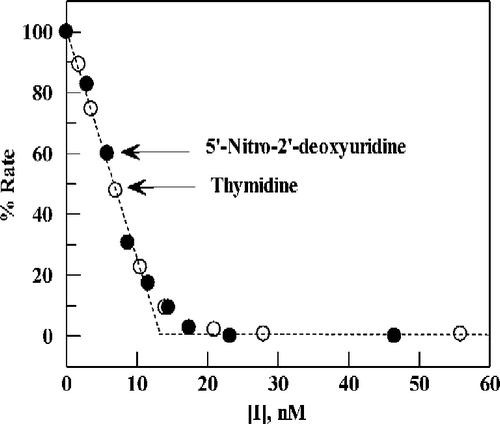
Data for TPI inhibition using thymidine as substrate were plotted on the same scale as those for 5-nitro-2′-deoxyuridine () by normalizing the velocities at zero inhibitor concentration to 100. Clearly for these two different substrates the data can be superimposed, indicative of tight inhibition for each of the substrate conditions used.
Figure 4 Plot of percent activity for E. coli TP versus the concentration of TPI, for substrates 5-nitro-2′-deoxyuridine (0.13 mM) (∘) or thymidine (20 μM)(•) in 0.1 M potassium phosphate (pH 7.4) at 25 °C. Points are experimental; the dashed line (angled, then horizontal) is the theoretical result expected for stoichiometric, instantaneous inhibition of enzyme present at 13 nM.
Further evidence that both TPI and 3 act as tight-binding stoichiometric inhibitors was obtained by using these compounds to titrate the same stock solution of TP. The titration inhibition-data could be superimposed for these inhibitors of different structures (), indicating that both were acting as tight-binding inhibitors. We have also studied the 5-bromo analogue of TPI (2) as well as the chloro (4) analogue of 3. Both 2 and 4 were also found to exhibit stoichiometric inhibition of the E. coli enzyme.
Figure 5 Inhibition of TP by TPI and 3. Percent activity is plotted versus concentrations of TPI and 3 using the same stock solution of TP and 0.13 mM 5-nitro-2′-deoxyuridine as substrate in 0.1 M potassium phosphate buffer at pH 7.4 and 25 °C.
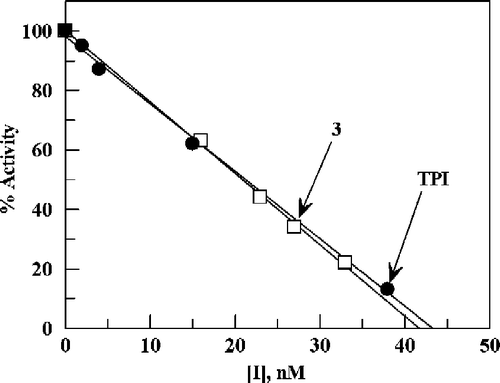
Note: it is important to point out that in some batches of enzyme the inhibition plots of did not level off at zero activity, but showed a real residual activity of a few percent. We have been unable to elucidate the origin of this, which presumably arises from some component of the enzyme preparation supplied. Pre-incubation of the enzyme with TPI or increasing the TPI concentration to 1 μM did not abolish the residual activity. Whilst it confounds the absolute rigor of the active-site titration process, the titration experiment itself alerts one to this background process in a particular sample. In practice, the great steepness of the titration curve means that the active enzyme concentration determined by extrapolation to zero activity even in such a (low) background situation is very close to the true value.
In separate studies [Citation24] we have determined the pKa value of the N(1)-H site in TPI to be 6.14 ± 0.04 by UV spectrophotometric titration, making it likely that the form of TPI that binds to the active site is the zwitterion. This zwitterion can adopt a conformation which mimics the shape and charge distribution of some of the elements of the putative transition state, consistent with our earlier proposals [Citation14,Citation19,Citation24], which although shown with oxacarbenium ion character still clearly involved transition-state interaction of both attacking and departing groups.
It is not yet possible to get an accurate value of Ki from the data for TPI or 3 because of the stoichiometric nature of the inhibition concentration-profile, and the sensitivity of the assay meaning that the enzyme concentration cannot be decreased to the region of the putative Ki value. We can estimate that the IC50 must be at least an order of magnitude lower than 0.6 nM, the lowest value determined in the active site determinations (enzyme concentration 1.2 nM in ), giving a Ki value of < 0.06 nM. Birck and Schramm [Citation30] have estimated that Km/Ki(TPI) for human TP is only 3 × 103 making TPI a relatively poor mimic of the transition-state. Our estimate of Km/Ki(TPI) for E. coli TP is >4 × 106 (this is an underestimate in view of the means that had to be used to set limits on the value of Ki). This is consistent with partial transition state mimicry as its mode of action against the E. coli enzyme. Comparable studies using TPI and 3 with human TP (both for an enzyme sample from Sigma-Aldrich, and for a glutathione transferase-fusion TP protein from AstraZeneca PLC) indicated that these inhibitors did not show stoichiometric inhibition behaviour: plots of percent inhibition versus inhibitor concentration did not show linear titration regions, and plots of inhibition data followed linear competitive kinetics (Sigma-Aldrich enzyme tested), consistent with the literature reports for TPI [Citation18] with human TP (Ki 17 nM). It may be that TPI binds to human and E. coli TP using a fundamentally different conformational structure (for TPI and/or enzyme) or that TPI binding follows a different pathway for the two species through the conformational changes that this enzyme is known to undergo (this would modify the true Ki values by a function reflecting the conformational equilibria). Alternatively, the difference may be merely apparent, the binding interactions simply being quantitatively better for one species than the other, but essentially the same qualitatively. In the human enzyme X-ray defined structure[Citation17], the hydrogen atoms (added by computer modelling software) and the carbon atom of the TPI C6-methylene group, are in too-tight or very-tight contact with surrounding enzyme residues. The resulting pressure and its partial relief has pushed the opposite side of the uracil ring into very-tight contact with the hydrogen-bonding groups of the enzyme that interact with the exocyclic oxygens and, particularly, N3-H. The binding site in the E. coli enzyme is likely to be more relaxed across those dimensions since it accommodates inhibitors with very much larger groups at C6 than does a mammalian enzyme [Citation31]. An indication of greater steric problems in the region around uracil C6 for the human enzyme compared to E. coli comes from comparing IC50 ratios for pairs of inhibitors using recently [Citation24] published data. For 6-(4′-amino-1′-imidazolylmethyl)-5-X-uracils, which are non-tight binding analogues of TPI (and corresponding 2-aminoimidazoles) and probably bind to the enzyme in a similar way, the human/E. coli IC50 ratio is smallest with X = hydrogen, increases for X = chlorine and is largest for X = bromine. The ratios mirror the sizes of the 5-substituents. Note this is not the case for the 2- or 4-nitroimidazole analogues (they are much too weakly basic to be protonated at pH 7.4), which we believe to be bound to an open form of the enzyme [Citation24]. The correlation with size in the (partially-protonated) 4-aminoimidazole case is suggested to arise because the smaller C5-substituents allow the C6-methylene to be pushed towards the ‘C5-X space’ in the human enzyme, thereby reducing adverse steric congestion. This subtle change in size of the active-site pocket may underlie the apparent species recognition difference for TPI.
| Abbreviations | ||
| TP: | = | thymidine phosphorylase (EC 2.4.2.4) |
| TPI: | = | 5-chloro-6-(2-imino-pyrrolidin-1-yl)methyl-uracil hydrochloride |
Acknowledgements
We are grateful to Dr Simon Barry of AstraZeneca PLC (UK) for helpful discussions, access to the X-ray data for the human enzyme and for supplies of human TP as a glutathione transferase construct, and to the Libyan Cultural Affairs Office (London) for financial support (AG).
References
- Brown NS, Bicknell R. Biochem J 1998; 334: 1–8
- Cole C, Foster AJ, Freeman S, Jaffar M, Murray PE, Stratford IJ. Anti-Cancer Drug Design 1999; 14: 383–392
- Toi M, Atiqur Rahman M, Bando H, Chow LWC. Lancet Oncol 2005; 6: 158–166
- Focher F, Spadari S. Curr Cancer Drug Targets 2001; 1: 141–153
- Akiyama SI, Furukawa T, Sumizawa T, Takebayashi Y, Nakajima Y, Shimaoka S, Haraguchi M. Cancer Sci 2004; 95: 851–857
- Folkman J. J Natl Cancer Inst 1996; 88: 1091–1092
- Griffiths L, Stratford IJ. Br J Cancer 1997; 76: 689–693
- Yano S, Kazuno H, Sato T, Suzuki N, Emura T, Wierzba K, Yamashita JI, Tada Y, Yamada Y, Fukushima M, Asao T. Bioorg Med Chem 2004; 12: 3443–3450
- Matsushita S, Nitanda T, Furukawa T, Sumizawa T, Tani A, Nishimoto K, Akiba S, Miyadera K, Fukushima M, Yamada Y, Yoshida H, Kanzaki T, Akiyama Si. Cancer Res 1999; 59: 1911–1916
- Walter MR, Cook WJ, Cole LB, Short SA, Koszalka GW, Krenitsky TA, Ealick SE. J Biol Chem 1990; 265: 14016–14022
- Pugmire MJ, Ealick SE. Structure 1998; 6: 1467–1479
- Esteban-Gamboa A, Balzarini J, Esnouf R, De Clercq E, Camarasa MJ, Perez-Perez MJ. J Med Chem 2000; 43: 971–983
- Liekens S, Bilsen F, De Clercq E, Priego EM, Camarasa MJ, Perez-Perez MJ, Balzarini J. FEBS Lett 2002; 510: 83–88
- Cole C, Marks DS, Jaffar M, Stratford IJ, Douglas KT, Freeman S. Anti-Cancer Drug Des 1999; 14: 411–420
- Pugmire MJ, Cook WJ, Jasanoff A, Walter MR, Ealick SE. J Mol Biol 1998; 281: 285–299
- Rick SW, Abashkin YG, Hilderbrandt RL, Burt SK. Poteins: Structure, Function, and Genetics. 1999; 37: 242–252
- Norman RA, Barry ST, Bate M, Breed J, Colls JG, Ernill RJ, Luke RWA, Minshull CA, McAlister MSB, McCall EJ, McMiken HHJ, Paterson DS, Timms D, Tucker JA, Pauptit RA. Structure 2004; 12: 75–84
- Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, Yamada Y, Asao T. Biochem Pharmacol 2000; 59: 1227–1236
- Cole C, Reigan P, Gbaj A, Edwards PN, Douglas KT, Stratford IJ, Freeman S, Jaffar M. J Med Chem 2003; 46: 207–209
- Nuessler V, Pogrebniak A, Pelka-Fleischer R, Boos K, Wilmanns W. Chromatographia 1999; 49: 173–178
- Gan TE, Hallam L, Pilkington GR, Van der Weyden MB. Clin Chim Acta 1981; 116: 231–236
- Friedkin M, Roberts D. J Biol Chem 1954; 207: 245–256
- Wataya Y, Santi DW. Anal Biochem 1981; 112: 96–98
- Reigan P, Edwards PN, Gbaj A, Cole C, Barry ST, Page KM, Ashton SE, Luke RWA, Douglas KT, Stratford IJ, Jaffar M, Bryce RA, Freeman S. J Med Chem 2005; 48: 392–402
- Robbins D, Odom OW, Lynch J, Kramer G, Hardesty B, Liou R, Ofengand J. Biochemistry 1981; 20: 5301–5309
- Giziewicz J, Wnuk SF, Robins MJ. J Org Chem 1999; 64: 2149–2151
- Nakayama C, Wataya Y, Meyer RB, Jr, Santi DV, Saneyoshi M, Ueda T. J Med Chem 1980; 23: 962–964
- Bradford MM. Anal Biochem 1976; 72: 248–255
- Schwartz M. Eur J Biochem 1971; 21: 191–198
- Birck MR, Schramm VL. J Am Chem Soc 2004; 126: 2447–2453
- Baker BR, Hopkins SE. J Med Chem 1970; 13: 87–89