Abstract
Aldose reductase (AR) is implicated to play a critical role in diabetes and cardiovascular complications because of the reaction it catalyzes. Our data reveal that peroxisome proliferator WY 14,643, follows a pure non-competitive inhibition pattern in the aldehyde reduction activity as well as in the alcohol oxidation activity of AR. This finding communicates for the first time a novel feature of WY 14,643 in regulating AR activity. In addition, this observation indicates that AR, AR-like proteins and aldo-keto reductase (AKR) members may be involved in the WY 14,643 mechanism of action when it is administered as PPAR agonist.
Introduction
One of the major physiological roles of aldose reductase (AR) is to produce a non-diffusible osmolyte in cells exposed to hypertonicity, as characterized by the renal medullary cells of the loop of Henle [Citation1]. AR plays an important role in the pathogenesis of a variety of diabetic complications, such as cardiovascular diseases, retinopathy, neuropathy and nephropathy. These complications are affected by abnormalities in glucose metabolism, such as an increase in polyol pathway flux mediated by AR under elevated blood glucose conditions Citation2-6. The conversion of glucose to sorbitol is catalyzed by AR in polyol pathway. Sorbitol that is synthesized by AR is transformed to fructose by the catalysis of sorbitol dehydrogenase (SDH). Under normal conditions only about 3% of glucose enters the polyol pathway because a majority of the cellular glucose is phosphorylated by hexokinase to glucose 6-phosphate [Citation7]. However, under hyperglycemic conditions, the increased flux of glucose through the polyol pathway accounts for as much as one-third of the total glucose turnover because ambient glucose saturates the hexokinase [Citation1, Citation5, Citation8]. The marked rise in intracellular glucose seen in the diabetes mellitus condition causes increased production of sorbitol [Citation1, Citation9]. This increased flux and accumulation of sorbitol is damaging to cells and may result in some of the long-term complications of diabetes Citation1 Citation10-12.
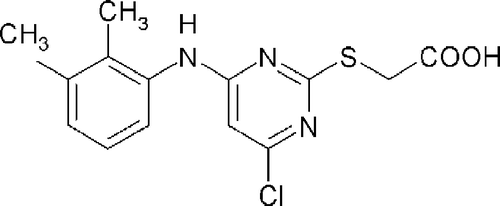
In particular the polyol pathway has been implicated in playing a role in myocardial ischemia-reperfusion injuries Citation13-15. Heart failure is a significant cause of morbidity and mortality in patients with diabetes [Citation16] and it's incidence is increased 2.4-fold in diabetic men and 5.1-fold in diabetic women [Citation17]. Although, many of these patients have coexistent hypertension or coronary artery disease, diabetes further predisposes them to the development of heart failure [Citation17, Citation18]. Diabetic patients also are more prone to develop heart failure as a result of acute myocardial infarction [Citation19, Citation20]. WY 14,643 () is a specific peroxisome proliferator-activated receptor α (PPARα) agonist with strong hypolipidemic effects. Here we show that WY 14,643 () is capable of inhibiting human AR activity.
Figure 1 Chemical structure of 4-Chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid, also known as WY 14,643, Pirinixic acid.
Materials and methods
Reagents, 2-mercaptoethanol, 3-acetylpyridine adenine dinucleotide phosphate (3-APADP+), DL-glyceraldehyde, imidazole, isopropyl β-D-thiogalactoside (IPTG) and WY 14,643 were obtained from Sigma. The source of all the other reagents are specified in parenthesis after their name.
Over-expression of human AR
Recombinant human AR (hAR) with his-tag was cloned into the bacterial expression vector pET15b (Novagen) and was expressed in E. coli BL21 cells by growing the culture in Luria-Bertani broth supplemented with ampicillin (50 μg/mL) at 37°C. AR was induced by the addition of 1 mM IPTG after the culture reached an attenuance (A600) of 0.7 − 0.8. The cells were harvested by centrifugation, washed, re-suspended in 50 mM sodium phosphate buffer (pH 7.0) containing 300 mM NaCl and 1 mM 2-mercaptoethanol, and lysed by sonication for 10 min. The centrifuged supernatant of the sonicated cell lysate was passed through 5 mL of Talon metal affinity matrix (Clontech), and eluted with 50 ml of 50 mM sodium phosphate buffer (pH 7.0) containing 300 mM NaCl, 1 mM 2-mercaptoethanol and 150 mM imidazole. The eluted protein was dialyzed overnight at 4°C in 50 mM sodium phosphate buffer (pH 7.0) containing 1 mM 2-mercaptoethanol, and the his-tag was removed using the thrombin cleavage kit (Novagen) as per the manufacturer's instruction. The his-tag removed-hAR was further purified by passing it through the DEAE Sephadex A25 column. As determined by the SDS-PAGE and enzyme activity the eluates that contained the AR were pooled and concentrated using Amicon Ultra 10,000 MW cutoff membrane tubes, and the concentration of the enzyme was adjusted to 100 μM using 20 mM Tris buffer (pH 8.5). Coomassie blue-stained SDS-PAGE gels along with the enzyme activity was used to evaluate the purity of the protein at each stage of purification. AR concentration was quantified by the Bradford method [Citation21].
Enzyme kinetics of aldehyde reduction activity by AR
Aldehyde reduction activity catalyzed by AR was determined at 25°C by the rate of decrease in the absorption of NADPH at 340 nm using a Beckman DU600 model spectrophotometer [Citation22]. Activity of the enzyme is represented as μmoles of NADPH oxidized/min/l. The assay mixture contained 0.2 M potassium phosphate buffer (pH 6.2), 0.05–10 mM DL-glyceraldehyde, 0–100 μM WY 14,643 (ethanol was used as the solvent), 0.15 mM NADPH and human AR (0.5 μM, 18 μg/ml).
The inhibition kinetic experiments included multiple inhibitor concentrations of WY 14,643, starting from the maximum concentration of [I]0 = 100 μM. In addition several serial dilutions (50.0, 10.0, 5.0, 1.0, 0.5, and 0.1 μM) as well as the negative control [I]0 = 0 μM, in the presence of six concentrations (0.05, 0.1, 0.5, 1.0, 5.0 and 10.0 mM) of the substrate DL-glyceraldehyde were incorporated. Each individual rate measurement was evaluated in duplicate. At least three independent determinations were performed for each kinetic constant.
Forward reaction enzyme kinetics data analysis
The Michaelis-Menten constant Km, the maximum velocity Vmax for DL-glyceraldehyde reduction, and the inhibition constant Ki for WY 14,643 were calculated. The untransformed kinetic data calculated from the mean of duplicates were fitted to a one-substrate Michaelis-Menten Equation (1). To determine the Ki corresponding to WY 14,643, competitive, non-competitive and uncompetitive models of inhibition were investigated using Equations (2), (3) and (4, respectively (where v = rate of reaction, Vmax = maximum initial velocity for the uninhibited reaction, Km = Michaelis constant, S = substrate concentration, I = inhibitor concentration, Kis = the slope inhibition constant and Kii = intercept inhibition constant). The Ki of WY 14,643 was calculated from the secondary plot of slope values of the double-reciprocal plot versus inhibitor concentration using the GraphPad Software Prism. Goodness of fit was assessed to be 0.9951 using the r2 test.
Enzyme kinetics of alcohol oxidation activity by human AR
Benzyl alcohol (Aldrich) and 3-APADP+ served as the substrate and the cofactor, respectively, to establish the alcohol oxidation activity of hAR. The oxidation of benzyl alcohol was monitored at 25°C as the rate of change in the absorption of 3-APADP+ at 363 nm [Citation23, Citation24] using a Beckman DU600 model spectrophotometer. The assay mixture contained (0.1, 0.5, 1.0, 5.0, 10.0 mM) benzyl alcohol, (0, 0.1, 0.5, 1.0, 5.0, 10.0, 50.0, 100.0 μM) WY 14,643, 0.10 mM 3-APADP+ and 0.5 μM hAR in 0.125 M MES-Tris-Hepes buffer (pH 8.5). The maximum concentration of [I]0 = 100 μM WY 14,643 was used in the experimental design for the reverse reaction. Each individual rate measurement was estimated in duplicate. At least three independent determinations were performed for each kinetic constant. The Michaelis constant Km and the maximum velocity Vmax for benzyl alcohol oxidation, the inhibition constant Ki and the mode of inhibition for WY 14,643 were calculated following the methods described for the aldehyde reduction reaction.
Results and discussion
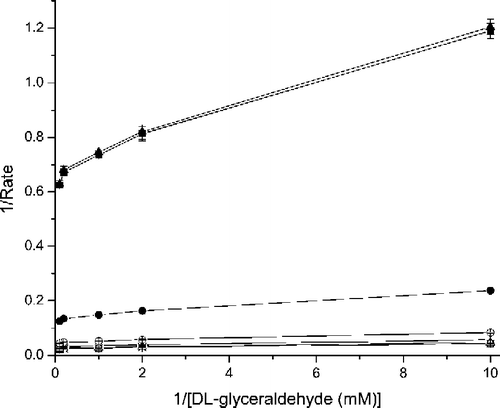
The reaction catalyzed by AR plays a crucial role in conditions such as hyperglycemia. Under high glucose conditions the majority of the glucose follows through the polyol pathway in which the rate determining step is the AR catalyzed reaction. The Km(NADPH, DL-glyceraldehyde) and Kcat DL-glyceraldehyde values for the uninhibited forward reaction catalyzed by hAR were 0.092 ± 0.01 mM and 45.5 ± 7 min− 1, respectively. For WY 14,643, the Kii (1.8 μM) = Kis (1.8 μM) for the forward reaction, indicating that WY 14,643 follows the classical non-competitive pattern of inhibition with respect to the reduction of DL-glyceraldehyde by the recombinant hAR ().
Figure 2 Lineweaver-Burk plot for the rate of reduction. Double reciprocal plot of the rate of reduction of DL-glyceraldehyde in the presence of various concentrations (□ - No inhibitor; × - 0.1 μM; △ − 0.5 μM; + − 1.0 μM; ○ − 5.0 μM; • − 10.0 μM; ▪ − 50.0 μM and ▴ − 100.0 μM) of WY 14,643. Error bar for 10.0 μM curve is not visible because of the size and the shading of the symbol •.
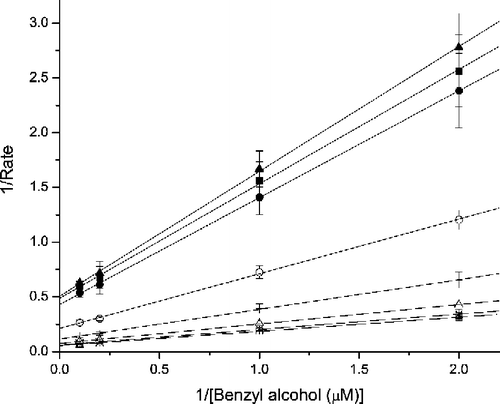
The reverse reaction, or the alcohol oxidation activity catalyzed by hAR, was determined using benzyl alcohol as the substrate and 3-APADP+ as the cofactor. The Km(3-APADP3, benzylalcohol) was 1.72 ± 0.14 mM and the kcat benzylalcohol was 14.4 ± 0.5 min− 1 mM− 1 for the hAR-catalyzed uninhibited reverse reaction (alcohol oxidation). For WY 14,643, Kis and Kii for the reverse direction were 1.67 μM and 1.63 μM, respectively. This implies that the inhibition pattern of WY 14,643 follows a pure non-competitive mode in the reverse reaction ().
Figure 3 Lineweaver-Burk plot for the benzyl alcohol oxidation reaction of AR in the presence of WY 14,643. Double reciprocal plot of the rate of oxidation of benzyl alcohol in the presence of various concentrations (□ - No inhibitor; × − 0.1 μM; △ − 0.5 μM; + − 1.0 μM; ○ − 5.0 μM; • − 10.0 μM; ▪ − 50.0 μM and ▴ − 100.0 μM) of WY 14,643.
The pure non-competitive inhibition pattern shown by WY 14,643 in the forward as well as the reverse reactions catalyzed by AR indicates that complex formation is not affected by the substrate concentration. WY 14,643 could bind to AR or AR√NADPH√DL-glyceraldehyde complex at a location other than the catalytic site. DL-glyceraldehyde binding may be unaltered, but the AR√NADPH√DL-glyceraldehyde√WY 14,643 complex cannot reduce the aldehyde like the AR√NADPH√DL-glyceraldehyde complex. Similarly the AR√NADP+√Benzyl alcohol√WY 14,643 complex will have limitations in oxidizing the alcohol. Considering the fact that both the forward and the reverse reactions catalyzed by AR require a cofactor, WY 14,643 is anticipated to alter the equilibrium between the cofactor-free AR, AR√NADPH complex and AR√NADP+ complex. This suggests that derivatives of WY 14,643 may have inhibitory effects on the activity of AR, AR-like proteins and AKR protein members. WY 14,643 belongs to the class of peroxisome proliferators and is considered a fibrate analog. However the chemical structure of WY 14,643 is unrelated to fibrates with the exception of the carboxylate moiety. We have previously shown that several members of fibrates are capable of inhibiting AR activity [Citation24].
Inhibition of AR activity is known to protect the rat heart from ischemic injury and improve functional recovery upon reperfusion [Citation13, Citation25, Citation26]. Furthermore studies have shown that inhibition of AR increases myocardial glycolysis as well as glucose oxidation in rats. These changes were also associated with attenuation of the cytosolic NADH/NAD+ ratio as well as reducing the obstruction of GAPDH function Citation26-28. It is known that the myocardium is primarily dependent on glycolysis to meet its energy demands during ischemia. Furthermore, improving glucose use is critical for rescuing ischemic myocardium [Citation29, Citation30]. Moreover hypertrophied heart has increased reliance on glucose as a fuel and depresses fatty acid oxidation, which is the dominant energy source for the normal heart Citation31-33. A key regulator of this substrate switching in the heart is postulated to be PPARα [Citation34]. Also, WY 14,643 increases fatty acid oxidation by increasing transcription of genes encoding peroxisomal and mitochondrial fatty acid β-oxidation enzymes [Citation35]. Moreover the ligand of PPARα, WY 14,643, causes a substantial reduction of myocardial infarct size in the rat [Citation36]. A large fraction of potentially eligible diabetic subjects with long-standing diabetes and peripheral neuropathy were found to have baseline cardiac functional abnormalities. This observation highlights the prevalence of cardiac abnormalities in high-risk diabetic subjects without clinically evident cardiac disease. The current study indicates that AR plays a central role in mediating the mechanism of WY 14,643 action when it is used under various scenarios presented by diabetes, its complications and other health conditions. Therefore AR-mediated pathways may be an added target of function for the WY 14,643 class of compounds beyond PPAR intervened action.
Acknowledgements
We thank Scott Kinnes and Steven Novak for their support to our research efforts. Rachel Perez and Richard Y. Calvo were supported through NCI CURE program. This work was supported by funding from the American Diabetes Association.
Notes
1Current address: Johnson & Johnson, San Diego, CA.
References
- Tomlinson DR, Stevens EJ, Diemel LT. Aldose reductase inhibitors and their potential for the treatment of diabetic complications. Trends Pharmacol Sci. 1994; 15: 293–297
- Gabbay KH. The sorbitol pathway and the complications of diabetes. N Engl J Med. 1973; 288: 831–836
- Jaspan J. Pharmacological inhibition of aldose reductase in human diabetic neuropathy. Drugs 1986; 32(Suppl 2)23–29
- Brankston ER, Corriveau R, Laroche C, Roy R, Savard C, Roy D, Kelly R, Forget P, Rivard M, Boulerice F. Effect of AR inhibition on heart rate variability in patients with severe or moderate diabetic autonomic neuropathy. Clin Drug Invest. 1998; 15: 111–121
- Hodgkinson AD, Sondergaard KL, Yang B, Cross DF, Millward BA, Demaine AG. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int. 2001; 60: 211–218
- Galvez AS, Ulloa JA, Chiong M, Criollo A, Eisner V, Barros LF, Lavandero S. Aldose reductase induced by hyperosmotic stress mediates cardiomyocyte apoptosis: Differential effects of sorbitol and mannitol. J Biol Chem. 2003; 278: 38484–38494
- Morrison AD, Clements RS, Jr, Travis SB, Oski F, Winegrad AI. Glucose utilization by the polyol pathway in human erythrocytes. Biochem Biophys Res Commun. 1970; 40: 199–205
- Gonzalez RG, Barnett P, Aguayo J, Cheng HM, Chylack LT, Jr. Direct measurement of polyol pathway activity in the ocular lens. Diabetes 1984; 33: 196–199
- Kicic E, Palmer TN. Increased white cell aldose reductase mRNA levels in diabetic patients. Diabetes Res Clin Pract. 1996; 33: 31–36
- Kinoshita JH, Nishimura C. The involvement of aldose reductase in diabetic complications. Diabetes Metab Rev. 1988; 4: 323–337
- Pugliese G, Tilton RG, Williamson JR. Glucose-induced metabolic imbalances in the pathogenesis of diabetic vascular disease. Diabetes Metab Rev. 1991; 7: 35–59
- Kawamura M, Heinecke JW, Chait A. Pathophysiological concentrations of glucose promote oxidative modification of low density lipoprotein by a superoxide-dependent pathway. J Clin Invest. 1994; 94: 771–778
- Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes 1997; 46: 292–300
- Ramasamy R, Trueblood N, Schaefer S. Metabolic effects of aldose reductase inhibition during low-flow ischemia and reperfusion. Am J Physiol. 1998; 275: H195–H203
- Oates PJ, Mylari BL. Aldose reductase inhibitors: Therapeutic implications for diabetic complications. Expert Opin Investig Drugs 1999; 8: 2095–2119
- Young LH, Russell RR, Chyun DA, Ramahi T. In Diabetes and Cardiovascular Disease. Humana Press, Totowa, NJ 2001; 281–298, Totowa, NJ
- Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: The framingham study. Am J Cardiol. 1974; 34: 29–34
- Levy D, Larson MG, Vasan RS, Kannel WB, Ho KK. The progression from hypertension to congestive heart failure. JAMA 1996; 275: 1557–1562
- Jaffe AS, Spadaro JJ, Schechtman K, Roberts R, Geltman EM, Sobel BE. Increased congestive heart failure after myocardial infarction of modest extent in patients with diabetes mellitus. Am Heart J. 1984; 108: 31–37
- Nesto RW, Zarich S. Acute myocardial infarction in diabetes mellitus: Lessons learned from ACE inhibition. Circulation 1998; 97: 12–15
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248–254
- Nishimura C, Yamaoka T, Mizutani M, Yamashita K, Akera T, Tanimoto T. Purification and characterization of the recombinant human aldose reductase expressed in baculovirus system. Biochim Biophys Acta. 1991; 1078: 171–178
- Liu SQ, Bhatnagar A, Srivastava SK. Does sorbinil bind to the substrate binding site of aldose reductase?. Biochem Pharmacol 1992; 44: 2427–2429
- Balendiran GK, Rajkumar B. Fibrates inhibit aldose reductase activity in the forward and reverse reactions. Biochem Pharmacol. 2005; 70: 1653–1663
- Tracey WR, Magee WP, Ellery CA, MacAndrew JT, Smith AH, Knight DR, Oates PJ. Aldose reductase inhibition alone or combined with an adenosine A(3) agonist reduces ischemic myocardial injury. Am J Physiol Heart Circ Physiol. 2000; 279: H1447–H1452
- Hwang YC, Sato S, Tsai JY, Yan S, Bakr S, Zhang H, Oates PJ, Ramasamy R. Aldose reductase activation is a key component of myocardial response to ischemia. FASEB J. 2002; 16: 243–245
- Trueblood N, Ramasamy R. Aldose reductase inhibition improves altered glucose metabolism of isolated diabetic rat hearts. Am J Physiol. 1998; 275: 75–83
- Trueblood NA, Ramasamy R, Wang LF, Schaefer S. Niacin protects the isolated heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2000; 279: H764–H771
- Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997; 33: 243–257
- Taegtmeyer H, King LM, Jones BE. Energy substrate metabolism, myocardial ischemia, and targets for pharmacotherapy. Am J Cardiol. 1998; 82: 54K–60K
- Kagaya Y, Kanno Y, Takeyama D, Ishide N, Maruyama Y, Takahashi T, Ido T, Takishima T. Effects of long-term pressure overload on regional myocardial glucose and free fatty acid uptake in rats. A quantitative autoradiographic study. Circulation 1990; 81: 1353–1361
- Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994; 267: H742–H750
- Doenst T, Goodwin GW, Cedars AM, Wang M, Stepkowski S, Taegtmeyer H. Load-induced changes in vivo alter substrate fluxes and insulin responsiveness of rat heart in vitro. Metabolism 2001; 50: 1083–1090
- Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. 2000; 10: 238–245
- Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem. 1998; 273: 5678–5684
- Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, Cuzzocrea S, Pisano B, Chatterjee PK, Thiemermann C. Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J. 2002; 16: 1027–1040