Abstract
A series of 15 previously reported N4-substituted isatin-3-thiosemicarbazones 3a-o has been screened for cytotoxic, antibacterial, antifungal and urease inhibitory activities. Compounds 3b, 3e and 3n proved to be active in cytotoxicity assay; 3e exhibited a high degree of cytotoxic activity (LD50 = 1.10 × 10− 5 M). Compound 3h exhibited significant antibacterial activity against B. subtilis, whereas compounds 3a, 3k and 3l displayed significant antifungal activity against one or more fungal strains i.e. T. longifusus, A. flavus and M. canis. In human urease enzyme inhibition assay, compounds 3g, 3k and 3m proved to be the most potent inhibitors, exhibiting relatively pronounced inhibition of the enzyme. These compounds, being non-toxic, could be potential candidates for orally effective therapeutic agents to treat certain clinical conditions induced by bacterial ureases like H. pylori urease. This study presents the first example of inhibition of urease by isatin-thiosemicarbazones and as such provides a solid basis for further research on such compounds to develop more potent inhibitors.
Introduction
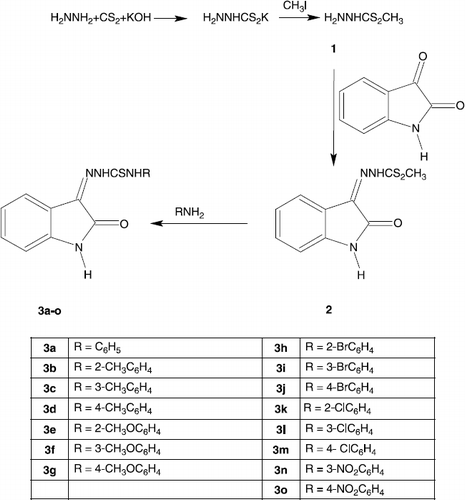
Thiosemicarbazones are a class of compounds, which has been found to possess a wide spectrum of medicinal properties [Citation1]. Isatins-derived thiosemicarbazones have also been found to possess a variety of physiological properties including antibacterial, antifungal, antineoplastic, antiulcer, antiviral and enzymatic inhibition [Citation2,3]. As a result of the significant pharmacological effects of isatins-thiosemicarbazones, there is an increasing interest in synthesizing and bio-testing of these derivatives [Citation4–10]. Prompted by this and in continuation of our earlier work [Citation11–15] in search of medicinally important organic and metallo-organic compounds, we reported very recently [Citation16] the synthesis of a series of 15 N4- substituted isatin-3-thiosemicarbazones based on the reactions of methyl 2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbodithioate (a common intermediate, prepared by condensing isatin with methyl 1-hydrazinecarbodithioate) with the readily available amines in essentially a one-step reaction (Scheme ). Since a few of these compounds as reported earlier by other workers have shown good antiviral activity [Citation17,18], it was considered worthwhile to study their other potential aspects such as cytotoxic, antibacterial, antifungal and enzymatic activities, particularly the urease inhibitory activity. During random screening of the organic compounds prepared in our laboratories, we found that isatins-thiosemicarbazones can inhibit this enzyme. To the best of our knowledge, this series of compounds has been scarcely investigated previously for their activity against urease, a nickel-dependent metallo-enzyme, the primary environmental role of which is to allow organisms to use external and internally generated urea as nitrogen source [Citation19]. In plants, urease may also participate in the systemic nitrogen transport pathways and possibly act as a defense protein [Citation20]. Nickel-dependent ureases are found in a wide array of different organisms, many have been isolated from various bacteria, fungi and higher plants. Ureases and urea catabolism may also have more practical uses. In an agricultural setting, rapid hydrolysis of urea (the most widely used nitrogen fertilizer in the world) by soil bacterial ureases results in unproductive volatilization of nitrogen and may cause ammonia toxicity or alkaline-induced damage to germinating seeds, seedlings and young plants [Citation21–24]. Urease inhibitors have been proposed to control urea hydrolysis in soil [Citation19,25 –30]. In particular, phosphoroamides have received considerable attention as urease inhibitors [Citation28,30]. Nonetheless, the efficiency of the presently available inhibitors is low, and negative effects on the environment have been reported [Citation19,31]. Medically, bacterial ureases are important virulence factors. Various bacterial ureases have been implicated in the pathogenesis of many clinical conditions such as pyelonephritis, ammonia and hepatic encephalopathy, hepatic coma, urinary catheter encrustation, peptic ulceration, and are directly associated with the formation of infection-induced urinary stones [Citation19,31,32]. Various urease inhibitors such as hydroxamic acids may be an answer for treatment of the listed conditions. However, early studies [Citation19,32–34] show adverse side effects, which include depressed bone marrow biosynthesis, inhibition of DNA synthesis, and high doses have been shown to be teratogenic. Therefore, studies on the synthesis of more potent and safe urease inhibitors continue to be of considerable interest. It has been unambiguously proved by spectroscopic studies that hydroxamic acids are good metal chelators and their mechanism of inhibition involves binding to the metal ions of the active site of enzyme. Nevertheless, regardless of the class of the compounds, it is reported [Citation35] that only a few functional groups with electronegative atoms such as oxygen, nitrogen and sulphur act either as bidentate, tridentate, or as ligand chelator to form octahedral complexes with two slightly distorted octahedral nickel atoms of the enzyme. These observations stimulated our interest to study the urease inhibitory activity of the synthesized isatin-thiosemicarbazones (derivatives of thiourea, a substrate-like urease inhibitor) as they might act as chelating agents coordinating through imino nitrogen, carbonylo oxygen and thiolato sulphur atoms. The present article, therefore, describes the cytotoxic, antibacterial, antifungal and more particularly urease inhibitory properties of these compounds.
Scheme 1 Synthesis of tilte compounds.
Meterials and methods
General
All reagents and solvents were used as obtained from the suppliers or recrystallized / redistilled as necessary. The isatin-thiosemicarbazones were synthesized by treating methyl 2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbodithioate with the readily available amines. The details of the reactions along with the physical, analytical and spectral data were reported elsewhere [Citation16]. In vitro biological testing of the synthesized compounds was made at the Department of Chemistry, The Islamia University of Bahawalpur, Pakistan and H.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Pakistan.
Biological testing
Cytotoxicity (in vitro)
Brine shrimp (Artemia salina leach) eggs were hatched in a shallow rectangular plastic dish (22 × 32 cm) filled with artificial sea water, which was prepared with a commercial salt mixture (Instant Ocean, Aquarium System, Inc., Mentor, Ohio, USA) and double-distilled water. An unequal partition was made in the plastic dish with the help of a perforated device. Approximately 50 mg of eggs were sprinkled into the large compartment, which was darkened, while the smaller compartment was opened to ordinary light. After two days, nauplii were collected by a pipette from the lighted side. A sample of the test compound was prepared by dissolving 2 mg of each compound in 2 mL of methanol. From this stock solution, 500, 50 and 5 μL were transferred to 9 vials, three for each dilution, and one vial was kept as control having 2 mL of methanol. The solvent was allowed to evaporate overnight. After two days, when shrimp larvae were ready, 1 mL of sea water and 10 shrimps were added to each vial (30 shrimps/ dilution) and the volume was adjusted with sea water to 5 mL per vial. After 24 h, the number of survivors was counted. Data were analyzed by a Finney computer program to determine the LD50 values [Citation36].
Antibacterial activity (in vitro)
The disc diffusion technique was adopted to determine the antibacterial activity of the test compounds [Citation37]. The test compounds 3a-o were dissolved in DMSO to get 10 mg/mL solution. A known volume (10 μL) of the solution was applied with the help of a micropipette on to the sterilized filter paper discs. The discs were dried at room temperature overnight and stored in sterile dry containers. Discs soaked with 10 μL of DMSO and dried in air at room temperature were used as the negative control. The standard antibiotic discs used as positive control were either purchased from manufacturer or prepared as above in the laboratory by applying a known concentration of the standard antibiotic solution. Bacterial cultures were grown in nutrient broth medium at 37°C overnight and spread on to solidified nutrient agar medium in Petri plates using sterilized cotton swabs in standard microbiological working environment. Test and control discs were then applied to the solidified medium surface with the help of sterilized forceps. The plates were incubated at 37°C for 12-15 h. The results were recorded by measuring the zone of inhibition in mm against each compound. As reference compound, sparaxin was used and the experiments were carried out in triplicate and the values obtained were statistically analyzed.
Antifungal activity (in vitro)
Antifungal activities of all the compounds were studied against six fungal cultures. Sabouraud dextrose agar (Oxoid, Hampshire, England) was seeded with 105 cfu mL− 1 fungal spore suspensions and transferred to petri plates. Discs soaked in 20 μL (200 μg / mL in DMSO) of all the compounds were placed at different positions on the agar surface. The plates were incubated at 27–29°C for seven days. The results were recorded [Citation38] as zone of inhibition (mm) and compared with standard drugs, miconazole and amphotericin B.
Urease inhibitory activity (in vitro)
Urease inhibitory activity of the synthesized compounds 3a-o was determined by a modified Berthelot (phenol-hypochlorite) method [Citation39]. Briefly, one unit of the Human urease (Gesellschaft für Biochemica und Diagonistica mbH, Germany) in 200 μL of the reagent 1 (120 mmol phosphate buffer pH 7.0, 60 mmol sodium salicylate, 5 mmol sodium nitroprusside and 1 mmol EDTA/L) was mixed with 600 μL of phosphate buffer and activated at 25°C for 10 min. This was followed by addition of 20 μL of the test solution containing 0.01μM - 100 μM of the test compound in DMF. DMF (20 μL) tested alone as a control did not have any inhibitory effect on the activity of the enzyme. The mixture was allowed to stand for 10 min to allow for interaction of the test compound with the enzyme. In order to achieve a final concentration of 1.5 mM urea per reaction, 150 μL of 20 mM urea in phosphate buffer (pH 7.0) was then added to each reaction mixture except the calibration mixture, where the same volume of the phosphate buffer alone was added. The urea-blank mixture was used to normalize against OD contribution by the test compound itself. The reaction mixture was incubated for an additional 10 min at 25°C to accomplish urea hydrolysis. The reaction was stopped by adding 1 mL of the reagent 2 (120 mmol phosphate buffer pH < 13 and ∼0.6 g hypochlorite / L). The ammonia liberated was allowed to complex with the hypochlorite and salicylate for 25 min and estimated by taking absorbance at 578 nm. Results were compared with thiourea, a standard inhibitor of urease. The percentage inhibition was calculated as the difference of absorbance values with and without the test compounds [Citation40].
Results and discussion
The present work describes the in vitro determination of the cytotoxic, antibacterial, antifungal and urease inhibitory effects of 15 N4-substituted isatin-3-thiosemicarbazones 3a-o, the synthesis of which has been reported elsewhere [Citation16].
Cytotoxicity (in vitro)
The synthetic isatin-thiosemicarbazones 3a-o were tested for their cytotoxic effects by the brine shrimp bioassay. From the data obtained, it is evident that only three compounds of this series i.e. 3b, 3e and 3n displayed promising cytotoxicity (LD50 = 1.10 × 10− 5 M − 2.52 × 10− 4 M) against Artemia salina. The remaining compounds gave values of LD50 > 2.67 × 10− 4 M − 3.38 × 10− 4 M in this assay and, therefore, can be considered to be almost inactive. It may be noted that compound 3e with a methoxy substituent at the ortho position of the phenyl ring showed maximum activity (LD50 = 1.10 × 10− 5 M) and, therefore, proved to be the most potent compound in the present series. Next potent was 3b (LD50 = 8.10 × 10− 5 M) having methyl substituent at the ortho position of the phenyl ring. Compound 3n with a nitro substituent at the meta position was found to be less potent, giving value of LD50 = 2.52 × 10− 4 M. These structure-activity relationships may serve as a basis for further chemical modifications directed towards the development of certain cytotoxic agents of clinical application.
Antibacterial activity (in vitro)
Antibacterial activity of the compounds 3a-o was tested against four Gram-negative (Escherichia coli, Pseudomonas aeruginosa, Salmonella typhi, Shigella flexneri) and two Gram-positive (Bacillus subtilis, Staphylococcus aureus) bacterial strains () at 10 mg/mL in DMSO. The results indicate that our compounds are, in general, weakly or moderately active. Only one compound i.e. 3h, out of the fifteen compounds tested, showed significant antibacterial activity against B. subtilis. Overall, this compound proved to be the most active one. The findings also indicated that compounds 3a and 3h exhibited a better antibacterial profile than the remainder.
Table I. Antibacterial activity (in vitro) of compounds 3a-o*,** (zone of inhibition in mm).
Antifungal activity (in vitro)
The synthetic compounds 3a-o were also screened for their antifungal activity against Trichophyton longifusus, Aspergillus flavus, Candida albicans, Microsporum canis, Fusarium solani and Candida glabrata () at 200 μg/mL in DMSO. Of these, compounds 3a, 3d, 3k and 3 l were found to be active against one or more selected fungi, exhibiting a range of inhibition (10–90%). Compound 3a with no substituent at the phenyl ring displayed 65% inhibition against T. longifusus and 85% inhibition against M. canis. Among the compounds 3b-d having methyl substituents at the different positions of the phenyl ring, only compound 3d with a para substituent was found to be active exhibiting 40% inhibition of A. flavus. In the case of chloro compounds 3k-m, variation in activity was observed with the position of the chloro group at the phenyl ring. Compound 3k having a chloro group at the ortho position of the phenyl ring displayed 70%, 85% and 80% inhibition of T. longifusus, A. flavus and M. canis, respectively. Similarly, compound 3 l with chloro group at the meta position of the phenyl ring exhibited 90% inhibition of T. longifusus; however, it was totally inactive against the other fungi viz. A. flavus and M.canis. Nevertheless, it displayed 10% inhibition of F. solani. To the contrary, compound 3m having a chloro substituent at the para position of the phenyl ring was found to be completely inactive against all these four fungal strains. Conclusively, three out of the four active compounds showed significant activity against one or more fungi, thus demonstrating the potential of isatins-thiosemicarbazones to be developed as antifungal agents. Therefore, the negative findings or exhibition of weak or moderate activities against certain selected fungi in the present assay do not preclude from further investigations of these compounds against other fungi.
Table II. Antifungal activity (in vitro) of the compounds 3a-o*,** (% inhibition).
Urease inhibitory activity (in vitro)
All the synthetic thiosemicarbazones 3a-o were further screened for their Human urease inhibitory potential. Of these, compounds 3c-o were found to exhibit varying degree of inhibitory effect at one or more tested concentrations. Compounds 3e, 3k and 3 l inhibited the activity of the enzyme at all the tested concentrations (100, 10, 1, 0.1 and 0.01 μM). Next to these were compounds 3 g, 3m and 3f, 3o, which inhibited the activity of the enzyme at four (100, 10, 1, 0.1 μM) and three (100, 10, 1 μM) concentrations, respectively. Similarly, compounds 3c, 3h-j and 3d, 3n displayed inhibitory effect at only two (100, 10 μM) and one (100 μM) concentrations, respectively (). Overall, compound 3 g having methoxy substituent at para position and 3k and 3m with chloro substituents at ortho and para positions of the phenyl ring proved to be the most potent inhibitors in the present series, as they exhibited relatively much greater activity at greater number of tested concentrations. However, the percentage of inhibition was not linear with the concentration of the compounds. Ureases obtained from different sources contain, in addition to the nickel metal, one to three protein subunits present in varying stoichiometric ratios [Citation32]. A urease inhibitor can, therefore, interact either with the metal or the protein component to interfere with the enzyme activity. Sulfenamide group, for example, has been reported to inhibit activity of the H. pylori urease by interacting with the SH group present in its protein components [Citation41,42]. A wide variety of mechanisms including competitive, non-competitive, un-competitive or cooperative binding are known to be involved in the interaction of an inhibitor with an enzyme. Although, the exact mechanism of urease inhibition by our test compounds 3c-o is not known, it is intriguing to investigate detailed kinetics of such interaction. These compounds could employ a mechanism of action by exploiting a common transition catalysis state and acting as ligand chelators to form octahedral complexes with the two slightly distorted octahedral Ni ions of the enzyme. Further, since the average plasma concentration of any drug is about 5 μg/mL, our antiurease compounds having no cytotoxicity are potential candidates for orally effective therapeutic agents to be used for the treatment of certain clinical conditions introduced by different microorganisms like H. pylori.
Table III. Inhibition of Human urease by compounds 3a-o.
In conclusion, the present investigations suggest that only three compounds of the series showed cytotoxic effect. Also, most of the compounds of the present series displayed weak to moderate activity against certain tested bacterial strains and only a few have shown significant antifungal activity against one or more selected fungi. The negative findings or display of weak and moderate activities, however, do not exclude from further investigations of these compounds against other microbial species. Further, structure-activity relationship studies may serve as a basis for chemical modifications directed towards the development of potential bioactive compounds of clinical interest. In the enzyme-inhibition assay, thirteen out of fifteen tested compounds demonstrated maximum urease inhibitory activity at the highest tested concentration (100 μM). A few of them proved to be potent inhibitors and could be potential candidates for orally effective therapeutic agents to treat certain clinical conditions caused by microbial ureases. Although the efficiency of these compounds is not high, to the best of our knowledge, this is the first example of inhibition of this enzyme by isatin-derived thiosemicarbazones. The present study provides a solid basis for further research on such compounds as urease inhibitors. Extensive studies to elucidate mechanism are required to contribute to a better understanding of the mechanism of action of these compounds.
References
- H Beraldo, and D Gambino. (2004). Wide pharmacological versatility of semicarbazones, thiosemicarbazones and their metal complexes. Mini-Rev Med Chem 4:159–165. (and references therein)
- JFM da Silva, SJ Garden, and AC Pinto. (2001). The chemistry of isatin: A review from 1975 to 1999. J Braz Chem Soc 12:273–324. (and references therein)
- SN Pandeya, S Smitha, M Jyoti, and SK Sridhar. (2005). Biological activities of isatin and its derivatives. Acta Pharm 55:27–46. (and references therein)
- N Karali. (2002). Synthesis and primary cytotoxicity evaluation of new 5-nitroindole-2,3-dione derivatives. Eur J Med Chem 37:909–918.
- I Chiyanzu, E Hansell, J Gut, PJ Rosenthal, JH McKerrow, and K Chibale. (2003). Synthesis and evaluation of isatins and thiosemicarbazone derivatives against cruzain falcipain-2 and rhodesain. Bioorg Med Chem Lett 13:3527–3530.
- TR Bal, B Anand, P Yogeeswari, and D Sriram. (2005). Synthesis and evaluation of anti-HIV activity of isatin β-thiosemicarbazone derivatives. Bioorg Med Chem Lett 15 (4451):4455.
- I Chiyanzu, C Clarkson, PJ Smith, J Lehman, J Gut, PJ Rosenthal, and K Chibale. (2005). Design, synthesis and anti-plasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorg Med Chem 13:3249–3261.
- A Rai, SK Sengupta, and OP Pandey. (2005). Lanthanum (III) and praseodymium (III) complexes with isatin thiosemicarbazones. Spectrochim Acta 61A:2761–2765.
- N Terzioglu, N Karali, A Gursoy, C Pannecouque, P Leysen, J Paeshuyse, J Neyts, and E de Clercq. (2006). Synthesis and primary antiviral activity evaluation of 3-hydrazono-5-nitro-2-indolinone derivatives. Arkivoc 109–118.
- DC Quenelle, KA Keith, ER Kern. In vitro and in vivo evaluation of isatin-β-thiosemicarbazone and marboran against vaccinia and cowpox virus infections. Antiviral Res 2006; 71: 24–30.
- ZH Chohan, H Pervez, KM Khan, A Rauf, and CT Supuran. (2004). Binding of transition metal ions [cobalt, copper, nickel and zinc] with furanyl-, thiophenyl-, pyrrolyl-, salicylyl- and pyridyl-derived cephalexins as potent antibacterial agents. J Enz Inhib Med Chem 19:51–56.
- ZH Chohan, H Pervez, KM Khan, A Rauf, GM Maharvi, and CT Supuran. (2004). Antifungal cobalt (II), copper (II), nickel (II) and zinc (II) complexes of furanyl-, thiophenyl-, pyrrolyl-, salicylyl- and pyridyl-derived cephalexins. J Enz Inhib Med Chem 19:85–90.
- ZH Chohan, H Pervez, KM Khan, A Rauf, GM Maharvi, and CT Supuran. (2004). Antifungal mono- and di-substituted symmetrical and unsymmetrical tirazine-derived Schiff bases and their transition metal complexes. J Enz Inhib Med Chem 19:161–168.
- ZH Chohan, H Pervez, KM Khan, A Rauf, and CT Supuran. (2004). Isatin-derived antibacterial and antifungal compounds and their transition metal complexes. J Enz Inhib Med Chem 19 (417):423.
- ZH Chohan, H Pervez, A Rauf, KM Khan, and CT Supuran. (2006). Antibacterial cobalt (II), copper (II), nickel (II) and zinc (II) complexes of mercaptothiadiazole-derived furanyl, thienyl, pyrrolyl, salicylyl and pyridinyl Schiff bases. J Enz Inhib Med Chem 21:193–201.
- H Pervez, MS Iqbal, MY Tahir, MI Choudhary, KM Khan. Synthesis of some N4-substituted isatin-3-thiosemicarbazones Nat Prod Res 2007, 21:1178–1186.
- RS Varma, PK Garg, HN Verma, and LP Awasthi. (1981). Potential biologically agents XXXII: Synthesis and antiviral activity of some 3-(arylthiosemicarbazono)-2-indolinones. Arch Pharm (Weinheim) 314:918–922.
- A-M ME Omar, NH Eshba, and HM Salama. (1984). Syntheses of some substituted isatin-β-thiosemicarbazones and isatin-β-hydrazonothiazoline derivatives as potential antiviral and antimicrobial agents. Arch Pharm (Weinheim) 317:701–709.
- HLT Mobley, and RP Hausinger. (1989). Microbial urease: Significance, regulation and molecular characterization. Microbiol Rev 53:85–108.
- JC Polacco, and MA Holland. (1993). Roles of urease in plant cells. Int Rev Cytol 145:65–103.
- RJ Radel, J Gautney, and GE Peters. Urease inhibitor developments In. B Bock, and D Kissel. Ammonia volatilization from urea fertilizers. Tennessee Valley Authority, Muscle Shoals, A.L, 1988111–136.
- GE Peters, RJ Radel, and RJ Medina. (1988). Synthesis of phenoxyaminocyclotriphosphazatrienes. J Agric Food Chem 36:384–390.
- JKR Gasser. (1964). Urea as fertilizer. Soils Fert 27:175–180.
- TE Tomlinson. (1970). Urea: Agronomic applications. Proc Fert Soc 113:1–76.
- KL Sahrawat. (1980). Control of urea hydrolysis and nitrification in soils by chemicals: Properties and problems. Plant Soil 57:335–352.
- RL Mulvaney, and JM Bremner. (1981). Control of urease transformation in soils. Soil Biochem 5:153–196.
- RD Hauck. InRD Hauck. editor. Nitrogen in crop production. Madison, WI, American Society of Agronomy; (1984) p 551–560.
- DA Martens, and JM Bremner. (1984). Effectiveness of phosphoroamides for retardation of urea hydrolysis in soils. Soil Sci Soc Am J 48:302–305.
- JM Bremner, and MJ Krogmeier. (1988). Elimination of the adverse effects of urea fertilizer on seed germination, seedling growth, and early plant growth in soil. Proc Natl Acad Sci USA 85:4601–4604.
- GW McCarty, JM Bremner, and JS Lee. (1990). Inhibition of plant and microbial ureases by phosphoroamides. Plant Soil 127:269–283.
- MJ Krogmeier, GW McCarty, and JM Bremner. (1989). Potential phytotoxicity associated with the use of soil urease inhibitors. Proc Natl Acad Sci USA 86:1110–1112.
- HLT Mobley, MD Island, and RP Hausinger. (1995). Molecular biology of microbial ureases. Microbiol Rev 59:451–480.
- DP Griffith. (1978). Struvite stones. Kidney Int 13:372–382.
- E Sim, A Jones, and L Stanley. (1985). Acetohydroxamate, a urease inhibitor, inhibits the covalent binding reaction of complement proteins C3 and C4. Acta Pharmacol Toxicol 57:304–306.
- Z Amtul, Siddiqui RA Atta-ur-Rahman, and MI Choudhry. (2002). Chemistry and mechanism of urease inhibition. Curr Med Chem 9:1323–1348. (and references therein)
- DJ Finney. Probit analysis3rd ed. London: Cambridge University Press; (1971).
- JG Cappuccino, and N Sherman. Microbiology: A laboratory manual4th ed. Harlow: Addison-Wesley Longman; (1999).
- M Hussain, MT Hussain, NH Rama, S Hameed, A Malik, and KM Khan. (2003). Synthesis and antimicrobial activities of some isocoumarin and dihydroisocoumarin derivatives. Nat Prod Res 17:207–214.
- MW Weatherburn. (1967). Phenol-hypochlorite reaction for determination of ammonia. Anal Chem 39:971–974.
- K Nagata, T Mizuta, Y Tonokatu, Y Fukuda, H Okamura, T Hayashi, T Shimoyama, and T Tamura. (1992). Monoclonal antibodies against the native urease of Helicobacter pylori: Synergistic inhibition of urease activity by monoclonal antibody combinations. Infect Immunol 60:4826–4831.
- K Nagata, H Satoh, T Iwahi, T Shimoyama, and T Tamura. (1993). Potent inhibitory action of the gastric proton pump inhibitor lansoprazole against urease activity of Helicobacter pylori: Unique action selective for H. pylori cells. Antimicrob Agents Chemother 37:769–774.
- TC Kuhler, J Fryklund, NA Bergman, J Weilitz, A Lee, and H Larsson. (1995). Structure-activity relationship of omeprazole and analogues as Helicobacter pylori urease inhibitor. J Med Chem 38:4906–4916.