Abstract
In continuation of our previous study on the urease inhibition by a number of chalcones, 2,3-dihydro-1,5-benzothiazepines and 2,3,4,5-tetrahydro-1,5-benzothiazepines, FlexX docking has been exploited to get a deeper insight into the mechanism of their inhibitory action. A comparison of the IC50 values of the active compounds reveals that, of the three classes of compounds studied, 2,3-dihydro-1,5-benzothiazepines were the most potent urease inhibitors. An in silico examination of these compounds showed that the activity is related to the interaction of ligand with the nickel metallocentre, its interaction with two amino acid residues, Asp224 and Cys322, in addition to the orientation of rings A and B in the catalytic core of the enzyme. The most active compound 2,3-dihydro-1,5-benzothiazepine (4) anchor tightly through a network of interactions with Ni701 and Ni702. This includes a number of hydrogen bonds and hydrophobic contacts with the amino acid residues in its vicinity. For their reduced analogs, the difference in the activity of different diastereomers has been observed to be configuration-dependent. This may be ascribed mainly to the difference in the orientation of ring B of the two stereoisomers and the extent of their interaction with Asp224 and Cys322 present in the catalytic core of the enzyme.
Introduction
Urease (EC 3.5.1.5), amidohydrolase, is a multi-subunit, nickel containing enzyme that catalyzes the hydrolysis of urea to carbamate and ammonia as a result of catabolism of nitrogen-containing compounds [Citation1]. The ammonia generated thereby causes severe metabolic disorders besides causing severe damage to gastric epithelium through its interactions with the immune system in human beings [Citation2,Citation3]. In agriculture, high urease activity causes significant environmental and economical problems by releasing abnormally large amount of ammonia into the atmosphere during urea fertilization. Moreover, it induces plant damage primarily by depriving plants of their essential nutrients and by ammonia toxicity which increases the pH of the soil [Citation4]. It is therefore extremely important to find urease inhibitors that may provide remedy to the above cited problems.
Several types of compounds known to be urease inhibitors are used both in medicine and agriculture, e.g. hydroxamic acid and its derivatives [Citation5], imidazole derivatives [Citation6], phosphorodiamidates, phosphazenes, Ecabet sodium and N-substituted hydroxy ureas [Citation7] more recently literature chiral boric acid [Citation8], dicoumarols [Citation9], biscoumarins [Citation10], α-hydroxy ketones [Citation11], polyphenols [Citation12] and a number of organobismuth compounds [Citation13] have also been evaluated for their effect on urease inhibition. A diterpene ester having a myrsinol type skeleton isolated from Euphorbia decipiens [Citation14] has recently been found to be a non-competitive urease inhibitor. Although some α,β-unsaturated ketones have been reported as urease inhibitors [Citation15], and Chalcones, their structural analogues were not generally found to be potential urease inhibitors. We have reported earlier the synthesis of a number of unsubstituted-, 2-hydroxy- and a few 4-amino-substituted chalcones but they were not found to have any significant activity against jack bean urease [Citation16]. More recently we have synthesized a parallel library of twenty five ferrocenyl chalcones, another library of thirty biphenyl-yl chalcones and a still larger library of 3-hydroxy chalcones but none of the members of any of these libraries was found to be potential urease inhibitor [Citation17].
1,5-Benzothiazepines, an important class of heterocycles compounds with oxygen and sulfur as heteroatoms in a seven-member ring, are synthetically accessible through a one pot reaction of chalcones with 2-aminobenzenethiol by a [4 + 3] annulation procedure. Although they are known to possess an impressive array of pharmacological activities [Citation18,Citation19], but they were not found to be tested for their potential as urease inhibitors. Therefore, we synthesized a number of substituted chalcones 1–24, converted them to 2,3-dihydro-1,5-benzothiazepines 24–35 followed by their reduction to 2, 3, 4, 5-tetrahydro-1, 5-benzothiazepines 36–40 [Citation16]. Since these tetrahydrobenzothiazepines have two chiral centers in the seven member thiazepine ring, two diastereomers were found in each case which was separated through fractional crystallization. All these compounds were screened for their potential as urease inhibitors and IC50 were calculated. It was interesting to note that (with the exception of one), none of the chalcones was found to possess any significant activity (consistent with our recent findings). However, the derived 2, 3-dihydrobenzothiazepines were found to have significant potential as urease inhibitors. The activity decreased again upon reduction to 2, 3, 4, 5-tetrahydrobenzothiazepines. Furthermore, the activity was found to be configuration dependant as one of the diastereomers in each case was found to be active and the other inactive. These findings called for a systematic study of the mechanism of urease inhibition of these three types of compounds by conducting molecular docking simulation studies on the active compounds 1–7 shown in .
Scheme 1. General scheme for the synthesis of compounds 1–7 and their in vitro Jack bean urease inhibitory activities.
Chalcone 1 was synthesized by the reaction of 3,4-methylenedioxybenzaldehde with 2-hydroxyacetophenone under Claisen-Schmidt conditions. 2,3-Dihydro-1,5-benzothiazepines (2–5) were synthesized by the condensation of the corresponding chalcones with 2-aminobenzenethiol based on a [4 + 3] annulation procedure. 2, 3, 4, 5-Tetrahydro-1, 5-benzothiazepines (6–7) were prepared by the reduction of the corresponding dihydrobenzothiazepines with lithium aluminum hydride. All the synthesized compounds were characterized through their physicochemical parameters; their spectral data have already been reported [Citation16].
Since the reduced benzothiazepines 6 and 7 contain two chiral centers in a 7-member ring at C-2 & C-4, the resulting two diastereomers 6a & 6b as well as 7a & 7b were separated through fractional crystallization. The relative configuration at the two chiral centres C-2 & C-4 in 6a and 6b was found to be (2S, 4R) and (2R, 4R) respectively based on their XRD analysis [Citation20] (supporting information). Such data could not be obtained for the diastereomers 7a and 7b due to their inability to crystallize.
Urease inhibition studies were carried out on all the compounds synthesized in a previous study but only compounds 1–7 shown in were found to be active with IC50 values from 19.07–93.74 μM (). In the current study thiourea was used as standard. As is evident from the data shown in , 2,3-dihydro-1,5-benzothiazepines (2–5) are the most potent urease inhibitors although the parent chalcones from which they have been derived are inactive except chalcone 1 while 2,3,4,5-tetrahydro-1,5-benzothiazepines (6–7) were found to be comparatively less active. Therefore, a detail study of the urease inhibition by these compounds has been carried out through molecular docking simulation studies.
Although urease was the first enzyme crystallized its mechanism of inhibition is still not well understood. There are many reports available that use the molecular docking simulation techniques to study the inhibition mechanism of a number of enzymes [Citation21]. Therefore, we exploited this important tool to explain the mechanism of urease inhibition by compounds 1–7. A unique feature of urease is the presence of a bimetallic centre having two nickel ions Ni701 and Ni702 in its active site which offer an opportunity for tight anchoring of the ligand by making non-covalent interactions such as hydrogen bonding and π-cation interactions.
Results and discussion
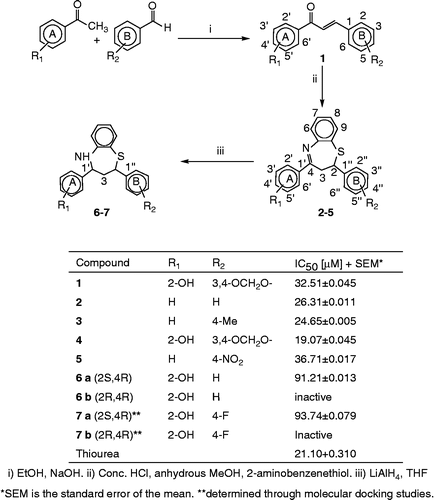
Current molecular docking studies on compounds 1–7 reveal that the fitting-in mechanism of chalcone 1, 2,3-dihydro-1,5-benzothiazepines 2–5 and 2,3,4,5-tetrahydrobenzothia-zepines 6–7 in the active site of the enzyme is quite similar. Chalcone 1 found to be active; it fits itself through ring A into the active site and coordinates via a 2-hydroxy group on ring A with the two nickel atoms, Ni701 and Ni702 at a distance of 1.97 and 2.50 Å respectively. The same functionality strengthens the enzyme-ligand complex by interacting through a week hydrogen bond with His139 at 3.31 Å which in turn forms another but strong hydrogen bond with Ala170 at 2.32 Å leading to further stabilization. Moreover, a network of histidine residues His137, His139, His222, His249, His275, His323 and other amino acids Asp363, Arg339, Ala170 present in the active site enable a number of hydrophobic contacts with chalcone as shown in . A combined effect of all these interactions suffices to provide a rationale for the only chalcone found to have significant potential as urease inhibitor with an IC50 value of 32.5 μM.
Figure 1. Compound 1 into the active site of urease. The molecule in gray is the ligand with the active site residues. Metals are represented in green.
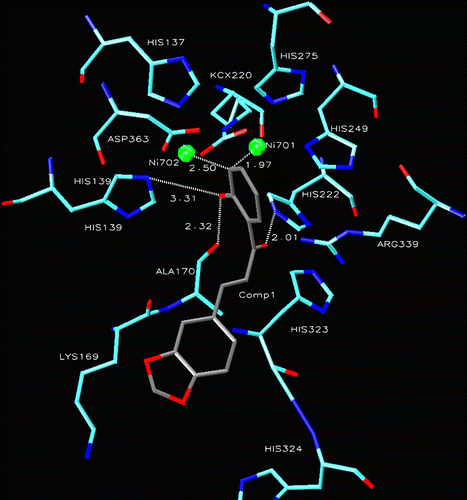
Comparatively to the fitting-in mechanism of chalcone, dihydrobenzothiazepine 4 anchors more tightly into the active site by coordinating through one of the two oxygens of the dioxymethylene moiety fused to ring B with both Ni701 and Ni702 at much stronger distances (1.75 and 2.17 Å respectively). The same oxygen also makes strong hydrogen bonds at 3.24 and 3.90 Å respectively with His249 and His137; the latter receives another hydrogen bond from His275 at 2.60 Å. A number of hydrophobic contacts with His249, Asp363, His275, Asp224, His323 and Ala170 are also established with the ligand in the active site, thereby, strengthening the enzyme-ligand complex. As mentioned earlier, thiourea was used as standard urease inhibitor in the present study; however, its structure cocrystallized with jack bean urease is not available in the protein data bank. Therefore, urease cocrystallized with acetohydroxamic acid has been taken as reference to control the performance of the docking approach. A comparison of compound 4 with that of acetohydroxamic acid-inhibited urease reveales a similarity in the fitting-in mechanism of the two ligands; while 4 made a pentavalent coordination pattern through one of the oxygens of the dioxymethylene functionality, acetohydroxamic acid shows an analogous pentavalent interaction with its oxygen of the hydroxyl group [Citation22]. Further, the same set of amino acid residues is found to stabilize the enzyme-ligand complex by building hydrophobic contacts ().
Figure 2. Compound 4 into the active site of urease. The lignad is represented as a ball and stick model.
The fitting-in mechanism of 2,3-dihydrobenzothiazepine 5 substituted with a 4-nitro group on the ring is found to be the same with respect to its anchoring in the active site through ring B and a pentavalent coordination pattern with only one of the oxygens of the nitro group as observed in 4. However, the observed difference in their activity may be ascribed to the absence of any strong interactions with Ni metals. The other dihydrobenzothiazepines 2 and 3, although lacking hydrophilic groups, are found to establish quite strong hydrophobic linkages with a number of amino acid residues such as His222, Glu223, Asp224, His249, Cys322, His323, Asp363, Ala366 and Met367 present in the active site may be considered responsible for their strong potential as urease inhibitors.
As is evident from the IC50 values given in , of the three series of compounds screened, tetrahydrobenzothiazepines 6 and 7 are found to be relatively weak urease inhibitors. This decrease in their affinity for urease may be ascribed either to a decrease in their flexibility to get into the active site or a loss of hydrogen bonding capability or a combined effects of the two.
As already mentioned, both diastereomers of tetrahydrobenzothiazepines 6 and 7 have been screened and in both cases only one of the two isomers is found to be active. As is evident from the XRD drawing of the two diastereomers (supporting information), the relative configuration at the two chiral centres in diastereomers 6a and 6b is 2S,4R and 2R,4R respectively, the former being active (IC50 91.21) while the latter inactive.
This difference in their urease inhibitory potential may be ascribed to their interaction with two important amino acid residues: Cys322 and Asp224 in the active site of the enzyme. The distance of two amino acid residues in case of 6a are smaller than those in 6b justifying the activity of the former stereoisomers.
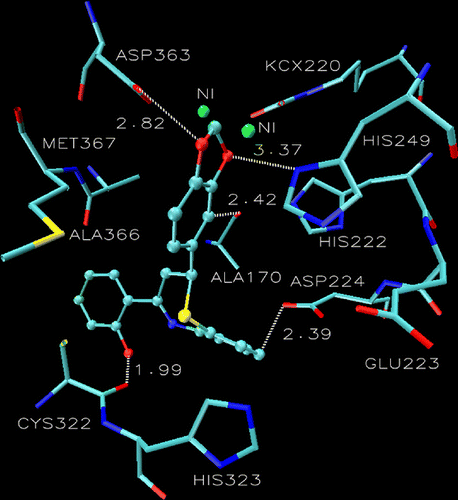
Another argument in favor of the difference in their behavior as urease inhibitors may be due to the difference in the orientation and stereochemistry of the ring B; while the ring B in 6a is completely sandwiched between Asp224, Cys322 and His323 making a flat structure that allows its easy and strong fitting into the catalytic core and hence greater activity (). On the contrary, the ring B in 6b is completely flipped and makes a cup shape which hinders its anchor into the active site of the enzyme, thereby providing a rationale for its inactivity as urease inhibitor ().
Figure 3. Superimposition of the docking pose of 6a (tan), 6b (gray), 7a (green) 7b (white) showing the difference in the orientation of ring B in the catalytic core of urease. Additionally the active site of urease clearly demonstrating the role of Asp224 and Cys322. The ligands are represented as a ball and stick model.
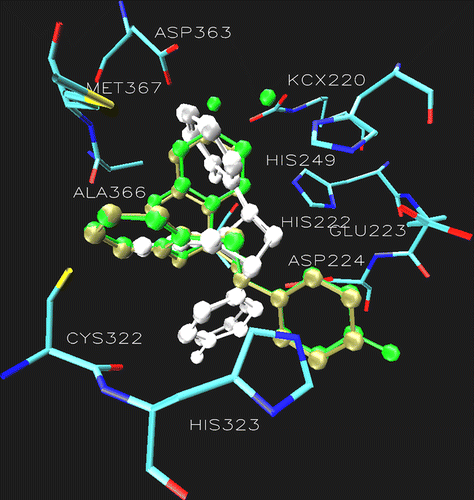
Almost similar situation was observed in thiazepine 7 having a 4-fluoro substituent on ring B, the urease inhibitory potential of the active diastereomer 7a (IC50 93.7) is about the same as that of 6a (IC50 91.2) but the distances from Asp224 and Cys322 in the active diastereomer 7a & the inactive isomer 7b are not much different compare to compound 6. The argument developed for 6 based on the distance from Asp 224 and Cys 322 and the difference in the orientation and stereochemistry of ring B making a flat structure (as explained in case of 6) plays an overriding role in making 7a as the active and 7b as the inactive isomer. This is clearly observed by doing the superimposition of the docking pose of two stereoisomers 7a and 7b as shown in .
As mentioned earlier, the relative configuration at C-4 and C-6 in diastereomers 6a and 6b was determined from their XRD data, but no such information was available for the two diastereomers 7a and 7b because they could not be crystallized for carrying out their XRD analysis. However, our docking studies have provided strong evidence that the relative configuration at the two chiral centres in the active isomer 7a is 2S, 4R and 2R, 4R in the inactive isomer 7b similar to that in 6a and 6b respectively.
The goal of the present study is to explore the possible binding modes of chalcones, 2, 3-dihydro-1, 5-benzothiazepines and 2,3,4,5-tetrahydro-1, 5-benzothiazepines which have already shown the ability as urease inhibitors. A sound docking technique has been used to figure out the difference in their urease inhibitory potential. One major observation based on the computational docking studies is that the most active 2, 3-dihydrobenzothiazepine 4 anchors tightly into the catalytic core through a network of hydrogen bonding with the nickel metallocentre and a number of amino acid residues in its vicinity. In the case of diastereomeric tetrahydrobenzothiazepines 6–7, the fitting-in mechanism is different and is configuration dependent. The superimposition of the docking pose of the two diastereomers 6a & 6b as well as 7a & 7b reveals that the difference in the orientation of the ring B and its interaction with Asp224 and Cys322 is responsible for the difference in the activity of the two diastereomers.
In order to understand the interaction of the ligands 1–7 with urease active site, they were subjected to molecular docking simulation studies that were conducted by using FlexX under Tripos SYBYL environment. Since the structure of thiourea cocrystallized with jack bean urease was not available in protein data bank, therefore, urease cocrystallized with acetohydroxamic acid was taken for a comparison to control the performance of the docking approach. The three dimensional structures of each ligand was constructed using commercially available molecular modeling package SYBYL 6.9 [Citation24]. In order to determine the binding mode of these compounds to urease, the FlexX implemented in SYBYL was applied to dock ligands with most of the default parameters (otherwise specified), in the catalytic core of the enzyme [Citation25]. An important point worth mentioning here is that SYBYL 6.9 is not completely parameterized for nickel ions, hence the atomic parameters for nickel have to be assigned by approximation. Shukla et al [Citation26] have reported that such a replacement of metal atoms during molecular docking has little or no effect on the structure of protein. Accordingly the nickel atoms of urease have been replaced by zinc during this study. A radius of 6.5 Å was used to define the interaction points in the active site. The energy minimization was performed using the tripos force field [Citation27] with a distance gradient algorithm with convergence criterion of 0.05 kcal/(mol Å) and a maximum of 1000 interactions, respectively. The docking results were analyzed by VMD [Citation28]. All computational studies were performed by using a dual processor 1.5 GHz Intel based PC running in the LINUX SUSE 10.2 operating system.
Acknowledgements
Authors thankfully acknowledge the financial support provided by the Higher Education Commission, Pakistan. Authors are also pleased to acknowledge Prof. Dr. Bernd M. Rode, (University of Innsbruck, Austria) for his valuable technical support.
Related Research Data
References
- HL Mobley, MD Island, and RP Hausinger. (1995). Molecular biology of microbial ureases. Microbiol Rev 59:451–480.
- C Preininger, and OS Wolfbeis. (1996). Disposable cuvette test with integrated sensor layer for enzymatic determination of heavy metals. Biosensors and Bioelectronics. 11:981–990.
- M Suzuki, S Miura, M Suematsu, D Fukumura, I Kurose, H Suzuki, A Kai, Y Kudoh, M Ohashi, and M Tsuchiya. (1992). Helicobacter pylori-associated ammonia production enhances neutrophil-dependent gastric mucosal cell injury. Am J Physiol 263:719–725.
- JM Bremner. (1995). Recent research on problems in the use of urea as a nitrogen fertilizer. Fert Res 42:321–329.
- EMF Muri, H Mishra, SM Stein, and JS Williamson. (2004). Molecular modeling, synthesis and biological evaluation of heterocyclic hydroxamic acids designed as Helicobacter pylori urease inhibitors. Lett Drug Design & Discov 1:30–34.
- TC Kuhler, J Fryklund, NA Bergman, J Weilitz, A Lee, and H Larsson. (1995). Structure-Activity relationship of omeprazole and analogues as Helicobacter pylori urease inhibitors. J Med Chem 38:4906–4916.
- Atta-ur-Rahman Amtul Z, RA Siddiqui, and MI Choudhary. (2002). Chemistry and mechanism of urease inhibition. Curr Med Chem 14:1323–1348.
- S Benini, WR Rypniewski, S Wilson, S Mangani, and S Ciurli. (2004). Molecular details of urease inhibition by boric acid: Insight into the catalytic mechanism. J Am Chem Soc 126:3714–3715.
- MK Khan, MA Lodhi, MG Maharvi, S Perveen, and MI Choudhary. (2004). Synthesis and urease enzyme inhibitory effects of some dicoumarols. J Enz Inhib Med Chem 19:367–371.
- MK Khan, S Iqbal, MA Lodhi, MG Maharvi, Z Ullah, and MI Choudhary. (2004). Atta-ur-Rahman. Perveen S. Biscoumarin: New class of urease inhibitors; economical synthesis and activity. J Bioorg Med Chem 12:1963–1968.
- T Tanaka, M Kawase, and S Tani. (2004). α-Hydroxyketones as inhibitors of urease. Bioorg Med Chem 12:501–505.
- Z-P Xiao, D-H Shi, H-Q Li, L-N Zhang, C Xu, and H-L Zhu. (2007). Polyphenols based on isoflavones as inhibitors of Helicobacter pylori urease. Bioorg Med Chem 15:3703–3710.
- T Murafuji, T Azuma, Y Miyoshi, M Ishibashi, AFMM Rahman, K Migita, Y Sugihara, and Y Mikata. (2006). Inhibition of jack bean urease by organobismuth compounds. Bioorg Med Chem Lett 16:1510–1513.
- MA Lodhi, J Hussain, AM Abbasi, RA Jassbi, MI Choudhary, and VU Ahmed. (2006). A new Bacillus pasteurii urease inhibitor from Euphorbia decipiens. J Enz Inhib Med Chem 21:531–535.
- T Tanaka, M Kawase, and S Tani. (2003). Urease activity of simple α,β-unsaturated ketones. Life Sci 73:2985–2990.
- FL Ansari, S Umbreen, L Hussain, T Makhmoor, SA Nawaz, MA Lodhi, SN Khan, F Shaheen, and Atta-ur-Rahman Choudhary MI. (2005). Synthesis and biological activities of chalcone and 1,5-Benzothiazepine derivatives: Promising new free-redical scavengers, and esterase, urease, and α-glucosidase inhibitors. Chem Biodiv 2:487–496. (b) Ullah A, Ansari FL, Ihsan-ul- Haq, Nazir S, Mirza B. Combinatorial synthesis, lead identification and antitumor study of a chalcone-based positional-scanning library. Chem Biodiv 2007; 4: 203-214
- FL Ansari, RA Khera, A Ullah. Green synthesis of ferrocenyl chalcones as leishmanicidal agents (Manuscript in preparation).
- VS Karthikeyan, and S Perumal. (2007). A facile protocol for the regioselective synthesis of novel thienobenzothiazepines. Tetrahedron Lett 48:2261–2265.
- P Cosmos, L Fodor, J Sinkkonen, K Pihlaja, and G Bernath. (2006). New isomers of 4,1-benzothiazepines. The first evidence for the desmotropy of seven-membered heterocycles. Tetrahedron Lett 47:5665–5667.
- FL Ansari, C Fedorchuk, M Parvez, S Umbreen, and S Saghir. (2002). Diastereomers of 4-(2-hydroxyphenyl)-2-phenyl-2,3,4,5-tetrahydro-1,5-benzothiazepine. Acta Cryst 58:422–425.
- Y Guo, J Xiao, Z Guo, F Chu, Y Cheng, and S Wu. (2005). Exploration of a binding mode of indole amide analogues as potent histone deacetylase inhibitors and 3D-QSAR analyses. J Bioorg Med Chem 13:5424–5434. (b) Musiani F, Arnofi E, Casadio R, Ciurli S. Structure-based computational study of the catalytic and inhibition mechanism of urease. J Biol Inorg Chem 2001; 6: 300-314
- M Ramiro, and K Muller. (1990). Molecular modeling studies on the urease active site and the enzyme-catalyzed urea hydrolysis. J Comp Aided Mol Design. 4:355–367.
- MW Weatherburn. (1967). Phenol-hypochlorite reaction for determination of ammonia Anal Chem 39:971–974.
- SYBYL, Version. 6.5. Molecular modeling software Tripos Associate Ltd St Louis MO, 1998.
- M Rarey, B Kramer, T Lengauer, and G Klebe. (1996). A fast flexible docking method using an incremental construction algorithm. J Mol Biol 261:470–489.
- KL Shukla, TM Gund, and SR Meshnick. (1995). Molecular modeling studies of the artemisinin (qinghaosu)-hemin interaction: Docking between the antimalarial agents and its putative receptors. J Mol Graphics 13:215–222. (b) Cheng F, Jianhua S, Xiamin L, Weiliang Z, J iande G, Ruyum J, Hualiang J, Kaixian C. Molecular docking and 3-D QSAR studies on the possible antimalarial mechanism of artemisinin analogues. Bioorg Med Chem 2002; 10: 2883-2891
- M Clark, RD Cramer, and VN Opdenbosh. (1989). Validation of the general purpose tripos 5.2 force field. J Comput Chem 10:982.
- W Humphrey, A Dalke, and K Schulten. (1996). VMD: Visual molecular dynamics. J Mol Graphics 14:33–38.