Abstract
Four series of pyrazolylbenzenesulfonamide derivatives were synthesized and evaluated for their anti-inflammatory activity using cotton pellet induced granuloma and carrageenan-induced rat paw edema bioassays. Moreover, COX-1 and COX-2 inhibitory activity, ulcerogenic effect and acute toxiCIT000y were also determined. Furthermore, the target compounds were screened for their in-vitro antimicrobial activity against Eischerichia coli, Staphylococcus aureus and Candida albicans. Compounds 4-(3-Phenyl-4-cyano-1H-pyrazol-1-yl)benzenesulfonamide 9a and 4-(3-Tolyl-4-cyano-1H-pyrazol-1-yl)benzenesulfonamide 9b were not only found to be the most active dual anti-inflammatory antimicrobial agents in the present study with good safety margin and minimal ulcerogenic effect but also exhibited good selective inhibitory activity towards COX-2. A docking pose for 9a and 9b separately in the active site of the human COX-2 enzyme was also obtained. Therefore, these compounds would represent a fruitful matrix for the development of dual anti-inflammatory antimicrobial candidates with remarkable COX-2 selectivity.
Introduction
Inflammatory diseases continue to be treated primarily by non steroidal anti-inflammatory drugs (NSAIDs). However, long term use of NSAIDs has been associated with gastrointestinal mucosal damage, bleeding [Citation1,Citation2], intolerance [Citation3], renal toxiCIT000y [Citation4] and contributing to an estimated 16500 deaths each year among rheumatoid arthritis and osteoarthritis patients in the USA alone [Citation5]. Consequently, extensive work has been made to improve the pharmacological profile of NSAIDs, which resulted in a major breakthrough in anti-inflammatory research with the discovery that cyclooxygenase (COX) exists in three isoforms COX-1, COX-2 and COX-3 that are regulated differently [Citation6,Citation7]. The discovery of the inducible enzyme COX-2 has stimulated the search for anti-inflammatory agents that specifically target COX-2 and avoid the undesirable side-effects associated with classical non selective NSAIDs. Much effort has been addressed to the diaryl-heterocycle class. Among this class of COX-2 inhibitors that comprise the pyrazole nucleus; celecoxib occupies a unique position as a potent and gastrointestinal (GI) safe anti-inflammatory and analgesic agent. Celecoxib is considered as a typical model of diaryl-heterocycles template that is known to inhibit selectively COX-2 enzyme [Citation8].
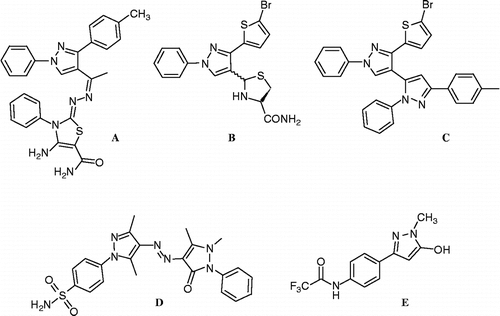
From pharmacoeconomic point of view, and to better patient compliance, an anti-inflammatory antimicrobial agent with minimum adverse effects and high safety margin is highly desirable. Drugs that serve dual anti-inflammatory antimicrobial functions will have greater potential than multi drugs regimen to treat inflammatory conditions that might be associated with microbial infections especially in patients with impaired liver or kidney functions. Compounds containing pyrazole functionality have been reported to exhibit promising anti-inflammatory activity Citation9–13. Moreover, several pyrazole derivatives exhibited antimicrobial activity Citation14–16. Taken together, the above reasons stimulated the research in our lab for agents that would have a dual anti-inflammatory antimicrobial effect. Several lead compounds containing a pyrazole moiety attached to various heterocyclic rings have been reported from our lab Citation17–23. Some of these reported compounds A [Citation19], B [Citation20], C [Citation18,Citation21] D [Citation18], and E [Citation23] showed pronounced dual anti-inflammatory and antimicrobial activities ().
Figure 1. Structure of reported active pyrazole derivatives A, B, C, D and E.
In extension to our efforts in the discovery of novel anti-inflammatory antimicrobial agents; we explored a new approach to, design new compounds by incorporating some pharmacophoric assemblies in celecoxib represented by an aminosulfonylphenyl group attached to a pyrazole ring bearing a lipophilic moiety at position 3, in addition to hydroxyiminomethyl or cyano group at position 4. In order to acquire new insights on the structure-activity relationship of this class of compounds, the dimethylaminomethyleneaminosulfonyl derivatives were pharmacologically evaluated to investigate the influence of this moiety on the anti-inflammatory activity compared to the free aminosulfonyl derivatives. The structural changes in such moiety might be expected to improve the pharmacokinetic parameters of these compounds. In addition to the targeted anti-inflammatory and antimicrobial activities, the ulcerogenic and acute toxiCIT000y profiles of the active compounds were determined.
Material and methods
Melting points were determined in open glass capillaries using a Gallenkamp melting point apparatus and are uncorrected. Infra-red (IR) spectra was recorded on Perkin-Elmer 1430 infrared spectrophotometer using the KBr disc technique. 1H NMR spectra was recorded on JEOL 300 MHz, 400 MHz or 500 MHz NMR spectrometer (DMSO-d6), and the chemical shifts are given in δ (ppm) downfield from tetramethylsilane (TMS) which served as an internal standard. Splitting patterns were designated as follows: s: singlet; d: doublet; m: multiplet. Elemental analyses were performed on Vario El Fab-Nr elemental analyzer and were found within ± 0.4% of the theoretical values. Follow up of the reactions and checking the purity of the compounds was made by thin layer chromatography (TLC) on silica gel-pre-coated aluminium sheets (Type 60 GF254, Merck) and the spots were detected by exposure to UV-lamp at λ = 254 nm for few seconds.
Synthesis
4-(2-Arylethylidenehydrazono)benzenesulfonamide 3a-e
A mixture of the appropriate acetophenone derivative (1a-e) (16.6 mmol), 4-hydrazinobenzenesulfonamide hydrochloride (2) (16.6 mmol) and anhydrous sodium acetate (1.36 g, 16.6 mmol) in ethanol (15 mL) was heated under reflux for 2–3 h. The reaction mixture was allowed to attain room temperature. The separated solid product was filtered, washed with ethanol, dried and crystallized from the proper solvent ( & ).
Table I. Physical data of compounds 3–9.
Table II. IR and 1H-NMR spectra of compounds 3–9.
Z-N,N-Dimethylaminomethylene-4-(3-aryl-4-formyl-1H-pyrazol-1-yl)benzenesulfonamide 4a-e
To an ice cold dimethylformamide (6.54 g, 6.93 ml, 89.5 mmol), a phosphorous oxychloride (2.42 g, 1.47 mL, 15.8mmol) was added drop-wise with stirring over a period of 30 min and stirring was continued for further 45 min with the reaction mixture kept at 0°C. The appropriate hydrazone (3a-e) (6.88 mmol) was then added and the reaction mixture was allowed to attain room temperature. The mixture was heated at 70°C for 2–3 h, allowed to cool to room temperature and then poured onto crushed ice and water. The mixture was boiled and the copious white precipitate obtained after cooling was filtered, dried and crystallized from the proper solvent ( & ).
4-(3-Aryl-4-formyl-1H-pyrazol-1-yl)benzenesulfonamide 5a-e
A solution of the proper aldehyde (4a-e) (5.24 mmol) in concentrated HCl (15 mL) was heated under reflux for 3 h. The reaction mixture was left to attain room temperature. The precipitated solid was filtered, washed with water, dried and crystallized from the proper solvent ( & ).
(Z,E)-N,N-Dimethylaminomethylene-4-(3-aryl-4-hydroxyliminomethyl-1H-pyrazol-1-yl)benzenesulfonamide 6a-e and E-4-(3-Aryl-4-hydroxyliminomethyl-1H-pyrazol-1-yl) benzenesulfonamide 7a-e
A solution of the selected aldehyde 4a-e or 5a-e (6.1 mmol), hydroxylamine hydrochloride (0.42 g, 6.1 mmol) and anhydrous sodium acetate (0.5 g, 6.1 mmol) in ethanol (15 mL) was heated under reflux for 4 h. The reaction mixture was concentrated and left to attain room temperature. The separated solid product was filtered, washed with water, dried and crystallized from the proper solvent ( & ).
Z-N,N-Dimethylaminomethylene-4-(3-aryl-4-cyano-1H-pyrazol-1-yl)benzenesulfonamide 8a-e
A solution of the appropriate oxime 6a-e (5.85 mmol) in acetic anhydride (10 mL) was heated under reflux for 2–3 h. The reaction mixture was left overnight at room temperature. The separated solid product was filtered, washed with water, dried and crystallized from the proper solvent ( & ).
4-(3-Aryl-4-cyano-1H-pyrazol-1-yl)benzenesulfonamide 9a-e
A solution of the selected oxime (7a-e) (2.92mmol) in phosphorous oxychloride (4.48 g, 2.72ml, 29.2 mmol) and triethylamine (0.44 g, 0.61 ml, 4.35mmol) was heated at 80°C for 2 h. The reaction mixture was left to attain room temperature, then poured onto ice water. The separated solid was filtered, dried and crystallized from the proper solvent ( & ).
Anti-inflammatory testing
Cotton pellet induced granuloma bioassay [Citation24]
Adult male Sprague-Dawley rats (120–140 g; sourced from Medical Research Institute, Alexandria University) were used to investigate the anti-inflammatory efficacy of the synthesized compounds. The rats were acclimated one week prior to use and were given ad libitum access to standard rat chow and water. Prior to the start of experiment, the animals were randomly divided into groups of six rats each. Cotton pellet (35 ± 1 mg) cut from dental rolls were impregnated with 0.2 mL (containing 0.01 mmol) of a solution of the test compound in chloroform or acetone and the solvent was allowed to evaporate. Each cotton pellet was subsequently injected with 0.2 ml of an aqueous solution of antibiotics (1 mg penicillin G and 1.3 mg dihydrostreptomycin/ml). Two pellets were implanted subcutaneously, one in each axilla of the rat, under mild general anaesthesia. Two groups of animals were treated as above with the substitution of tested compounds with standard reference (indomethacin or celecoxib) and the antibiotics at the same level. Pellets containing only the antibiotics were similarly implanted in the control rats. After seven days, the animals were sacrificed and the two cotton pellets, with adhering granulomas, were removed, dried for 48 h at 60 °C and weighed. The increment in dry weight (difference between the initial and final weights) was taken as a measure of granuloma ± SEM. This was calculated for each group and the percentage reduction in dry weight of granuloma from control value was also calculated. The ED50 values were determined through dose-response curves, using doses of 4, 7, 10 and 15 μmol for each compound. All experiments were conducted within Institute guidelines laid down for animal experimentation.
Carrageenan-induced rat paw edema bioassay [Citation25]
Male albino rats weighing 120–150 g (Medical Research Institute, Alexandria University) were used to investigate the carrageenan-induced rat paw edema. The rats were kept in animal house under standard conditions of light and temperature with free access to food and water. The animals were randomly divided into groups of six rats each. The paw edema was induced by subplantar injection of 50 μL of 2% carrageenan solution in saline (0.9%). Indomethacin, celecoxib and test compounds were dissolved in dimethylsulfoxide (DMSO) and injected subcutaneously in a dose of 10 μmol/ kg body weight, one hour prior to carrageenan injection. DMSO alone was injected to the control group. The volume of paw edema (mL) was determined by means of water plethysmometer immediately after injection of carrageenan and 4 h later. The increase in paw volume between time 0 to 4 h was measured [Citation25]. The percentage protection against inflammation was calculated as follows:
where Vc is the increase in paw volume in absence of test compound (control) and Vd is the increase of paw volume after injection of the test compound. Data were expressed as the mean ± SEM. Significant difference between the control and treated groups was performed using Student's t-test and P values were considered significant at P < 0.001. The anti-inflammatory activity of the test compounds relative to that of indomethacin was also calculated.
Human COX-1 and COX-2 enzymatic assay
Human COX-1 and COX-2 activities were determined according to the literature protocol [Citation30]. COX-1 assay was carried out using platelets microsome fraction. Human platelets were prepared from NSAID-free normal human volunteers according to the method of Hammarström and Falardeau [Citation27]. COX-2 assay was performed utilising human recombinant COX-2 (hrCOX-2) purchased from Sigma-Aldrich. Human COX-1 (0.3 mg protein/assay) or COX-2 (1 mg protein/assay) was suspended in 0.2 mL of 100 mmol Tris/HCl buffer (pH 8) containing hematin (2 mmol) and tryptophan (5 mmol) as cofactors. The reaction mixture was pre-incubated with each test compound for 5 min at 24 °C. [14C] Arachidonic acid (100.00 dpm, 30 mmol) was added to the mixture and then incubated for 2 min (COX-1) or 5 min (COX-2) at 24 °C. The reaction was stopped by addition of 400 μL of a stop solution composed of Et2O/MeOH/1M CIT000ric acid (30:4:1, v/v/v). After centrifugation of the mixture at 1700 × g for 5 min. at 4 °C, 50 μL of the upper phase was applied to a thin-layer chromatography (TLC) plate. TLC was performed at 4 °C with a solvent system consisting of Et2O/MeOH/AcOH (90:2:0.1, v/v/v). Enzyme activity was calculated from the percent conversion of arachidonic acid to PGH2 and its decomposition products, using a radiometric photographic system. The concentration of the compound causing 50% inhibition (IC50) was calculated.
Ulcerogenic effects
All synthesized compounds were evaluated for their ulcerogenic potential in rats [Citation28]. Indomethacin was used as a reference standard. Male albino rats (100–120 g) were fasted for 12 h prior to administration of the compounds. Water was given ad libitum. The animals were divided into groups of six rats each. Control group received 1% gum acacia orally. Other groups received indomethacin or test compounds orally in two equal doses at 0 and 12 h for three successive days at a dose of 30 μM/kg per day. Animals were sacrificed by diethyl ether 6 h after the last dose and the stomach was removed. An opening at the greater curvature was made and the stomach was cleaned by washing with cold saline and inspected with a 3X magnifying lens for any evidence of hyperemia, hemorrhage, definite hemorrhagic erosion or ulcer. An arbitrary scale (Normal stomach: Score 0.0, Petechial hemorrhage or hyperemia: Score 1, Definite hemorrhagic erosions: Score 2, Very small ulcers ( < 1 mm long): Score 3, Small ulcers (from 1 to < 2 mm long): Score 4, Medium ulcers (from 2 to < 3 mm long): Score 5, Large ulcers (from 3 to < 4 mm long): Score 6, Very large ulcers (>4 mm long: Score 7) was used to calculate the ulcer index which indicates the severity of stomach lesions [Citation28]. The percentage ulceration for each group was calculated as follows:
Acute toxiCIT000y
The biologically significant compounds (7a, 7b, 9a and 9b) were further investigated for their oral acute toxiCIT000y in male mice (each 20 g, supplied by Medical Research Institute, Alexandria University) as reported in previous reports [Citation26,Citation29]. Groups of mice each of six animals were used for the assay. The compounds were given orally suspended in 1% gum acacia, in doses of 1, 10, 100, 200, 250, 300 mg/kg. After 24 h, the % mortality in each group/compound was recorded. Moreover, these compounds were further tested for their parenteral acute toxiCIT000y using groups of mice each of six animals. The compounds or their vehicle, propylene glycol (control), were given by intraperitoneal injection in doses of 10, 25, 50, 75, 100 mg / kg. Survival of the mice was followed up to 7 days [Citation20].
Modeling Studies
Computer-assisted simulated docking experiments were carried out under an MMFF94X force field in (PDB ID: 1CX2) using Chemical Computing Group's Molecular Operating Environment (MOE-Dock 2005) software, Montréal, Canada [Citation31].
Docking Protocol
The coordinates from the X-ray crystal structure of human COX-2 used in this simulation were obtained from the Protein Data Bank (PDB ID 1CX2), where the active site is bound to the selective COX-2 inhibitor SC-588. The ligand molecules were constructed using the builder module and were energy minimized. The active site of COX-2 was generated using the MOE-Alpha Site Finder, and then ligands were docked within this active site using the MOE-Dock. The lowest energy conformation was selected and the ligand interactions (hydrogen bonding and hydrophobic interaction) with COX-2 were determined.
In vitro antimicrobial activity
The microdilution susceptibility test in Müller-Hinton Broth (Oxoid) and Sabouraud Liquid Medium (Oxoid) were used for determination of antibacterial and antifungal activity [Citation32]. Test organisms were Escherichia coli ATCC 25922 as Gram-negative bacteria, Staphylococcus aureus ATCC 19433 as Gram-positive bacteria and Candida albicans as a yeast-like fungi. Ampicillin trihydrate and clotrimazole were used as standard antibacterial and antifungal agents, respectively. Solutions of the test compounds, ampicillin trihydrate and clotrimazole were prepared in DMSO at concentration of 1600 μg/mL. From this stock solution, serial dilutions of the compounds (800, 400,…, 6.25 μg/mL) were prepared. The microorganism suspensions at concentration of 106 CFU/ml (colony forming unit/ml) were inoculated to the corresponding wells. Plates were incubated at 36 °C for 24–48 h. The incubation chamber was kept sufficiently humid during the incubation period and at the end of the incubation period, the minimal inhibitory concentrations (MIC) were determined. Controls with DMSO and uninoculated media were run parallel to the test compounds under same conditions.
Results and discussion
Chemistry
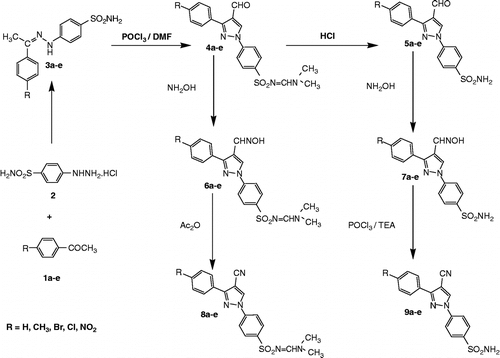
The target compounds were synthesized according to the steps outlined in Scheme . Condensation of the appropriate acetophenone derivatives 1a-e with 4-hydrazinobenzenesulfonamide hydrochloride 2 and anhydrous sodium acetate in ethanol afforded the corresponding hydrazones 3a-e. The hydrazones 3a-e were subjected to Vilsmeyer Haack reaction [Citation33], to yield the dimethylaminomethyleneamino aldehydes 4a-e. The structures of these compounds were confirmed by IR spectra that showed lack of the absorption bands characteristic for NH2 group. Moreover, 1H-NMR spectra for these compounds revealed two singlets at δ 2.9 and 3.15 ppm characteristic for two methyl groups, in addition to a singlet at δ8.23–8.27 ppm characteristic for methine proton. The latter signal appeared at high chemical shift indicating presence of compounds 4a-e as Z-isomers [Citation34]. These results are in accordance to the findings of Sen and Mukhopadhyay [Citation35] who reported the synthesis of dimethylaminomethyleneaminosulfonyl derivative by reaction of sulfonamido derivative with Vilsmeyer reagent. Compounds 4a-e were hydrolyzed to the free aminosulfonyl derivatives 5a-e by heating under reflux with concentrated HCl for 3 h. Reaction of the selected aldehyde 4a-e and 5a-e with hydroxylamine hydrochloride in ethanol containing anhydrous sodium acetate yielded the corresponding oximes 6a-e and 7a-e respectively. Inspection of 1H-NMR spectra for compounds 6a-e and 7a-e revealed iminomethyl proton at δ = 7.40–7.55 ppm confirming existence of these compounds as E-isomers [Citation36]. Dehydration of the oximes 6a-e with acetic anhydride [Citation21] afforded the cyano derivatives 8a-e. 1H-NMR spectra of the latter compounds showed methine proton at δ = 8.24–8.27 ppm indicating their existence as Z-isomers similar to their precursors 4a-e. Dehydration of the oximes 7a-e was accomplished using POCl3 in presence of triethylamine [Citation37] rather than acetic anhydride to avoid acetylation of the sulfonamido group. This method resulted in the corresponding cyano derivatives 9a-e in high yield.
Scheme 1. Synthesis of test compounds 3-9a–e
Anti-inflammatory activity
Cotton pellet-induced granuloma bioassay
The anti-inflammatory activity of the synthesized compounds 6, 7, 8, 9 was evaluated by applying the cotton-pellet granuloma bioassay in rats [Citation24] using celecoxib and indomethacin as reference standards. The ED50 values were determined for each compound and expressed as the mean ± SEM. Significant differences between the control and the treated groups was obtained using student's t-test and P values. The difference in results was considered significant at P < 0.001 (). All test compounds showed anti-inflammatory activity higher than celecoxib (ED50 = 16.74 μmol), whereas, compounds 7a, 7b, 9a and 9b (ED50 = 9.46, 9.38, 8.58 and 8.94 μmol respectively) possessed anti-inflammatory activity comparable to that of indomethacin (ED50 = 9.68 μmol). In general, the free aminosulfonyl pyrazole derivatives were more active than their dimethylaminomethyleneamino analogues. The replacement of hydroxyliminomethyl group at position 4 of the pyrazole ring with a cyano group (compounds 8a, 8e and 9a-c) enhanced the anti-inflammatory activity. Within the active series 7 and 9, the introduction of a methyl group at position 4 of the phenyl moiety didn't alter the activity. However the bromo, chloro and nitro groups exhibited reduced anti-inflammatory activity. Compounds 9a and 9b were found to be the most active anti-inflammatory agents in the present study (ED50 = 8.58 and 8.94 μmol, respectively).
Table III. The anti-inflammatory activity (ED50, μmol / cotton pellet)a and ulcerogenic activitya.
Carrageenan-induced rat paw edema bioassay
Compounds 7a, 7b, 9a and 9b that showed anti-inflammatory activity comparable to that of indomethacin and higher than celecoxib in the cotton pellet–induced granuloma bioassay, were further evaluated for their in-vivo systemic effect using the carrageenan–induced paw edema bioassay in rats [Citation25]. The results () revealed that compounds 9a and 9b exhibited systemic anti-inflammatory activity (% protection = 76.5 and 73.4, respectively) comparable to that of indomethacin and celecoxib (% protection = 74.4 and 77.5, respectively) whereas compounds 7a and 7b (% protection = 69.3 and 67.3) were relatively less active.
Table IV. Effect of compounds 7a, 7b, 9a and 9b on carrageenan-induced rat paw edemaa (mL) at a single dose (10 μmol/kg), % protection and activity relative to indomethacin.
Human COX-1 and COX-2 enzymatic assay
Compounds 7a, 7b, 9a and 9b that exhibited potent anti-inflammatory profiles in the pre-mentioned animal models were further tested for their ability to inhibit human COX-1 and COX-2 enzymes in-vitro applying the methodology of Wakitani et al [Citation30]. The concentration of the compound causing 50% enzyme inhibition (IC50 μmol) is shown in . The test compounds exhibited much weaker inhibitory activity against COX-1 enzyme (IC50 values range 76.41–>100 μmol) compared with indomethacin (IC50 = 0.26 μmol). In addition, the test compounds showed higher inhibitory profile against COX-2 (IC50 values were between 0.78 and 1.35 μmol) compared with indomethacin (IC50 = 2.63 μmol). This led to a very high approximate selectivity ratio (COX-1/COX-2) for the test compounds (ranged between 60.6–128.2) compared with indomethacin (selectivity ratio is 0.098). However, all test compounds showed approximate selectivity ratio (COX-1/COX-2) lower than that of celecoxib. In general, oxime derivatives (compounds 7a, 7b) showed lower selectivity towards COX-2 enzyme than cyano derivatives (compounds 9a, 9b). Compound 9a was the most selective COX-2 inhibitor (approximate selectivity ratio = 128.20) in the present study, but the proximate selectivity was >40% of that found with celecoxib (approximate selectivity ratio >333).
Table V. In vitro human COX-2a and COX-1b enzymes inhibitory activities of compounds 7a, 7b, 9a and 9b.
Ulcerogenic effects
The synthesized compounds were evaluated for their ulcerogenic potential in rats [Citation28]. All test compounds proved to have superior gastrointestinal safety profiles (0–20% ulceration) in the population of the tested animals at oral doses of 30 μmol/kg per day, compared with indomethacin which was found to cause 100% ulceration under the same experimental conditions (). Compounds having bromo, chloro or nitro group 6c, 7c, 7d, 8c, 8d, 9d, 9e showed a slight ulcerogenic effect (10–20%). Gross observation of the isolated rat stomachs showed a normal stomach texture for the other compounds.
Acute ToxiCIT000y
Compounds 7a, 7b, 9a and 9b were further evaluated for their oral acute toxiCIT000y in male mice according to established methods [Citation26,Citation29]. The results indicated that most of the tested compounds proved to be non-toxic and well tolerated by experimental animals up to 140 mg/kg. Moreover, these compounds were tested for their toxiCIT000y through parenteral route [Citation20]. The results revealed that all compounds were non-toxic up to 65 mg /kg.
Docking Studies
Molecular docking as well as conformational alignment studies of compounds (9a, 9b) were performed using Molecular Operating Environment's (MOE-Dock 2005) module [Citation31] in order to rationalize the obtained biological results. Molecular docking studies further helps in understanding the various interactions between the ligands and enzyme active sites in detail.
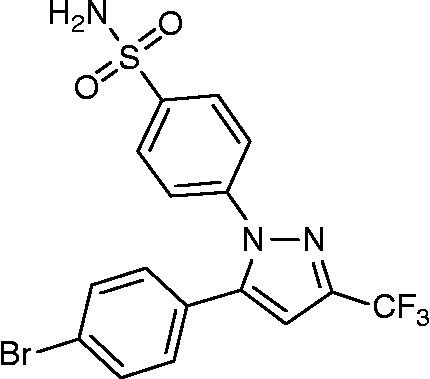
The determination of the three-dimensional co-crystal structure of COX-2 complexed with a selective inhibitor, SC-558 (; PDB ID: 1CX2) has led to the development of a model for the topography of the NSAIDs binding site in human COX-2 [Citation38]. Therefore herein, we performed the docking studies using this COX-2 co-crystal with SC-558 as a template.
Figure 2. Structure of SC-558.
and shows the binding interactions of both compounds 9a, 9b to the active site of COX-2, respectively where both of them exhibited some similar interactions as SC-558. In general, compounds 9a, 9b showed a hydrogen bonding interactions with His90, Ile517, Phe518, Ser146, Ser38, Glu671 and a hydrophobic interaction with: Met113, Val116, Val349, Leu352, Leu359, Phe518, Val523, Leu531.
Figure 3. 3D View from a molecular modeling study, of the minimum-energy structure of the complex of 9a docked in COX-2 (PDB ID: 1CX2). Viewed using Molecular Operating Environment (MOE) module.
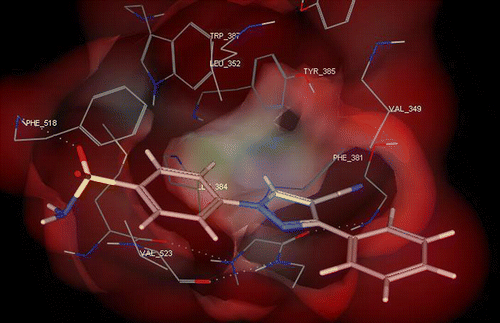
Figure 4. 3D View from a molecular modeling study, of the minimum-energy structure of the complex of 9b docked in COX-2 (PDB ID: 1CX2). Viewed using Molecular Operating Environment (MOE) module.
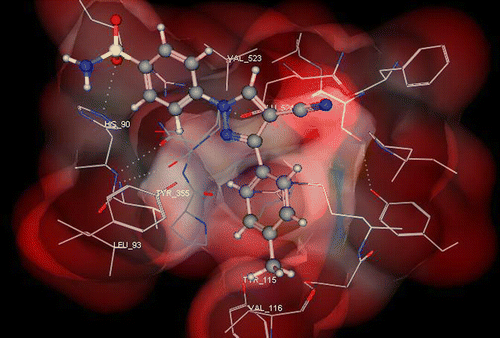
In a similar pattern to SC-588, one of the O-atoms of the SO2NH2 substituent of both 9a and 9b forms a hydrogen bond with the NH hydrogen of His90 () and the other oxygen forms hydrogen bonds with backbone amide hydrogens of Phe518 (). Furthermore, the phenyl ring bearing the sulfonamide is surrounded by a hydrophobic pocket of Val349, Leu352, Phe518, and the more important Val523 (found only in COX-2). On the other hand, the conformational alignment of SC-558 and 9b showed an overlapping between the phenylsulfonamide moiety of both SC-558 and compound 9b while the 3-tolyl ring was inserted into the trifluoromethyl zone.
From the abovementioned data, the docking results of the examined compounds (9a, 9b) provide a good explanation for the potent inhibitory activity of both compounds and give an insight about the COX-2 binding of the pyrazole nucleus with a 1,3-diaryl substituted pattern as a suitable scaffold for the development of selective COX-2 inhibitors.
Antimicrobial activity
The designed compounds 6a-e, 7a-e, 8a-e and 9a-e were evaluated for their antimicrobial activity. The microdilution susceptibility test in Müller-Hinton Broth (Oxoid) and Sabouraud Liquid Medium (Oxoid) were used for determination of antibacterial and antifungal activities [Citation32]. The minimal inhibitory concentration (MIC) revealed that all test compounds have no antifungal activity (). The antibacterial activity of the test compounds 6d, 7b, 7d, 8b and 9b against E. coli was similar to the reference antimicrobial compound (ampicillin). Compounds 7e, 8a, 8e, 9a and 9c-e showed half the antimicrobial activity of ampicillin against E. coli. In general cyano derivatives 8a-c, 8e, 9a and 9c possessed better antimicrobial activity than their oxime precursors 6a-c, 6e, 7a and 7c against E. coli. Moreover, aminosulfonyl derivatives 7b, 7c and 7e had higher activity than their dimethylaminomethyleneamino analogs 6b, 6c and 6e may be attributed to the benzenesulfonamido moiety. Furthermore, the antibacterial activity against E. coli was enhanced when dimethylaminomethyleneamino group of 8c and 8d was hydrolyzed to the corresponding aminosulfonyl analogs 9c and 9d. Regarding the antibacterial activity against S. aureus, all test compounds showed weak or no antibacterial activity except for compounds 7c and 9b which showed half the activity of ampicillin.
Table VI. Minimal inhibitory concentrations (MIC μg/mL) of test compounds.
Conclusion
The investigated compounds 7a, 7b, 9a and 9b were found to possess anti-inflammatory activity comparable to that of indomethacin and celecoxib. Meanwhile, they displayed higher selective inhibitory activity to COX-2 when compared to indomethacin. Moreover, compounds 7b and 9b exhibited similar antimicrobial activity to that of ampicillin against E. coli. Compounds 9a and 9b showed anti-inflammatory activity profile lower than previously reported compound A [Citation19] and comparable to that of compounds B [Citation20], C [Citation18,Citation21], D [Citation18] and E [Citation23]. In addition, compounds 9a and 9b exhibited COX-1/COX-2 selectivity ratio higher than compound A and lower than compound C. Compound 9a displayed antimicrobial activity against E. coli. comparable to compounds A, B, C and E, whereas, the antimicrobial activity of 7b and 9b was comparable to compound D. The antimicrobial activity of compounds 7a, 7b and 9a against S. aureus was lower than compounds A, B, C and D, while the activity of 9b was comparable to compound D. Overall, it can be concluded that compounds 9a and 9b proved to be the most active anti-inflammatory agents (ED50 = 8.58 and 8.94 μmol) in the present study with no ulcerogenic effect and good safety margin. However, the antibacterial activity of compound 9b was twice that found with compound 9a against E. coli and S. aureus. The antibacterial activity of compound 9b against E. coli was equivalent to that of ampicillin, whereas, the antibacterial activity against S. aureus is about 50% that of ampicillin.
Acknowledgements
The authors wish to thank Dr. Elsayed Aboulmagd, Department of Microbiology, Faculty of Pharmacy, University of Alexandria, Alexandria 21521, Egypt and Dr. Azza Baraka, department of Pharmacology, Faculty of Pharmacy, University of Alexandria for their assistance during biological testing.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- JM Scheiman. (2005). Effects of nonsteroidal anti-inflammatory drugs, including COX-2 specific inhibitors on the GI tract. Clin Update 12:1–4.
- S Fiorucci, L Santucci, and E Distrutti. (2007). NSAIDs, coxibs, CINOD and H2S-releasing NSAIDs: What lies beyond the horizon. Digest Liver Dis 39:1043–1051.
- DC Kosegarten, EF LaSala, SF Long In: , L Shargel, AH Mutnick, PF Souncy, LN Swanson, and LH Block. editors. Comprehensive pharmacy review. Philadelphia: Williams and Wilkins; (2001). p 289–291.
- MR Griffin, A Yared, and WA Ray. (2000). Nonsteroidal Antiinflammatory Drugs and Acute Renal Failure in Elderly Persons. Am J Epidemiol 151:488–496.
- G Singh, and G Triadafilopoulos. (1999). Epidemiology of NSAID induced gastrointestinal complications. J Rheumatol 26:18–24.
- Y Song, DT Connor, R Doubleday, RJ Sorenson, AD Sercel, PC Unangst, BD Roth, RB Gilbertsen, K Chan, DJ Schrier, A Guglietta, DA Bornemeier, and RD Dyer. (1999). Synthesis, structure-activity relationships, and in vivo evaluations of substituted Di-tert-butylphenols as a novel class of potent, selective, and orally active Cyclooxygenase-2 inhibitors 1. Thiazolone and Oxazolone Series. J Med Chem 42:1151–1160.
- NV Chandrasekharan, H Dai, KL Turepu Roos, NK Evanson, J Tomsik, TS Elton, and DL Simmons. (2002). COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure and expression. PNAS 99:13926–13931.
- A Palomer, F Cabre, J Pascual, J Campos, MA Trujillo, A Entrena, MA Callo, L Garcia, D Mauleon, and A Espinosa. (2002). Identification of novel Cyclooxygenase-2 selective inhibitors using pharmacophore models. J Med Chem 45:1402–1411.
- AK Gadad, BS Kittur, SG Kapsi, CS Mahajanshetti, and SB Rajur. (1996). Synthesis, analgesic and anti-inflammatory activities of some 1-acyl/aracyl-5-aminopyrazole derivatives. Arzneim Forsch 46:1082–1085.
- TD Penning, JJ Talley, SR Bertenshaw, JS Carter, PW Collins, S Docter, MJ Graneto, LF Lee, JW Malecha, JM Miyashiro, RS Rogers, DJ Rogier, SS Yu, GD Anderson, EG Burton, JN Cogburn, SA Gregory, CM Koboldt, WE Perkins, K Seibert, AW Veenhuizen, YY Zhang, and PC Isakson. (1997). Synthesis and biological evaluation of the 1,5-Diarylpyrazole class of Cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J Med Chem 40:1347–1365.
- K Tsuji, K Nakamura, T Ogino, N Konishi, T Tojo, T Ochi, N Seki, and M Matsuo. (1998). Studies on anti-inflammatory agents. VI. Synthesis and pharmacological properties of 2,3-diarylthiophenes. Chem Pharm Bull 46:279–286.
- SA Beers, EA Malloy, W Wu, M Wachter, J Ansell, M Singer, M Steber, A Barbone, T Kirchner, D Ritchie, and D Argentieri. (1997). N-(5-substituted) thiophene-2-alkylsulfonamides as potent inhibitors of 5-lipoxygenase. Bioorg Med Chem Lett 5:779–786.
- DH Boschelli, DT Connor, DA Bornemeier, RD Dyer, JA Kennedy, PJ Kuipers, GC Okonkwo, DJ Schrier, and CD Wright. (1993). 1,3,4-oxadiazole 1,3,4-thiadiazole, and 1,2,4-triazole analogs of the fenamates: In vitro inhibition of cyclooxygenase and 5-lipoxygenase activities. J Med Chem 36:1802–1810.
- A Tanitame, Y Oyamada, K Ofuji, Y Kyoya, K Suziki, H Ito, M Kawasaki, K Nagai, M Wachi, and J Yamagishi. (2004). Design, synthesis and structure-activity relationship studies of novel indazole analogues as DNA gyrase inhibitors with Gram-positive antibacterial activity. Bioorg Med Chem Lett 14:2857–2862.
- A Tanitame, Y Oyamada, K Ofuji, K Suziki, H Ito, M Kawasaki, M Wachi, and J Yamagishi. (2004). Potent DNA gyrase inhibitors; novel 5-vinylpyrazoleanalogues with Gram-positive antibacterial activity. Bioorg Med Chem Lett 14:2863–2866.
- A Tanitame, Y Oyamada, K Ofuji, M Fujimoto, N Iwai, Y Hiyama, K Suziki, H Ito, H Terauchi, M Kawasaki, K Nagai, M Wachi, and J Yamagishi. (2004). Design, synthesis and antibacterial activity of a novel series of potent DNA gyrase inhibitor. Pyrazole derivatives. J Med Chem 47:3693–3696.
- AM Farghaly, AA Bekhit, and JY Park. (2000). Design and synthesis of some oxadiazolyl, thiadiazolyl, thiazolidinyl, and thiazolyl derivatives of 1H-pyrazole as anti-inflammatory antimicrobial agents. Arch Pharm Pharm Med Chem 333:53–57.
- AA Bekhit, and T Abdel-Azeim. (2004). Design, synthesis and biological evaluation of some pyrazole derivatives as anti-inflammatory-antimicrobial agents. Bioorg Med Chem 12:1935–1945.
- AA Bekhit, HTY Fahmy, ShAF Rostom, and AM Baraka. (2003). Design and synthesis of some substituted 1H-pyrazolyl-thiazolo[4,5-d]pyrimidines as anti-inflammatory-antimicrobial agents. Eur J Med Chem 38:27–36.
- AA Bekhit, and HTY Fahmy. (2003). Design and synthesis of some substituted 1H-pyrazolyl-oxazolidines or 1H-pyrazolyl-thiazolidines as anti-inflammatory-antimicrobial agents. Arch Pharm Pharm Med Chem 336:111–118.
- Bekhit AA. Egyptian Patent GATT/TRIPS Treaty, Appl. No. 1117 12 2003..
- AA Bekhit, HMA Ashour, and A Guemei. (2005). Novel Pyrazole Derivatives as Potential Promising Anti-inflammatory Antimicrobial Agents. Arch Pharm Chem Life Sci 338:167–174.
- AA Bekhit, HM Abdel-Rahman, and A Guemei. (2006). Synthesis and biological evaluation of some hydroxypyrazole derivatives as anti-inflammatory-antimicrobial agents. Arch Pharm Chem Life Sci 339:81–87.
- R Meier, W Schuler, and P Desaulles. (1950). On the mechanism of cortisone inhibition of connective tissue proliferation. Experientia 6:469–471.
- M Di Rosa, and DA Willoughby. (1971). Screens for anti-inflammatory drugs. J Pharm Pharmacol 23:297–298.
- K Wakitani, T Nanayama, M Masaki, and M Matsushita. (1998). Profile of JTE-522 as a human cyclooxygenase-2 inhibitor. Jpn J Pharmacol 78:365–371.
- S Hammarström, P Falardeau. Resolution of prostaglandin endoperoxide synthase and thromboxane synthase of human platelets PNAS 1977;74:3691–3695.
- MS Abou Zeit-Har, T Verimer, and JP Long. (1982). Effect of long term estrogen and lithium treatment on restraint induced gastric erosion in intact and ovariectomized rats. Pharmazie 37:593–595.
- M Verma, M Tripathi, AK Saxena, and K Shanker. (1994). Anti-inflammatory activity of novel indole derivatives. Eur J Med Chem 29:941–946.
- JT Litchfield, and F Wilcoxon. (1949). A simplified method of evaluating dose-effect experiments. J Pharmacol Exp Ther 96:99–113.
- Molecular Operating Environment (MOE). Montreal, Canada: Chemical Computing Group, Inc; http://www.chemcomp.com.
- GL Woods, and AW. Washington. In: PR Murray, EJ Baron, MA Pfaller, FC Tenover, and RH Yolken. editors. Manual of Clinical Microbiology. Washington, D.C: Am Soc Microbiol; (1995).
- M Bernard, E Hulley, H Molenda, K Stochla, and U Wrzeciono. (1986). Azole. XVII: β-(4-Pyrazol)acryl- bzw. -propionsäure und ihre antiphlogistische Wirkung. Pharmazie 41:560–562.
- E Pretsch, P Bühlmann, and C Affolter. Structure Determination of Organic Compounds. Berlin Heidelberg New York: Springer-Verlag; (2000) p212.
- AK Sen, and AK Mukhopadhyay. (1984). Synthesis of compounds related to 4(5)-aminoimidazole-5(4)-carboxamides: Part VI – synthesis of 3-(6-methoxy-8-quinolyl)-7-methylpurin-6(3H)-one. Indian J Chem Sect B 28B (9):870–873.
- E Pretsch, P Bühlmann, and C Affolter. editors Structure Determination of Organic Compounds. Berlin Heidelberg New York: Springer-Verlag; (2000). p 211.
- VD Piaz, MP Giovannoni, C Castellana, JM Palacios, J Beleta, T Domench, and V Segarra. (1998). Heterocyclic-fused 3(2H)-pyridazinones as potent and selective PDE IV inhibitors: Further structure-activity relationships and molecular modelling studies. Eur J Med Chem 33:789–797.
- RG Kurumbail, AM Stevens, JK Gierse, JJ McDonald, RA Stegeman, JY Pak, D Gildehaus, JM Iyashiro, TD Penning, K Seibert, PC Isakson, and WC Stallings. (1996). Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 384:644–648.