Abstract
The glutamate receptor system is implicated in the development and maintenance of epileptic seizures and it has been reported that compounds showing high affinity for both AMPA and KA binding sites are more potent anticonvulsants than compounds having selective affinity toward AMPA or KA receptor. These outcomes make such inhibitors future potential antiepileptic drugs. So, the pair wise binding affinity for AMPA and KA receptors inhibition was proposed by using the addition between biological activities of ligands. This approach for evaluation of pair wise binding affinity was exemplified using set of triazolo [1,5-a] quinoxaline for AMPA and KA receptors. The biological activity towards AMPA and KA receptors (expressed as -log IC5O) was taken as a dependent variable for building CoMFA and CoMSIA models. The resulting models show the ways of increasing binding affinity to both AMPA and KA receptors as potential target for epilepsy. The statistically significant results show that pair wise CoMFA and CoMSIA models are better then individual models. The resulting cross-validated r2CV value 0.806 for CoMFA is greater then 0.780 for CoMSIA pair wise model. The non-cross validated run giving a coefficient of determination r2 value of 0.946 and 0.908 for CoMFA and CoMSIA respectively, provided a good correlation between the observed and computed affinities of the compounds.
Introduction
It is well known that to obtain good binding affinity toward particular receptor, is still challenging task. This can be done only through insilco drug design techniques. We hereby propose a term- pair wise binding affinity to designate the affinity to simultaneously bind with given pair of receptor subtypes of same family. In this work AMPA and KA, the subtype of ionotropic glutamate receptors (iGluR- ligandgated ion channels) have been considered as a typical pair for binding affinity.
Endogenous ligand (S)-glutamate (Glu) is the major excitatory neurotransmitter in the central nervous system (CNS), which is responsible for basal excitatory synaptic transmission and many forms of synaptic plasticity. On the other hand, glutamatergic hyperactivity may lead to neurotoxicity. In fact, excessive endogenous Glu is implicated in a number of acute and chronic neurodegenerative pathologies such as epilepsy, cerebral ischaemia and parkinson’s diseases [Citation1]. Glutamate (Glu) activates specific receptors that belong to the classes of metabotropic receptors (mGluRs, coupled to G-protein) and ionotropic receptors (iGluRs, ligandgated ion channel). The latter are divided in to N-methyl-D-aspartic acid (NMDA) receptor, the kainic acid (KA) receptor and the (R,S)-2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)-propionic acid (AMPA) receptor subtype [Citation2, Citation3]. In this paper, AMPA and KA receptors is described as potential targets for antiepileptic therapeutic intervention. Different series of compounds with inhibitory activity toward AMPA and KA receptors have been developed. Most of these inhibitors are structurally derived from AMPA, quinoxaline, quinoxalinedione or 2,3-benzodiazepine and limited number of KA receptor inhibitors contain quinoxalinedione or decahydroisoquinoline scaffold [Citation1, Citation4–9]. Early studies showed that AMPA and KA receptor antagonists were capable of blocking seizures in rodent models of epilepsy [Citation10–11]. In spite of promising anticonvulsant activity in various animal model studies, no AMPA/KA receptor inhibitors are in clinical use against epilepsy today. It has been reported that compounds showing high affinity for both AMPA and KA binding sites are more potent anticonvulsant than the compounds having selective affinity toward AMPA receptor [Citation11, Citation12]. Thus AMPA and KA receptors can be considered as the potential target for therapeutic prevention of epileptic seizers [Citation8, Citation12]. So, we proposed an approach for increasing the pair wise binding affinity of ligands toward both receptor subtypes (AMPA and KA) that may help in the development of potential antiepileptic agents.
The comparative molecular field analysis (CoMFA) [Citation13] and comparative molecular similarity indices (CoMSIA) [Citation14] are widely used tool for predicting biological activity of ligands. They enable designing novel ligands with increased affinity to given receptors. To address the pair wise binding affinity, set of triazolo [1,5-a] quinoxaline with the biological activities expressed as 2logIC50 i.e. negative logarithm of concentration necessary for 50% inhibition of AMPA and KA receptors, was taken as dependent variable for building CoMFA and CoMSIA models. The resulting model would directly suggest the ways of increasing pair wise binding affinity for AMPA and KA receptors.
Experimental
Dataset for study
Reported in vitro AMPA receptor inhibition data on a series of triazolo [1,5-a] quinoxaline derivatives were used for this study () [Citation15–18]. The IC50 values of 35 molecules were segregated into group of 30 and 5 as training set and test set respectively. To get homogeneity of the data Ki (μM) values were converted to IC50 (μM) by Cheng-Prusoff equation [Citation19] and next all IC50 values converted to 2log IC50 (pIC50).
Table 1. Actual and predicted values of triazolo [1,5-a] quinoxaline derivatives.
Molecular modeling
The 3D-QSAR was performed using SYBYL [Citation20] version 6.9 installed on IBM server and Linux as operating system. The most active analogue was subjected to conformational search. The least energy conformer thus obtained as the bioactive conformation was taken as the template and rest of the molecules were built from it. The geometry of all molecules was optimized using the tripose force field and powell’s conjugated gradient with the Gasteiger-Hückel charges. The minimum energy difference of 0.001 kcal mol−1 was set as convergence criterion.
Alignment
CoMFA and CoMSIA studies require the coordinate of molecule to be aligned according to reasonable bioactive conformation. All the molecules were aligned with respect to template molecule No. 22 by the Atom Fit method in SYBYL (version 6.9) molecular modeling software to ensure pharmacophore matching. The alignment of molecules is shown in .
Figure 1. Alignment of triazolo [1,5-a] quinoxaline derivatives.
![Figure 1. Alignment of triazolo [1,5-a] quinoxaline derivatives.](/cms/asset/fe37ac0e-c595-46a7-b1a9-19a0c0e3b662/ienz_a_356965_f0001_b.gif)
CoMFA interaction energy
The van der Waals potential and Coulombic term representing the steric and electrostatic fields respectively was calculated using standard tripose force fields. A distance-dependent dielectric constant of 1.00 was used. A sp3 carbon with a charge of +1 served as a probe atom. CoMFA field descriptors were calculated at each lattice interaction of a regularly space of 2.0 å in all three dimensions with in defined region. The steric and electrostatic fields were truncated at ±30 kcal mol−1. The CoMFA QSAR equations were derived with the partial least squares (PLS) algorithm.
CoMSIA interaction energy
The recently reported CoMSIA method is based on molecular similarity descriptors. CoMSIA calculates the similarity descriptors by way of a grid lattice. For a molecule j with atoms i at the grid point q, the CoMSIA similarity indices AF are calculated by the equation as follows:
AFq (j) = −Σ Wprobe.k Wik exp (αr2iq)
where ωik is the actual value of the physicochemical property k of atom i, ωprobe.k is the property of probe atom with pre-set charge (+1 in this case), radius (1.53 å), and hydrophobicity of 1and riq is the mutual distance between the probe atom at grid point q and atom i of the molecule. In the CoMSIA calculations, five physicochemical properties (steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor) were determined for all of the molecules. The value of attenuation factor was set to 0.3. Similarity descriptors can be calculated at all grid points inside as well as outside the molecule. This generally follows the CoMFA protocols and evaluated by PLS analysis.
PLS analysis
Partial Least Squares analysis (PLA) as performed to obtain a 3D QSAR models after all of the CoMFA and CoMSIA descriptors were calculated. The PLS method has been used to correlate the activity (dependent variable) with various physicochemical properties (independent variables). The CoMFA and CoMSIA standard scaling and column filtering of 0.5 were used in PLS analysis.
Internal cross-validations in PLS were done by the leave-one-out procedure to find out the optimal number of components in building the regression models and to check statistic significance of models. The leave-one-out technique provides a good way to quantitatively evaluate the internal predictive ability of a model by removing one compound out at a time and then building the QSAR model and calculating the activity of the compound using the newly model constructed from remaining compounds in the data set.
The high value of cross-validated coefficient of determination (r2CV) and lowest standard error of prediction resulted in optimum number of components considered for further analysis. The optimal number of components obtained is then used to derive the final QSAR model using all the compounds (without cross-validation). The conventional coefficient of determination (r2) and F value are used to measure the quality of the model.
An external cross-validation was also performed in addition to the internal cross-validation using molecules of the test set (). PLS analysis was used to predict the activity of given test set molecules.
Table 3. Prediction of test series by CoMFA and CoMSIA methods.
Results and discussion
QSAR Models
CoMFA and CoMSIA models were developed with derivatives of triazolo [1,5-a] quinoxaline from inhibition constants (pIC50) with respect to the AMPA and KA receptors, while the third one was built using the addition between pIC50 for AMPA and KA as a target property. Various 3D-QSAR (CoMFA and CoMSIA) models were generated and the best one was selected using the statistically significant parameters. Actual and predicted values obtained after the CoMFA and CoMSIA analysis of triazolo [1,5-a] quinoxaline derivatives are given in .
The PLS analysis for different number of components was tried using 0.0 kcal mol−1 column filtering value for CoMFA and CoMSIA (AMPA, KA and pair wise) models. Based on better statistical values optimum number of components were selected. For the CoMFA pair wise model cross- validated coefficient of determination (r2CV) = 0.806 with six component, non cross-validated coefficient of determination (r2) = 0.944, F value = 67.25 and boot straped r2 = 0.964 were found. The value of standard error of prediction (SEP) and predicted residual sum of square (PRESS) obtained for CoMFA was 0.305 and 0.39 respectively.
The CoMSIA results were obtained using the same alignment and same training set molecules. As depicted in , for CoMSIA analysis individual models were developed taking different combination of fields i.e. steric-electrostatic (S-E), steric-electrostatic-hydrophobic (S-E-H), steric-electrostatic-hydrogen bond donor-hydrogen bond acceptor (S-E-D-A) and using all (S-E-D-A-H) fields. Out of these, model having combination of S, E, D, A fields was slightly better then other models. However, the all field model is the one providing the most descriptive information and hence was used for the prediction of training and test set molecules. The cross-validated r2CV = 0.780 with five component, non cross-validated r2 = 0.908, F value= 47.46 and boot straped r2 = 0.982 was obtained from the CoMSIA-all field model. The value of standard error of prediction (SEP) and predicted residual sum of square (PRESS) obtained for CoMSIA-all field model was 0.315 and 0.41 respectively.
Table 2. The PLS statistics results of CoMFA and CoMSIA 3D QSAR models.
When the standard deviation of the sum (or difference) of two independent random variables take place then standard deviation σ equals to σ /√2. Therefore the value of standard error of estimate 0.292 for CoMFA and 0.373 for CoMSIA would be: 0.292/√2 = 0.206 for CoMFA, and 0.373/√2 = 0.263 for CoMSIA pair wise model. The value of standard error of estimate thus obtained for CoMFA pair wise model 0.206, is slightly greater then 0.187 (AMPA model) and less than 0.216 (KA model), while the SEE value for CoMSIA pair wise model 0.263 is slightly greater then 0.223 (AMPA model) and less then 0.317 (KA model).
The results of 3D QSAR analysis reflect that the r2CV value for CoMFA and CoMSIA pair wise model is greater then AMPA and KA individual models. Non-cross validated run giving coefficient of determination values of 0.946 and 0.908 for CoMFA and CoMSIA respectively, providing good correlation between the observed and predicted affinities of the compound in the training set.
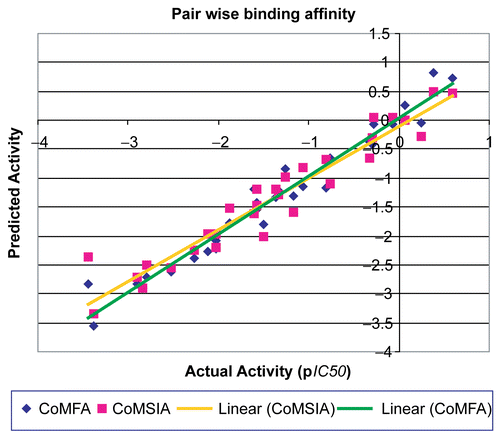
A comparative study data () of CoMFA and CoMSIA models show that the cross-validated r2CV and non cross-validated r2 from CoMFA pair wise model is slightly higher than CoMSIA-all field and SEDA pair wise model. All statistical parameter i.e. F value, SEE, and SEP suggest that CoMFA pair wise model is slightly better than CoMSIA pair wise model for prediction of training and test molecules. The Comparative graphical representation between predicted activity and actual activity related to AMPA and KA receptors obtained from pair wise CoMFA and CoMSIA study is shown in .
Figure 2. Graph between predicted and actual activity for pair wise models of CoMFA and CoMSIA.
The CoMFA and CoMSIA calculated electrostatic, steric, hydrophobic, hydrogen bond donor and acceptor properties are based on the grid built around these molecules. It is observed with the CoMFA results that, the steric and electrostatic contributions (0.524 and 0.476) are nearly equal than the individual models while with CoMSIA results, steric and electrostatic contributions (0.079 and 0.252) of pair wise model are similar to AMPA and different from KA model. CoMFA model shows the nearly equal significance of steric and electrostatic properties while in CoMSIA model electrostatic contribution are almost three fold of the steric contribution, indicating the importance of electrostatic fields in model generation. Remaining all other fields like hydrophobic, hydrogen bond donor and acceptor are virtually equal contributor in the CoMSIA models. Since CoMFA and CoMSIA models have almost equal statistically significant parameters, so either of the CoMFA or CoMSIA model could be used for prediction of activity but CoMSIA can be preferred to explain the importance of hydrophobic, hydrogen bond donor and acceptor properties together with stereo-electric parameter.
In addition to internal validation by LOO method external validation was also performed. For external validation test molecules were so selected, so as to include variety of substituents at position R7 and R8, and possessing triazolo [1,5-a] quinoxaline as common molecular scaffold (). Prediction of activity for test molecules was obtained from pair wise CoMFA and CoMSIA-all models with predicted residual sum of square (PRESS) value of 0.88 and 1.98, respectively.
Graphical interpretation of results
3D QSAR models depict the change in binding preference occurring upon the change in molecular fields around ligands. Counter maps were generated as scalar product of coefficients and standard deviation associated with each CoMFA column. The compound No. 22 (template), the most active molecule of the series is used for the presentation of contour maps.
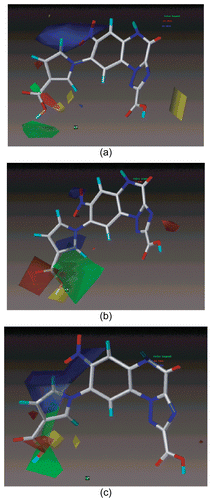
The CoMFA steric interactions are represented by green and yellow colored contours while electrostatic interactions are represented by red and blue colored contours. The bulky substituents are favored in green regions and disfavored in yellow regions. The increase in positive charge is favored in blue regions while the increase in negative charge is favored in red regions. Significant difference in stereo-electrostatic field is evident in contour plots for individual AMPA and KA CoMFA models (), and pair wise CoMFA model. In CoMFA pair wise model (), sterically favorable green contour and small electronegative favorable red contour near 1H-pyrrole-3-carboxylic acid group at position 8 of triazolo [1,5-a] quinoxaline ring support the incorporation of bulky and electronegative substituents specifically on heterocyclic group of position-8 for increasing the binding affinity. As is evident from the comparison, molecule No. 13 with 1H-pyrrole-3-carboxylic acid have more activity than molecule 9 with 1H-pyrrole-3-carbaldehyde substituents at R8 for AMPA and KA receptors. An electropositive favorable blue contour surrounding the position R7 and R8 of quinoxaline moiety suggest the addition of the positive charged substituents on the position R7 and R8 would increase pair wise binding affinity for both AMPA and KA receptors.
Figure 3. CoMFA STDEV*COEFF contour maps for steric and electrostatic fields of a) AMPA, b) KA, c) Pair wise Model. The most active molecule 22 is displayed in the background.
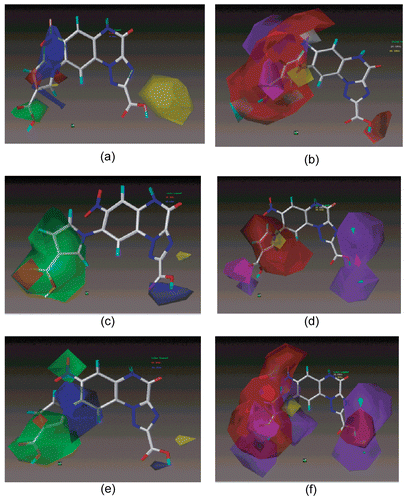
Considering the steric contours of CoMSIA, green contour indicate favorable regions while yellow contours indicate unfavorable regions for bulkier substituents. In the electrostatic contours, the introduction of electronegative substituents in red regions may increase the affinity while in blue regions decrease the affinity. In hydrophobic contours, grey regions favor hydrophobic group while yellow favor hydrophilic groups. The cyan and purple contours denote favorable and unfavorable regions for hydrogen bond donor groups while magenta and red contours denote the favorable and unfavorable regions for hydrogen bond acceptor groups. In CoMSIA pair wise model () the stereo-electric contours are almost similar with CoMFA excluding sterically favorable green color contour on the position-7 and 8 of substituted pyrrole ring attached to quinoxaline scaffold. The two contour maps for bulky and positive charged region are major contributors which imply presence of bulky electropositive charged substituents at position-7 and -8 while carboxyl group at position 2 might increase the binding affinity e.g. molecule 22. In the unfavorable hydrogen bond donor (purple) and hydrogen bond acceptor (red) contour maps share major portion at R7 and R8 of the quinoxaline ring indicate that absence of hydrogen bond donor and acceptor group near R7 and R8 respectively, may be responsible for increasing the pair wise binding affinity. One magenta color contour close to 3-carboxy-pyrol at position-8 of quinoxaline ring suggest that hydrogen bond acceptor group at this position possibly will increase the binding affinity. A small yellow contour on pyrole ring attached to position-8 of quinoxaline scaffold recommends the presence of hydrophilic group for increasing affinity. Apart from all, two heavy purple color contours and one small red contour near position-2 of triazolo-quinoxaline skeleton indicate the absence of hydrogen bond donor and presence of hydrogen bond acceptor group would be able to increase the pair wise affinity for the receptor subtypes. Thus CoMFA and CoMSIA models suggest that by modifying the structure of ligands particularly at position R7, R8 and R2, one could gain the higher pair wise binding affinity toward AMPA and KA receptors.
Figure 4. CoMSIA STDEV*COEFF contour maps for steric and electrostatic fields of a) AMPA, c) KA, e) Pair wise model. Hydrophobic, donor and acceptor field of b) AMPA, d) KA, f) Pair wise model. The most active molecule 22 is displayed in the background.
Conclusions
3D QSAR analysis makes it possible to relate chemical structures of ligands and their binding affinity with respect to different bio targets by using the CoMFA and CoMSIA techniques. Consequently it provides a direct view of factors expressed in terms of CoMFA and CoMSIA molecular fields (electrostatic, steric, hydrophobic, hydrogen bond donor and acceptor) affecting the binding affinity, which in turn could give the reasonably good prediction of binding affinity.
The statistical model for the pair wise binding affinity analysis cannot be directly derived from CoMFA or CoMSIA model of individual receptor. So, the pair wise model was proposed to predict the pair wise binding affinity with respect to both AMPA and KA receptors. These models could give reasonably good prediction of pair wise binding affinity rather then individual models. Finally, this analysis is helpful in suggesting some structural modification in known compounds for designing novel ligands, which might be more effective as anticonvulsive agents.
Acknowledgements
Author wishes to thank Dr. Harish Padh, Director, B. V. Patel Pharmaceutical Education and Research Development (PERD) Center for providing the software facility. This work was supported by the junior research fellowship provided from All India Council of Technical Education (AICTE) to the author.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bräuner-Osborne H, Egebjerg J, Nielsen Eø, Madsen U, Krogsgaard-Larsen P. Ligands for glutamate receptor: design and therapeutic prospects. J Med Chem (2000); 43:2609.
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev (1999); 51:7.
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci (1994); 17:103.
- Lees GJ. Pharmacology of AMPA/kainate receptor ligands and their therapeutic potential in neurological and psychiatric disorders. Drugs (2000); 59:33.
- Madsen U, Stensbøl TB, Krogsgaard-Larsen P. Inhibitors of AMPA and kainate receptors. Curr Med Chem (2001); 81:291.
- Nikam SS, Kornberg BE. AMPA receptor antagonists. Curr Med Chem (2001); 8:155.
- Stensbøl TB, Madsen U, Krogsgaard-Larsen P. The AMPA receptor binding site: focus on agonists and competitive antagonists. Curr Pharm Des (2002); 8:857.
- Auberson YP. Competitive AMPA antagonism: a novel mechanism for antiepileptic drugs. Drugs Fut (2001); 26:463.
- Donevan SD, Rogawski M. GYKI 52466, a 2, 3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron (1993); 10:51.
- Tortorella A, Halonen T, Sahibzada N, Gale K. A crucial role of the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype of glutamate receptors in piriform and perirhinal cortex for the initiation and propagation of limbic motor seizures. J Pharmacol Exp Ther (1997); 280:1401.
- Löscher W. Pharmacology of glutamate receptor antagonists in the kindling model of epilepsy. Prog Neurobiol (1998); 54:721.
- Löscher W, Lehmann H, Behl B, Seemann D, Teschendorf HJ, Hofmann HP, Lubisch W, Hoger T, Lemaire HG, Gross G. A new pyrrolyl-quinoxalinedione series of non-NMDA glutamate receptor antagonists: pharmacological characterization and comparison with NBQX and valproate in the kindling model of epilepsy. Eur J Neurosci (1999); 11:250.
- Desraju GR, Gopalakrishnan B, Jetti RK, Nagarjun A, Raveendra D, Sarma JA, Sobhia ME, Thiagavathi R. Computer-aided design of selective cox-2 inhibitors: comparative molecular field analysis, comparative molecular similarity indices analysis, and docking studies of some 1,2-diarylimidazole derivatives. J Med Chem (2002); 45:4847.
- TBohm M, Sturzebecher J, Klebe G. Three dimensional quantitative structure activity relationship analysis using comparative molecular field analysis and comparative molecular similarity search analysis to elucidate selectivity differences of inhibitor binding to trypsin, thrombin and factor Xa. J Med Chem (1999); 42:458.
- Catarzi D, Colotta V, Varano F, Cecchi L, Filacchioni G, Galli A, Costagli C, Carla V. 7-Chloro-4-5-dihydro-8-(1,2,4-triazole-4-yl)-4-oxo-1,2,4-triazolo[1,5-a]quinoxaline-2-carbo-xylates as novel highly selective AMPA receptor antagonists. J Med Chem (2000); 43:3824.
- Catarzi D, Colotta V, Varano F, Cecchi L, Filacchioni G, Calabri VR, Galli A, Costagli C, Carla V. Synthesis and biological evaluation of analogues of 7-chloro-4,5-dihydro-4-oxo-8-(1,2,4-triazol-4-yl)-1,2,4-triazolo[1,5-a] quinox-aline-2-carboxylic acid (TQX-173) as novel selective AMPA receptor antagonists. J Med Chem (2004); 47:262.
- Catarzi D, Colotta V, Varano F, Cecchi L, Filacchioni G, Galli A, Costagli C, Carla V. Synthesis, ionotropic glutamate receptor binding affinity, and structure-activity relationships of a new set of 4,5-dihydro-8-heteroaryl-4-oxo-1,2,4-triazolo[1,5-a] quinoxaline-2-carboxylates analogues of TQX-173. J Med Chem (2001); 44: 3157.
- Varano F, Catarzi D, Colotta V, Filacchioni G, Galli A, Costagli C, Carla V. Synthesis and biological evaluation of a new set of pyrazolo [1,5-c]quinazoline-2-carboxylates as novel excitatory amino acid antagonists. J Med Chem (2002); 45:1035.
- Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol (1973); 22:3099.
- Sybyl ver. 6.9, Tripos Inc., St. Louis, (MO) USA.