
Abstract
Nineteen new compounds containing tetrazole and/or cyanamide moiety have been designed and synthesised. Their structures were confirmed using spectroscopic methods and elemental analyses. Anti-inflammatory activity for all the synthesised compounds was evaluated in vivo. The most active compounds 4c, 5a, 5d–f, 8a and b and 9a and b were further investigated for their ulcerogenic liability and analgesic activity. Pyrazoline derivatives 9b and 8b bearing trimethoxyphenyl part and SO2NH2 or SO2Me pharmacophore showed equal or nearly the same ulcerogenic liability (UI: 0.5, 0.75, respectively), to celecoxib (UI: 0.50). Most of tested compounds showed potent central and/or peripheral analgesic activities. Histopathological investigations were done to evaluate test compounds effect on rat's gastric tissue. The obtained results were in consistent with the in vitro data on COX evaluation. Docking study was also done for all the target compounds inside COX-2-active site.
Introduction
Tetrazole represents an important class of heterocyclic compounds for biological and pharmacological applications. The tetrazole ring is a bioisostere of the carboxylic acid (–COOH) group so they have close pKa values, also, it has similar planar skeleton structure and nitrogen-rich multi-electron conjugated systemCitation1–4. At the same time, it does not have the acidic –COOH properties, and so, it helps in decreasing toxic properties of drugsCitation5. 1,2,3,4-Tetrazole derivatives possess potent pharmacological activities such as antimicrobialCitation6–9, antihypertensiveCitation10,Citation11, anticonvulsantCitation12,Citation13, anticancerCitation14,Citation15, analgesicCitation4,Citation16, antiulcerCitation17 and anti-inflammatoryCitation3,Citation18–22.
On the other hand, pyridine scaffoldCitation23–25 was the base of many bioactive molecules especially anti-inflammatory agents. Moreover, pyrazoline ringCitation26–29, hydrazone functionalityCitation30 and chalconesCitation31–33 are attractive motifs for anti-inflammatory drugs.
Non-steroidal anti-inflammatory drugs (NSAIDs) are of great importance in the treatment of inflammation and pain. They act by inhibiting cyclooxygenase enzymes (COXs) – a membrane-bound haeme protein – that control the conversion of arachidonic acid to prostaglandins and thromboxanes. Two distinct isoforms of COX enzyme are present, a constitutive form (COX-1), associated with several side effects such as haemorrhagia and gastrointestinal (GI) ulceration, and an inducible form (COX-2) which is different in the regulation and tissue distribution from COX-1Citation34–36. In 2012, Al-Hourani et al. reported a novel COX-1 splice variant termed as COX-3Citation37. COX-1 and COX-2 use identical catalytic mechanism to catalyse the same reactions by sharing the same substrates and producing the same products. They are very similar in their protein tertiary conformation as demonstrated from their X-ray crystal structuresCitation38. These similarities in the structure of both COX isoforms represent a great challenge for the development of selective COX-2 inhibitors. The space of selectivity pocket is the main difference between the two isoforms. It is reduced in COX-1 due to the presence of Ile523 rather than Val523 in COX-2. Conformational change is occurred as a result of the presence of Val523 in COX-2, leading to the formation of additional hydrophobic secondary internal pocket protruding off the primary binding siteCitation39. A large number of compounds have been synthesised and evaluated for their selective COX-2 inhibitory activity. Celecoxib (I), rofecoxib (II), valedecoxib (III) and etoricoxib (IV) are the most common approved selective COX-2 inhibitors. However, inhibition of COX-2 reduces urinary sodium excretion leading to increase in blood pressure as well as myocardial infarction, that is why rofecoxib and valdecoxib had been withdrawn from the market ()Citation40–42.
Figure 1. General structure of some known selective COX-2 inhibitors.
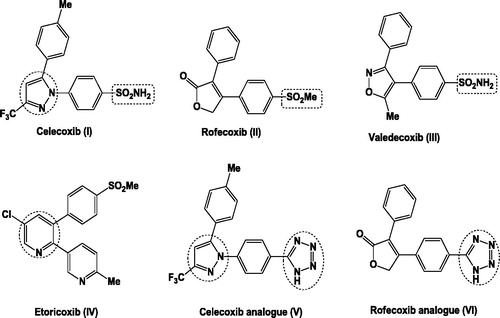
Until now, celecoxib has been one of the most popular selective COX-2 drugs. So, there is still a need for designing and developing new effective anti-inflammatory drugs with improved safety profiles.
New water-soluble, parentral COX-2 inhibitors containing tetrazole moiety in their structures, such as celecoxib and rofecoxib analogues (V, VI, respectively) were reported to have in vivo anti-inflammatory activity and exhibited a high COX-2/COX-1 selectivity (SI = 2.16, 2.11, sequentially) when compared to the reference celecoxib (SI = 1.68). Both of them lacked the side effect of gastric ulceration ()Citation43.
The majority of selective COX-2 inhibitors are of five-membered heterocyclic ring as pyrazole in celecoxib (I), or attached to pyridine six-membered ring as in etoricoxib (IV)Citation44. Moreover, pyrazoline present in antipyrin – the first pyrazoline derivative used in the treatment of pain and inflammation – and several related analogues as in felcobuzone, morazone and ramifenazone are also available as NSAIDsCitation45.
Furthermore, different important selective COX-2 pharmacophores such as aminosulphonyl and methylsulphonyl groups are essential for potent and selective anti-inflammatory activityCitation40,Citation41.
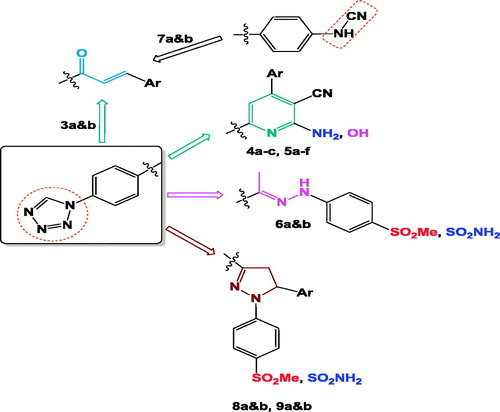
In view of the above-mentioned facts and as a continuation of our previous work on the development of selective COX-2 inhibitorsCitation36,Citation40,Citation46, four groups of compounds have been synthesised: (i) chalcone derivatives 3a and b and 7a and b, (ii) pyridine containing compounds 4a–c and 5a–f, (iii) hydrazone of methylsulphonyl and aminosulphonyl derivatives 6a and b and (iv) five-membered pyrazoline ring-bearing methylsulphonyl 8a and b group or aminosulphonyl 9a and b moiety, (. Most of the prepared compounds were linked to tetrazole ring aiming to generate novel molecular templates for safe anti-inflammatory agents. The synthesised compounds were subjected to in vitro evaluation as (COX-1/COX-2) inhibitors and in vivo (AI) activity. Analgesic activity and ulcer index (UI) have also been studied. Moreover, the effect of the most active synthesised compounds on rat’s gastric tissue was evaluated using histopathological study. Finally, docking study was performed on COX-2 enzyme to explore the possible binding mode of the designed compounds inside the enzyme.
Figure 2. Design for the newly synthesised compounds 3a&b: 9a&b.
Experimental section
Chemistry
Melting points were determined using a Griffin apparatus and were uncorrected. IR spectra were recorded on a Shimadzu IR-435 spectrophotometer using KBr discs and values were represented in cm−1. 1H NMR and 13 C NMR (DEPT-Q) were carried out on Bruker apparatus at 400 MHz for 1H NMR and 100 MHz for 13 C NMR spectrophotometer, (Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt), in DMSO-d6, D2O using TMS as an internal standard and chemical shifts were recorded in ppm on δ scale using DMSO-d6 (2.5) as a solvent. Coupling constant (J) values were estimated in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet, t, triplet; q, quartet; dd, doublet of doublet; m, multiplet. The electron impact (EI) mass spectra were recorded on Hewlett Packard 5988 spectrometer (Palo Alto, CA). Microanalysis was performed for C, H, N on Perkin-Elmer 2400 at the Microanalytical center, Cairo University, Egypt and was within ±0.4% of theoretical values. Analytical thin-layer chromatography (TLC): pre-coated plastic sheets, 0.2 mm silica gel with UV indicator (Macherey-Nagel) was employed routinely to follow the course of reactions and to check the purity of products. All other reagents, solvents and compound 1 were purchased from the Aldrich Chemical Company (Milwaukee, WI), were used without further purification.
General procedure for synthesis of compounds 3a and b
A mixture of 2 (2 g, 0.01 mol) and KOH (0.56 g, 0.01 mol) in absolute ethanol (20 mL), was stirred at room temperature for 30 min. Then, the corresponding aldehyde (0.01 mol) was added and the reaction mixture was stirred at room temperature for about 10–12 h. The solid separated was filtered, dried and crystallised from DMF/ethanol (1:2) to give 3a and b.
(ZE)-1-[4-(1H-Tetrazol-1-yl)phenyl]-3-(3,4-dimethoxyphenyl)prop-2-en-1-one (3a)
Yellow solid; (1.53 g, 86%) yield; mp 214–216 °C; IR (KBr) 1674 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.83 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 7.05 (d, J= 8 Hz, 1H, dimethoxyphenyl H-5), 7.45 (d, J= 8 Hz, 1H, dimethoxyphenyl H-6), 7.58 (s, 1H, dimethoxyphenyl H-2), 7.77 (d, J= 15.2 Hz, 1H, COCH = CH), 7.90 (d, J = =15.2, 1H, COCH = CH), 8.12 (d, J= 8 Hz, 2H, phenyl H-3, H-5), 8.42 (d, J = 8 Hz, 2H, phenyl H-2, H-6), 10.27 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 56.63 (OCH3), 60.53 (OCH3), 107.16 (dimethoxyphenyl C-2), 107.78 (dimethoxyphenyl C-5), 121.40 (–CO–CH = CH–), 121.46 (dimethoxyphenyl C-6), 121.55 (phenyl C-3, C-5), 130.52 (dimethoxyphenyl C-1), 130.93 (phenyl C-2, C-6), 137.17 (phenyl C-1), 138.59 (phenyl C-4), 142.90 (tetrazole C-5), 145.87 (–CO–CH = CH–), 153.58 (dimethoxyphenyl C-3), 188.60 (C = O); EIMS (m/z) 336 (M+., 31.93%), 145 (100%). Anal. Calcd for C18H16N4O3: C, 64.28; H, 4.79; N, 16.66. Found: C, 64.37; H, 4.65; N, 16.51.
(ZE)-1-[4-(1H-Tetrazol-1-yl)phenyl]-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (3b)
Yellow solid; (1.53 g, 79%) yield; mp 215–217 °C; IR (KBr) 1663 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.73 (s, 3H, p-OCH3), 3.88 (s, 6H, 2 m-OCH3), 7.28 (s, 2H, trimethoxyphenyl H-2, H-6), 7.77 (d, J = 15.2 Hz, 1H, COCH = CH), 7.95 (d, J = 15.2, 1H, COCH = CH), 8.16 (d, J = 8.4 Hz, 2H, phenyl H-3, H-5), 8.42 (d, J = 8.4 Hz, 2H, phenyl H-2, H-6), 10.27 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 56.65 (2 m-OCH3), 60.64 (p-OCH3), 107.25 (trimethoxyphenyl C-2, C-6), 121.41 (–CO–CH = CH–), 121.52 (phenyl C-3, C-5), 130.54 (trimethoxyphenyl C-1), 130.93 (phenyl C-2, C-6), 137.23 (trimethoxyphenyl C-4), 138.60 (phenyl C-1), 140.46 (phenyl C-4), 142.97 (tetrazole C-5), 145.86 (–CO–CH = CH–), 153.60 (trimethoxyphenyl C-3, C-5), 188.49 (C = O); Anal. Calcd for C19H18N4O4: C, 62.29; H, 4.95; N, 15.29. Found: C, 62.15; H, 5.03; N, 15.32.
General procedure for synthesis of 4a–c
Method A
To a mixture of 2 (2 , 0.01 mol) and the appropriate aromatic aldehyde (0.01 mol) in absolute ethanol (20 mL), malononitrile (0.66 g, 0.01 mol) and ammonium acetate (1.54 g, 0.02 mol) were added. The reaction mixture was refluxed for 20 h. The reaction mixture was cooled then poured into crushed ice. The obtained solid was filtered off, dried and crystallised from 95% ethanol to give 4a and b.
Method B (for preparation of 4c)
An ethanolic mixture of chalcone 3a (3.36 g, 0.01 mol) and malononitrile (0.66 g, 0.01 mol) in the presence of ammonium acetate (1.54 g, 0.02 mol) was refluxed for 8–10 h. After cooling, the obtained solid was filtered off, dried and crystallised from 95% ethanol to give 4c.
6-[4-(1H-Tetrazol-1-yl)phenyl]-2-amino-4-phenylnicotinonitrile (4a)
Yellow solid; method A; (1.62 g, 45%) yield; mp 238–240 °C; IR (KBr) 3229, 3117 (NH2), 2210 (C≡N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 6.93–6.94 (m, 3H, phenylnicotinonitrile H-3, H-4, H-5), 7.54–7.66 (m, 5H, phenylnicotinonitrile H-2, H-6, pyridine H-5, NH2, D2O exchangeable), 7.95 (d, 2H, J = 8.4 Hz, phenyl H-3, H-5), 8.08 (d, 2H, J = 8.4 Hz, phenyl H-2, H-6), 10.20 (s, 1H, tetrazole H-5); 13 C NMR (100 MHz, DMSO-d6) δ: 97.05 (pyridinecarbonitrile C-3), 107.93 (pyridinecarbonitrile C-5), 116.81 (C≡N), 121.61 (phenyl-6-yl C-2, C-6), 128.76 (phenyl-4-yl C-2, C-6), 129.31 (phenyl-4-yl C-3, C-5), 130.08 (phenyl-6-yl C-3, C-5), 130.98 (phenyl-4-yl C-4), 133.93 (phenyl-4-yl C-4), 135.83 (phenyl-6-yl C-1), 136.35 (phenyl-4-yl C-1), 142.79 (tetrazole C-5), 160.12 (pyridine C-4), 162.71 (pyridine C-6), 162.87 (pyridine C-2); EIMS (m/z) 339 (M+., 0.84%), 335 (100%). Anal. Calcd for C19H13N7: C, 67.25; H, 3.86; N, 28.89. Found: C, 67.41; H, 3.92; N, 28.74.
6′-[4-(1H-Tetrazol-1-yl)phenyl]-2′-amino-[3,4′-bipyridine]-3′-carbonitrile (4b)
Yellow solid; method A; (2.21 g, 57%) yield; mp 238–240 °C; IR (KBr) 3352, 3136 (NH2), 2210 (C≡N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 6.99 (s, 2H, NH2, D2O exchangeable), 7.03 (s, 1H, pyridinecarbonitrile H-5), 7.59 (dd, J = 4.8 Hz, 8 Hz, 1H, pyridine H-5), 7.96 (d, J = 8.4 Hz, 2 H, phenyl H-3, H-5), 8.09–8.13 (m, 3H, phenyl H-2, H-6 and pyridine H-6), 8.73 (d, J= 4.8 Hz, 1H, pyridine H-4), 8.86 (s, 1H, pyridine H-2), 10.20 (s, 1H, tetrazole H-5); 13 C NMR (100 MHz, DMSO-d6) δ: 95.09 (pyridinecarbonitrile C-3), 112.99 (pyridinecarbonitrile C-5), 116.24 (C≡N), 121.85 (phenyl C-2, C-6), 124.00 (pyridine C-5), 130.94 (phenyl C-3, C-5), 134.74 (phenyl C-4), 137.16 (phenyl C-1), 138.75 (pyridine C-6), 139.65 (pyridine C-1), 142.68 (tetrazole C-5), 149.07 (pyridine C-4), 150.91 (pyridine C-2), 154.46 (pyridinecarbonitrile C-6), 157.05 (pyridinecarbonitrile C-4), 162.14 (pyridinecarbonitrile C-2); Anal. Calcd for C18H12N8: C, 63.52; H, 3.55; N, 32.92. Found: C, 63.35; H, 3.42; N, 32.80.
6-[4-(1H-Tetrazol-1-yl)phenyl]-2-amino-4-(3,4-dimethoxyphenyl)nicotinonitrile (4c)
Yellow solid; method B; (2.06 g, 61%) yield; mp 230–232 °C; IR (KBr) 3210, 3121 (NH2), 2207 (C≡N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.82 (s, 6H, 2OCH3), 6.95 (s, 1H, dimethoxyphenyl H-2), 7.01–7.06 (m, 2H, dimethoxyphenyl H-5, H-6), 7.20 (s, 2H, NH2, D2O exchangeable), 7.78–7.88 (m, 3H, phenyl H-3, H-5, pyridine H-5), 8.15 (d, J = 8.4 Hz, 2H, phenyl H-2, H-6), 10.21 (s, 1H, tetrazole H-5); 13 C NMR (100 MHz, DMSO-d6) δ: 56.03 (OCH3), 56.35 (OCH3), 90.87 (pyridine C-3), 109.17 (pyridine C-5), 110.95 (dimethoxyphenyl C-5), 112.19 (dimethoxyphenyl C-2), 114.64 (C≡N), 117.59 (dimethoxyphenyl C-6), 121.50 (phenyl C-2, C-6), 130.59 (dimethoxyphenyl C-1), 130.89 (phenyl C-3, C-5), 134.53 (phenyl C-4), 139.64 (phenyl C-1), 142.85 (tetrazole C-5), 149.10 (dimethoxy phenyl C-3), 151.08 (dimethoxyphenyl C-4), 154.02 (pyridine C-6), 157.00 (pyridine C-4), 161.74 (pyridine C-2); EIMS (m/z) 399 (M+., 3.64%), 214 (100%). Anal. Calcd for C21H17N7O2: C, 63.15; H, 4.29; N, 24.55. Found: C, 63.44; H, 4.17; N, 24.38.
General procedure for synthesis of 5a–f
Method A
To a mixture of 2 (2 g, 0.01 mol) and the corresponding aromatic aldehyde (0.01 mol) in absolute ethanol (20 mL), ethyl cyanoacetate (1.13 g, 0.01 mol) and ammonium acetate (1.54 g, 0.02 mol) were added. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled and then poured into crushed ice. The obtained solid was filtered off, dried and crystallised from 95% ethanol to give 5a–d.
Method B (for preparation of 5e and 5f)
An ethanolic mixture of the selected chalcones 3a or 3b (0.01 mol) and ethyl cyanoacetate (1.13 g, 0.01 mol) in the presence of ammonium acetate (1.54 g, 0.02 mol) was refluxed for 5–6 h. After cooling, the obtained solid was filtered off, dried and crystallised from 95% ethanol to give 5e and 5f.
6-[4-(1H-Tetrazol-1-yl)phenyl]-2-oxo-4-phenyl-1,2-dihydropyridine-3-carbonitrile (5a)
Yellow solid; method A, (1.73 g, 48%) yield; mp 281–283 °C; IR (KBr) 3483 (OH), 2222 (C≡N), 1659 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 7.02 (s, 1H, pyridine H-5), 7.59–7.60 (m, 3H, phenylnicotinonitrile H-3, H-4, H-5), 7.76–7.78 (m, 2H, phenylnicotinonitrile H-2, H-6), 8.11 (d, J = 8.8 Hz, 2H, phenyl H-3, H-5), 8.22 (d, J = 8.8 Hz, 2H, phenyl H-2, H-6), 10.22 (s, 1H, tetrazole H-5), 12.94 (s, 1H, OH, D2O exchangeable); 13 C NMR (100 MHz, DMSO-d6) δ: 95.06 (pyridinecarbonitrile C-3), 116.35 (C≡N), 119.00 (pyridinecarbonitrile C-5), 121.43 (phenyl-6-yl C-2, C-6), 127.58 (phenyl-4-yl C-2, C-6), 128.87 (phenyl-4-yl C-3, C-5), 129.20 (phenyl-4-yl C-4), 130.02 (phenyl-6-yl C-3, C-5), 134.84 (phenyl-6-yl C-4), 137.82 (phenyl-6-yl C-1), 139.00 (phenyl-4-yl C-1), 142.88 (tetrazole C-5), 148.76 (pyridine C-4), 150.54 (pyridine C-6), 154.58 (pyridine C-2); Anal. Calcd for C19H12N6O: C, 67.05; H, 3.55; N, 24.69. Found: C, 66.89; H, 3.61; N, 24.53.
6′-[4-(1H-Tetrazol-1-yl)phenyl]-2′-hydroxy-[3,4′-bipyridine]-3′-carbonitrile (5b)
Yellow solid; method A; (2.06 g, 57%) yield; mp 287–289 °C; IR (KBr) 3333 (OH), 3132 (NH), 2214 (C≡N), 1652 (C = O); 1H NMR (400 MHz, DMSO-d6) δ: 7.19 (s, 1H, pyridine carbonitrile H-5), 7.64 (dd, J = 8, 4.8 Hz, 1H, pyridine H-5), 8.13 (d, J = 8.8 Hz, 2H, phenyl H-2, H-6), 8.20–8.26 (m, 3H, phenyl H-3, H-5 and pyridine H-6), 8.77 (dd, J = 4.8, 1.2 Hz, 1H, pyridine H-4), 8.97 (s, 1H, pyridine H-2), 10.23 (s, 1H, tetrazole H), 12.91 (s, 1H, OH, D2O exchangeable); 13 C NMR (100 MHz, DMSO-d6) δ: 95.14 (pyridinecarbonitrile C-3), 116.20 (C≡N), 119.05 (pyridinecarbonitrile C-5), 121.76 (phenyl C-2, C-6), 124.09 (pyridine C-5), 130.98 (phenyl C-3, C-5), 133.74 (phenyl C-4), 134.88 (phenyl C-1), 136.80 (pyridine C-6), 138.82 (pyridine C-1), 142.87 (tetrazole C-5), 147.22 (pyridinecarbonitrile C-6), 149.00 (pyridinecarbonitrile C-4), 149.25 (pyridine C-4), 150.83 (pyridine C-2), 154.53 (pyridinecarbonitrile C-2); EIMS (m/z) 341 (M+., 0.77%), 288 (100%). Anal. Calcd for C18H11N7O: C, 63.34; H, 3.25; N, 28.73. Found: C, 63.25; H, 3.13; N, 28.56.
6-[4-(1H-Tetrazol-1-yl)phenyl]-4-(4-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5c)
Yellow solid; method A; (1.92 g, 49%) yield; mp > 300 °C; IR (KBr) 3483 (OH), 3117 (NH), 2222 (C≡N), 1659 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.86 (s, 3H, OCH3), 6.97 (s, 1H, pyridine H-5), 7.14 (d, J = 8.8 Hz, 2H, Ar-H, p-methoxyphenyl H-3, H-5), 7.77 (d, J = 8.4 Hz, 2H, phenyl H-3, H-5), 8.00 (d, J = 8.8 Hz, 2H, p-methoxyphenyl H-2, H-6), 8.20 (d, J = 8.4 Hz, 2H, phenyl H-2, H-6), 10.22 (s, 1H, tertrazole H), 12.86 (s, 1H, OH, D2O exchangeable); 13 C NMR (100 MHz, DMSO-d6) δ: 55.93 (OCH3), 92.01 (pyridine C-3), 105.05 (pyridine C-5), 114.77 (p-methoxyphenyl C-3, C-5), 115.93 (C≡N), 121.78 (phenyl C-2, C-6), 128.78 (p-methoxyphenyl C-1), 129.97 (phenyl C3, C-5), 131.41 (p-methoxyphenyl C-2, C-6) 135.24 (phenyl C-4), 137.26 (phenyl C-1), 142.91 (tetrazole C-5), 153.83 (pyridine C-6), 158.55 (pyridine C-4), 162.32 (p-methoxyphenyl C-4), 164.00 (pyridine C-2); Anal. Calcd for C20H14N6O2: C, 64.86; H, 3.81; N, 22.69. Found: C, 64.72; H, 4.05; N, 22.58.
6-[4-(1H-Tetrazol-1-yl)phenyl]-4-[4-(dimethylamino)phenyl]-2-oxo-1,2-dihydropyridine-3-carbonitrile (5d)
Brown solid; method A; (1.67 g, 41%) yield; mp 263–265 °C; IR (KBr) 3445 (OH), 3129 (NH), 2210 (C≡N), 1705 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.03 (s, 6H, 2CH3), 6.84–6.86 (m, 3H, dimethylaminophenyl H-3, H-5, pyridine H-5), 7.72 (d, J = 7.6 Hz, 2H, dimethylaminophenyl H-2, H-6), 8.10 (d, J = 8.4 Hz, 2H, phenyl H-3, H-5), 8.17 (d, J = 8.4 Hz, 2H, phenyl H-2, H-6), 10.22 (s, 1H, tetrazole H), 12.65 (s, 1H, OH, D2O exchangeable); 13 C NMR (100 MHz, DMSO-d6) δ: 49.06 (2 NCH3), 93.76 (pyridine C-3), 101.07 (pyridine C-5), 112.01 (dimethylaminophenyl C-3, C-5), 117.87 (C≡N), 121.61 (phenyl C-2, C-6), 122.40 (dimethylaminophenyl C-1), 129.95 (dimethylaminophenyl C-2, C-6), 130.23 (phenyl C3, C-5), 135.69 (phenyl C-4), 138.69 (phenyl C-1), 142.82 (tetrazole C-5), 151.76 (pyridine C-6), 152.36 (pyridine C-4), 154.38 (dimethylaminophenyl C-4), 162.31 (pyridine C-2); EIMS (m/z) 383 (M+., 2.85%), 144 (100%). Anal. Calcd for C21H17N7O: C, 65.79; H, 4.47, N, 25.57. Found: C, 65.56; H, 4.53; N, 25.41.
6-[4-(1H-Tetrazol-1-yl)phenyl]-4-(3,4-dimethoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5e)
Yellow solid; method B; (2.28 g, 57%) yield; mp 272–274 °C; IR (KBr) 3325 (OH), 3132 (NH), 2218 (C≡N), 1686 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.86 (s, 6H, 2 OCH3), 6.99 (s, 1H, pyridine H-5), 7.01 (d, J = 8 Hz, 1H, dimethoxyphenyl H-5), 7.38–7.40 (m, 2H, dimethoxyphenyl H-2, H-6), 8.11 (d, J = 8.4 Hz, 2H, phenyl H-3, H-5), 8.20 (d, J = 8.4 Hz, 2H, phenyl H-2, H-6), 10.23 (s, 1H, tetrazole H), 12.86 (s, 1H, OH, D2O exchangeable); 13 C NMR (100 MHz, DMSO-d6) δ: 56.18 (OCH3), 56.21 (OCH3), 92.32 (pyridine C-3), 100.91 (pyridine C-5), 112.10 (dimethoxyphenyl C-5), 112.43 (dimethoxyphenyl C-2), 117.29 (C≡N), 121.61 (dimethoxyphenyl C-6), 122.05 (phenyl C-2, C-6), 122.40 (dimethoxyphenyl C-1), 128.43 (phenyl C-4), 130.08 (phenyl C3, C-5), 135.81 (phenyl C-1), 142.85 (tetrazole C-5), 146.05 (dimethoxyphenyl C-3), 149.09 (dimethoxyphenyl C-4), 151.29 (pyridine C-6), 153.63 (pyridine C-4), 161.67 (pyridine C-2); Anal. Calcd for C21H16N6O3: C, 62.99; H, 4.03; N, 20.99. Found: C, 63.04; H, 3.98; N, 20.79.
6-[4-(1H-Tetrazol-1-yl)phenyl]-4-(3,4,5-trimethoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5f)
Yellow solid; method B; (2.32 g, 54%) yield; mp > 300 °C; IR (KBr) 3426 (OH), 3121 (NH), 2226 (C≡N), 1659 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.76 (s, 3H, p-OCH3), 3.87 (s, 6H, 2 m-OCH3), 7.02 (s, 1H, pyridine H-5), 7.07 (s, 2H, trimethoxyphenyl H-2, H-6), 8.09 (d, J = 8.4 Hz, 2H, phenyl H-3, H-5), 8.22 (d, J= 8.4 Hz, 2H, phenyl H-2, H-6), 10.22 (s, 1H, tetrazole H-5), 12.94 (s, 1H, OH, D2O exchangeable); 13 C NMR (100 MHz, DMSO-d6) δ: 56.65 (2OCH3), 60.63 (OCH3), 97.20 (pyridine C-3), 103.82 (pyridine C-5), 106.60 (trimethoxyphenyl C-2, C-6), 115.19 (C≡N), 121.59 (phenyl C-2, C-6), 129.98 (phenyl C3, C-5), 131.89 (phenyl C-4), 132.75 (trimethoxyphenyl C-1), 135.67 (phenyl C-1), 140.84 (trimethoxyphenyl C-4), 142.84 (tetrazole C-5), 150.29 (pyridine C-6), 153.34 (pyridine C-4), 154.01 (trimethoxyphenyl C-3, C-5), 159.75 (pyridine C-2); EIMS (m/z) 431 (M + 1, 2.95%), 430 (M+., 10.09%), 136 (100%). Anal. Calcd for C22H18N6O4: C, 61.39; H, 4.22; N, 19.53. Found: C, 61.27; H, 4.29; N, 19.47.
General procedure for preparation of 6a and b
A mixture of acetophenone derivative 2 (1.88 g, 0.01 mol) and p-substituted sulphonylphenylhydrazine hydrochloride derivative (0.015 mol) in absolute ethanol (20 mL) was heated under reflux for 6–8 h (monitored by TLC). The obtained solid on hot was filtered, dried and crystalised from 95% ethanol to give 6a&b.
(ZE)-1-{4-[1-(2-(4-Methanesulphonylphenyl)hydrazono)ethyl]phe-nyl}-1H-tetrazole (6a)
Yellow solid; (1.53 g, 43%) yield; mp 258–260 °C; IR (KBr) 3440 (NH), 1277, 1126 (SO2) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 2.37 (s, 3H, CH3), 3.13 (s, 3H, SO2CH3), 7.44 (d, J = 8.8 Hz, 2H, methanesulphonylphenyl H-3, H-5), 7.77 (d, J = 8.8 Hz, methanesulphonylphenyl H-2, H-6), 7.96 (d, J = 8.8 Hz, 2H, phenyl H-3, H-5), 8.08 (d, J = 8.8 Hz, 2H, phenyl H-2, H-6), 10.05 (s, 1H, NH, D2O exchangeable), 10.15 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 13.68 (CH3), 44.71 (SO2CH3), 113.01 (methanesulphonylphenyl C-2, C-6), 121.42 (phenyl C-3, C-5), 127.49 (methanesulphonylphenyl C-3, C-5), 129.17 (phenyl C-2, C-6), 130.48 (methanesulphonylphenyl C-4), 133.60 (phenyl C-4), 140.25 (phenyl C-1), 142.62 (tetrazole C), 143.11 (methanesulphonylphenyl C-1), 150.11 (C = N-NH); EIMS (m/z) 356 (M+., 5.59%), 158 (100%). Anal. Calcd for C16H16N6O2S: C, 53.92; H, 4.52; N, 23.58. Found: C, 54.08; H, 4.35; N, 23.71.
(ZE)-4-{2-[1–(4-(1H-tetrazol)-1-yl)phenyl)ethylidene]hydrazinyl}benzenesulphonamide (6b)
Yellow solid; (1.74 g, 49%) yield; mp 255–257 °C; IR (KBr) 3449–3109 (NH & NH2), 1273, 1157 (SO2) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 2.35 (s, 3H, CH3), 7.11(s, 2H, NH2, D2O exchangeable), 7.38 (d, J = 8.8 Hz, 2H, benzenesulphonamide H-3, H-5), 7.70 (d, J = 8.8 Hz, 2H, benzenesulphonamide H-2, H-6), 7.95 (d, J = 8.4 Hz, 2H, phenyl H-3, H-5), 8.08 (d, J = 8.4 Hz, 2H, phenyl H-2, H-6), 9.90 (s, 1H, NH, D2O exchangeable), 10.15 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 13.48 (CH3), 112.71 (benzenesulphonamide C-2, C-6), 121.44 (phenyl C-3, C-5), 127.40 (benzenesulphonamide C-3, C-5), 127.73 (phenyl C-2, C-6), 133.39 (benzenesulphonamide C-4), 134.28 (phenyl C-4), 140.38 (phenyl C-1), 142.29 (benzenesulphonamide C-1), 142.54 (tetrazole C), 148.74 (C = N-NH); Anal. Calcd for C15H15N7O2S: C, 50.41; H, 4.23; N, 27.43. Found: C, 50.37; H, 4.15; N, 27.17.
General procedure for synthesis of 7a and b
To a solution of 3a or 3b (0.01 mol) in absolute ethanol (30 mL), urea or thiourea (0.01 mol) and KOH (0.56 g, 0.01 mol) were added. The reaction mixture was heated under reflux for 10–12 h (monitored by TLC). The solid obtained was filtered, dried and crystallised from 95% ethanol to afford 7a&b.
(ZE)-N-{4-[3–(3,4-Dimethoxyphenyl)acryloyl]phenyl}cyanamide (7a)
Orange solid; (1.23 g, 40%) yield; mp 112–114 °C; IR (KBr) 3121 (NH), 2203 (C≡N), 1674 (C = O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.82 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 5.45 (s, 1H, NH, D2O exchangeable), 7.03 (d, J= 8 Hz, 1H, Ar-H, dimethoxyphenyl H-5), 7.10 (d, J = 8 Hz, 2H, Ar-H, phenyl H-2, H-6), 7.38 (d, J= 8 Hz, dimethoxyphenyl H-6), 7.53 (s, 1H, dimethoxyphenyl H-2), 7.68 (d, J = 15.6 Hz, 1H, COCH = CH), 7.81 (d, J = 15.6, 1H, COCH = CH), 8.19 (d, J = 8 Hz, 2H, Ar-H, phenyl H-3, H-5); 13 C NMR (100 MHz, DMSO-d6) δ: 56.04 (OCH3), 56.16 (OCH3), 111.03 (dimethoxyphenyl C-2), 112.00 (dimethoxyphenyl C-5), 112.15 (C≡N), 115.43 (phenyl C-3, C-5), 119.79 (CO-CH = CH), 124.34 (dimethoxyphenyl C-6), 127.94 (dimethoxyphenyl C-1), 131.27 (phenyl C-2, C-6), 132.60 (phenyl C-1), 143.99 (phenyl C-4), 144.59 (CO-CH = CH), 149.42 (dimethoxyphenyl C-4), 151.65 (dimethoxyphenyl C-3), 187.82 (C = O); EIMS (m/z) 308 (M+., 11.81%), 120 (100%). Anal. Calcd for C18H16N2O3: C, 70.12; H, 5.23; N, 9.09. Found: C, 70.09; H, 5.16; N, 9.11.
(ZE)-N-{4-[3–(3,4,5-Trimethoxyphenyl)acryloyl]phenyl}cyanamide (7b)
Brown solid; (1.28 g, 38%) yield; mp 109–111 °C; IR (KBr) 3210 (NH), 2257 (C≡N), 1682 (C = O)cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.72 (s, 3H, p-OCH3), 3.87 (s, 6H, 2 m-OCH3), 5.40 (s, 1H, NH, D2O exchangeable), 7.03 (d, J = 8.8 Hz, 2H, phenyl H-2, H-6), 7.22 (s, 2H, trimethoxyphenyl H-2, H-6), 7.66 (d, J = 15.6 Hz, 1H, COCH = CH), 7.86 (d, J = 15.6, 1H, COCH = CH), 8.15 (d, J = 8.8 Hz, 2H, phenyl H-3, H-5); 13 C NMR (100 MHz, DMSO-d6) δ: 56.37 (2 m-OCH3), 60.62 (p-OCH3), 106.83 (trimethoxyphenyl C-2, C-6), 113.61 (C≡N), 115.78 (phenyl C-3, C-5), 121.62 (CO-CH = CH), 130.76 (trimethoxyphenyl C-1), 130.95 (phenyl C-2, C-6), 131.50 (phenyl C-1), 140.03 (trimethoxy C-4), 144.12 (CO-CH = CH), 146.52 (phenyl C-4), 153.56 (trimethoxyphenyl (C-3, C-5), 187.48 (C = O); EIMS (m/z) 338 (M+., 4.68%), 43 (100%). Anal. Calcd for C19H18N2O4: C, 67.44; H, 5.36; N, 8.28. Found: C, 67.35; H, 5.27; N, 8.09.
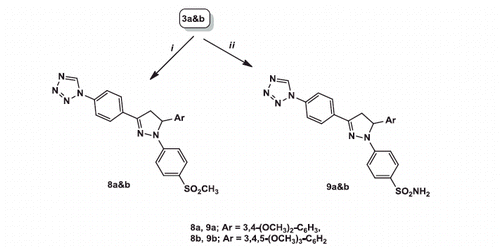
General procedure for preparation of 1,3,5-triaryl-4,5-dihydro-1H-pyrazoles 8a and b
To a solution of the appropriate chalcone 3a and b (0.01 mol) in absolute ethanol (20 mL), 4-methanesulphonylphenylhydrazine hydrochloride (0.33 g, 0.015 mol) was added and the reaction mixture was heated under reflux for 12 h. After completion of the reaction (monitored by TLC plates using chloroform/methanol 9.5:0.5 V/V), the obtained solid was filtered, dried and crystallised from 95% ethanol to give the respective triarylpyrazoles 8a and b.
1-{4-[5-(3,4-Dimethoxyphenyl)-1-(4-(methanesulphonyl)phenyl]-4,5-dihydro-1H-pyrazol-3-yl)phenyl}-1H-tetrazole (8a)
Red solid; (2.01 g, 40%) yield; mp 201–203 C; IR (KBr) 2925–2837 (CH aliphatic), 1294, 1136 (SO2) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.09 (s, 3H, SO2CH3), 3.29 (dd, J = 17.6, 5.6 Hz, 1H, pyrazoline H-4), 3.64 (s, 3H, OCH3), 3.70 (s, 3H, OCH3), 4.01(dd, J = 17.6, 12.4 Hz, 1H, pyrazoline H′-4), 5.63 (dd, J = 12.4, 5.6 Hz, 1H, pyrazoline H-5), 6.74 (d, J = 8.4 Hz, 1H, dimethoxyphenyl H-6), 6.91 (d, J= 8.4 Hz, 1H, dimethoxyphenyl H-5), 6.98 (s, 1H, dimethoxyphenyl H-2), 7.21 (d, J = 8.8 Hz, 2H, methanesulphonylphenyl H-3, H-5), 7.71 (d, J = 8.8 Hz, 2H, methanesulphonylphenyl H-2, H-6), 8.01–8.10 (m, 4H, phenyl H-2, H-3, H-5, H-6), 10.18 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 43.45 (pyrazoline C-4), 44.58 (SO2CH3), 55.96 (2OCH3), 63.05 (pyrazoline C-5), 110.16 (dimethoxyphenyl C-2), 112.67 (methanesulphonylphenyl C-2, C-6), 112.99 (dimethoxyphenyl C-6), 117.90 (dimethoxyphenyl C-5), 121.69 (phenyl C-3, C-5), 128.13 (methanesulphonylphenyl C-3, C-5), 129.00 (phenyl C-2, C-6), 129.92 (methylsulphonyl phenyl C-4), 133.29 (phenyl C-4), 134.09 (phenyl C-1), 134.34 (dimethoxyphenyl C-1), 142.62 (tetrazole C-5), 147.50 (dimethoxyphenyl C-4), 148.70 (dimethoxyphenyl C-3), 149.58 (methylsulphonyl phenyl C-1), 149.75 (pyrazole C-3); Anal. Calcd for C25H24N6O4S: C, 59.51; H, 4.79; N, 16.66. Found: C, 59.71; H, 4.85; N, 16.74.
1-{4-[1-(4-Methanesulphonyl)phenyl]-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-1H-tetrazole (8b)
Orange solid; (2.24 g, 42%) yield; mp 241–243 °C; IR (KBr) 2924–2855 (CH aliphatic), 1234, 1142 (SO2) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.10 (s, 3H, SO2CH3), 3.31–3.32 (m, 1H, pyrazoline H-4), 3.63 (s, 3H, p-OCH3), 3.71 (s, 6H, 2 m-OCH3), 4.03 (dd, J = 18, 12.4 Hz, 1H, pyrazoline H′-4), 5.58 (dd, J = 12.4, 6.4 Hz, 1H, pyrazoline H-5), 6.63 (s, 2H, trimethoxyphenyl H-2, H-6), 7.23 (d, J = 8.8 Hz, 2H, methanesulphonylphenyl H-3, H-5), 7.72 (d, J = 8.8 Hz, 2H, methanesulphonylphenyl H-2, H-6), 8.02–8.08 (m, 4H, phenyl H-2, H-3, H-5, H-6), 10.19 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 43.44 (pyrazoline C-4), 44.54 (SO2CH3), 56.35 (2 m-OCH3), 60.46 (p-OCH3), 63.61 (pyrazoline C-5), 103.29 (trimethoxyphenyl C-2, C-6), 113.06 (methanesulphonylphenyl C-2, C-6), 121.78 (phenyl C-3, C-5), 128.22 (methanesulphonylphenyl C-3, C-5), 129.04 (phenyl C-2, C-6), 129.97 (methylsulphonyl phenyl C-4), 133.22 (phenyl C-4), 134.35 (phenyl C-1), 137.20 (trimethoxyphenyl C-4), 137.65 (trimethoxyphenyl C-1), 142.59 (tetrazole C-5), 147.74 (methylsulphonyl phenyl C-1), 149.94 (pyrazole C-3), 153.85 (trimethoxyphenyl C-3, C-5); EIMS (m/z) 534 (M+., 0.76%), 43 (100%). Anal. Calcd for C26H26N6O5S: C, 58.41; H, 4.90; N, 15.72. Found: C, 58.32; H, 4.86; N, 15.78.
General procedure for preparation of 1,3,5-triaryl-4,5-dihydro-1H-pyrazoles 9a and b
To a solution of the appropriate chalcone 3a and b (0.01 mol) in absolute ethanol (20 mL), 4-benzenesulphonamidehydrazine hydrochloride (0.33 g, 0.015 mol) was added and the reaction mixture was heated under reflux for 18 h. After completion of the reaction (monitored by TLC plates using chloroform/methanol 9.5:0.5 V/V), the obtained solid was filtered, dried and crystallised from 95% ethanol to give the respective triarylpyrazoles 9a and b.
4-{3-[4-(1H-Tetrazol-1-yl)phenyl]-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulphonamide (9a)
Yellow solid; (2.57 g, 51%) yield; mp 226–228 °C; IR (KBr) 3429 (broad NH2), 2929 (CH aliphatic), 1259, 1151 (SO2) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.30 (dd, J = 18, 5.6 Hz, 1H, pyrazoline H-4), 3.70 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.99 (dd, J = 18, 12.4 Hz, 1H, pyrazoline H′-4), 5.61 (dd, J = 12.4, 5.6 Hz, 1H, pyrazoline H-5), 6.64 (d, J = 12.8 Hz, 1H, dimethoxyphenyl H-5), 6.99 (d, J = 12.8 Hz, 1H, dimethoxyphenyl H-6), 7.00 (s, 1H, Ar-H, dimethoxyphenyl H-2), 7.11 (s, 2H, NH2, D2O exchangeable), 7.34 (d, J = 8.8 Hz, 2H, benzenesulphonamide H-2, H-6), 7.62 (d, J = 8.8 Hz, 2H, benzenesulphonamide H-3, H-5), 8.00–8.16 (m, 4H, Ar-H, phenyl H-2, H-3, H-5, H-6), 10.18 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 43.66 (pyrazoline C-4), 56.10 (OCH3), 56.24 (OCH3), 63.12 (pyrazoline C-5), 110.19 (dimethoxyphenyl C-2), 112.82 (benzene sulphonamide C-2, C-6), 117.93 (dimethoxyphenyl C-6), 121.49 (dimethoxyphenyl C-5), 121.68 (phenyl C-3, C-5), 127.31 (phenyl C-2, C-6), 127.98 (benzenesulphonamide C-3, C-5), 130.85 (benzenesulphonamide C-4), 133.49 (phenyl C-4), 133.94 (phenyl C-1), 134.21 (dimethoxyphenyl C-1), 142.64 (tetrazole C-5), 146.27 (benzenesulphonamide C-1), 148.64 (dimethoxyphenyl C-4), 148.87 (dimethoxyphenyl C-3),149.56 (pyrazoline C-3); Anal. Calcd for C24H23N7O4S: C, 57.02; H, 4.59; N, 19.39. Found: C, 56.98; H, 4.55; N, 19.22.
4-{3-[4-(1H-Tetrazol-1-yl)phenyl]-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulphonamide (9b)
Yellow solid; (2.46 g, 46%) yield; mp 246–248 °C; IR (KBr) 3428 (broad NH2), 2940–2839 (CH aliphatic), 1279, 1125 (SO2) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 3.32–3.33 (m, 1H, pyrazoline H-4), 3.71 (s, 3H, p-OCH3), 3.88 (s, 6H, 2 m-OCH3), 4.01–4.03 (m, 1H, pyrazoline H′-4), 5.57–5.58 (m, 1H, pyrazoline H-5), 6.61 (s, 2H, trimethoxyphenyl H-2, H-6), 7.07–7.18 (m, 4H, benzenesulphonamide H-2, H-3, H-5, H-6), 7.64 (s, 2H, NH2, D2O exchangeable), 8.03–8.05 (m, 4H, phenyl H-2, H-3, H-5, H-6), 10.18 (s, 1H, tetrazole H); 13 C NMR (100 MHz, DMSO-d6) δ: 40.51 (pyrazoline C-4), 56.35 (OCH3), 60.44 (OCH3), 63.73 (pyrazoline C-5), 103.32 (dimethoxyphenyl C-2, C-6), 112.85 (benzene sulphonamide C-2, C-6), 121.73 (phenyl C-3, C-5), 127.61 (phenyl C-2, C-6), 128.05 (benzenesulphonamide C-3, C-5), 133.40 (benzenesulphonamide C-4), 134.04 (phenyl C-4), 134.21 (phenyl C-1), 137.16 (dimethoxyphenyl C-1), 137.81 (dimethoxyphenyl C-4), 142.61 (tetrazole C-5), 146.52 (benzenesulphonamide C-1), 149.05 (pyrazoline C-3), 153.82 (dimethoxyphenyl C-3, C-5); EIMS (m/z) 535 (M+., 0.73%), 43 (100%). Anal. Calcd for C25H25N7O5S: C, 56.06; H, 4.70; N, 18.31. Found: C, 55.99; H, 4.75; N, 18.37.
Biological evaluation
In vitro cyclooxygenase (COX) inhibition assay
The ability of the test compounds listed in to inhibit ovine COX-1 and COX-2 (IC50 value, μM) was tested using an enzyme immune assay (EIA) kit (Cayman Chemical, Ann Arbor, MI) was according to a reported methodCitation47,Citation48.
Table 1. In vitro COX-1 and COX-2 inhibition of tested compounds and reference drug, celecoxib.
In vivo assays
Female wister rats (150–200 g) were used in this study. The animals were kept at controlled conditions (temperature 23 ± 2 °C, humidity 60 ± 10%) and a 12/12-h light/dark cycle with access to food and water. All procedures relating to animal care and treatments were performed according to protocols approved by the Research Ethical Committee of Faculty of Pharmacy Beni-Suef University (2014-Beni-Suef, Egypt).
Anti-inflammatory activity
The anti-inflammatory activity of the synthesised compounds was determined in vivo by Carragenan-induced paw oedema method in ratsCitation49. Rats were divided into 21 groups of four animals each, and then, they were fasted overnight with free access to water before the experiment. Before any drug administration, thickness of the left hind paw of each rat was measured in millimetres. Group 1 served as a control and administered the vehicle (2.5% Tween 80). Groups (2–20) were orally administered (50 mg/kg) compounds 3a, 3b, 4a, 4b, 4c, 5a, 5b, 5c, 5d, 5e, 5f, 6a, 6b, 7a, 7b, 8a, 8b, 9a, 9b, respectively. Group 21 was administered celecoxib (50 mg/kg) as a reference standard. paw oedema was induced by subcutaneous injection of 1% carrageen in saline (0.02 mL/rat) into the left hind paw of each rat, one hour after administration of vehicle, test compounds or celecoxib. Paw thickness of each rat was measured after 1, 3, 5 h of Carragenan injection, and then, the change in thickness and % inhibition of paw oedema were calculated.
Ulcerogenic liability
The ulcerogenic effects of compounds 4a, 5a, 5d, 5e, 5f, 8a, 8b, 9a, 9b and celecoxib were evaluated and compared to that of indomethacin. Forty-eight rats were used in this study, divided into 12 groups and fasted for 18 h before drug administration. The control group received the vehicle (2.5% Tween 80). Other groups received test compounds, celecoxib or indomethacin at a dose of 50 mg/kg, then animals were fed after 2 h. Rats were given the specified dose orally for three successive days. Rats were sacrificed after 2 h of the last dose, then the stomach of each rat was removed and opened along the greater curvature for determination of the ulcer number and ulcer index according to the reported methodCitation50.
Analgesic activity
Hot plate method
This method was used to evaluate the central analgesic activity by determination of the delay in the latency time of pain responseCitation51. The analgesic activity of compounds 4a, 5a, 5d, 5e, 5f, 8a, 8b, 9a, 9b and celecoxib were evaluated. The mice were orally administered 10 mg/kg of either test compounds or celecoxib, while 2.5% Tween 80 solution was administered to the normal control group. After 60 min, the animals were placed on a hot plate maintained at 55 ± 0.5 °C. The reaction time was recorded as the time taken by the animals to blow or lick the fore or hind paw or jump off the plate.
Writhing method
The assay was performed as described by Koster et al.Citation52. Pain was induced by intraperitoneal injection of acetic acid, then counting the number of abdominal writhings. Mice were orally administered 10 mg/kg of compounds 4a, 5a, 5d, 5e, 5f, 8a, 8b, 9a, 9b and celecoxib 30 min before intraperitoneal injection of 0.6% acetic acid. After 5 min, the animals were observed and the number of abdominal writhings was recorded for 20 min.
Histopathological investigation
Whole rats stomach were collected, selected stomach samples from each rat were fixed in 10% neutral buffered formalin for 48 h and routinely processed for paraffin embedding. 5 μm Thickness sections were obtained then stained with haematoxylin and eosin for histopathological evaluationCitation53.
Statistical analysis
Significant difference among groups was assessed using one-way ANOVA followed by Dunnett’s test. Differences were considered significant at *p > .05, **p < .01 and ***p > .001. GraphPad Prism software version 5 (Canada) was used to carry out all statistical tests.
Docking study
Docking was performed to obtain prediction of conformation and also energy ranking between COX-2 receptor (PDB: 1CX2) and the designed compoundsCitation47. Molecular Operating Environment (MOE, Version 2005.06, Chemical Computing Group Inc., Montreal, Quebec, Canada) was used in docking studies.
The cocrystallised ligand was docked first to study the scoring energy, root mean standard deviation (RMSD) and amino acid interactions. RMSD, for COX-2 enzyme and the lead compound SC-558 was 3 A°.
Docking was performed using London dG force and refinement of the results was done using force field energy. Preparation of the synthesised compounds for docking was achieved via their 3 D structure built by MOE. Before docking, 3 D protonation of the structures, running conformational analysis using systemic search and selecting the least energetic conformer were performed. The same docking protocol used with ligand was also applying for the designed compounds. Amino acid interactions and the hydrogen bond lengths were calculated. The data obtained are summarised in .
Results and discussion
Chemistry
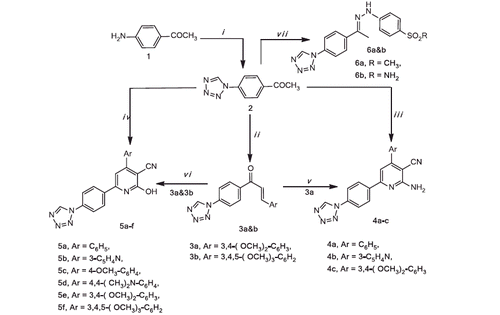
The synthetic routes of the target compounds are summarised in Schemes 1–3. 1-[4–(1H-Tetrazol-1-yl)phenyl]ethanoneCitation2 was obtained using 4-aminoacetophenone as the starting material according to the literatureCitation54.
Scheme 1. Reagent and conditions: (i) NaN3, TEOF, gl. HAc, reflux, 12 h, (ii) ArCHO, KOH, abs. EtOH, r.t., 10–12 h, (iii) ArCHO, CN(CH2)CN, NH4OAc, abs. EtOH, (iv) ArCHO, CNCH2COOEt, NH4OAc, abs. EtOH, (v) CN(CH2)CN, NH4OAc, abs. EtOH, (vi) CNCH2COOEt, NH4OAc, abs. EtOH, (vii) p-substitutedphenylhydrazine hydrochloride, abs. EtOH, reflux, 6–8 h.
Scheme 2. Reagents and conditions: (i) thiourea or urea, KOH, abs. ethanol, reflux 10–12 h.
Scheme 3. Reagents and conditions: (i) p-methanesulphonylphenyl hydrazine hydrochloride, abs. ethanol; (ii) p-benzene sulphonamide hydrazine hydrochloride, abs. ethanol.
Chalcone derivatives 3a and b were synthesised in high yields (79–86%) by a base catalysed Claisen–Schmidt condensation of acetophenone derivative 2 and substituted aryl aldehydes namely: 3,4-dimethoxybenzaldehyde and 3,4,5-trimethoxybenzaldehyde, respectively.
The prepared compounds have been characterised by IR, 1H NMR, 13 C NMR, mass spectra and elemental analyses.
IR spectra of 3a and b showed a sharp peak at 1674, 1663 cm−1 corresponding to C = O group. 1H NMR spectra of 3a and b revealed the presence of two doublet protons at δ 7.77 due to COCH = CH and at δ 7.90, 7.95 corresponding to COCH = CH proton with high J value (15.2 Hz). 13 C NMR spectra of 3a and b showed a peak at δ 188.49, 188.60 attributed to C of C = O.
Formation of aminocyanopyridine derivatives 4a–c and their isosteric hydroxycyanoypyridine derivatives 5a–f was achieved using two different methods (A and B). In method A, one-pot multicomponent synthetic approach was utilised. Thus, compound 2 reacted with the respective aromatic aldehyde, malononitrile or ethyl cyanoacetate in the presence of ammonium acetate to yield the respective derivatives 4a–c (45–61%, yield) and 5a–f (41–57%, yield).
The second procedure (B), stepwise reaction was applied, thus reaction of acetophenone derivative 2 with the respective aldehyde to give the corresponding chalcone 3a and b, followed by the reaction of ammonium acetate and malononitrile or ethyl cyanoacetate to afford 4c (39%, yield) and 5e and f (41–46%, yield).
Better yield and operational simplicity of the first method (A) encouraged us to prepare the remaining derivatives 4a and b and 5a–d using it.
IR spectra of cyanopyridine derivatives 4a–c and 5a–f showed the appearance of sharp peak at 2226–2207 cm−1 corresponding to C≡N group. In addition to a forked peak at 3352–3210 and 3136–3117 cm−1 due to NH2 in 4a–c, and a broad peak at 3483–3325 cm−1 attributed to OH group in case of 5a–f. Moreover, the absence of peak due to C = O group of the parents 2 and 3a and b confirmed the structure of 4a–c and 5a–f.
The 1H NMR spectra of 4a–c and 5a–f showed a singlet of one-proton intensity at δ 6.84–7.88 corresponding to pyridine H-5. Also, D2O exchangeable singlet peak at δ 6.99, 7.66 attributed to NH2 protons for the aminopyridine derivatives 4a–c, and a broad exchangeable peak at δ 12.65–12.94 corresponding to OH in case of hydroxypyridine derivatives 5a–f.
13 C NMR spectra of 4a–c and 5a–f confirmed the formation of pyridine ring by the presence of three peaks at δ 90.87–97.20, 100.91–119.05 and 114.64–117.87 corresponding to pyridine C-3, C-5 and C of C≡N. No evidence for the presence of C = O of the starting compounds 2 and 3a and b.
Condensation of compound 2 with 4-methanesulphonylphenyl hydrazine hydrochloride or 4-benzenesulphonamide hydrazine hydrochloride afforded 6a and b, respectively.
IR spectra of 6a and b showed two sharp peaks at 1277, 1273 and 1157, 1126 cm−1 corresponding to SO2, in addition to a peak at 3449 and 3440 cm−1 due to NH group. There was no evidence for the presence of C = O group that present in the parent compound 2.
1H NMR spectra of 6a and b revealed the presence of an exchangeable singlet signal at δ 9.90, 10.05 attributed to NH proton. Another D2O exchangeable singlet signal of two protons intensity appeared at δ 7.11 corresponding to SO2NH2 protons in 6b. Moreover, three protons of SO2CH3 group in 6a appeared as a singlet signal at δ 3.13.
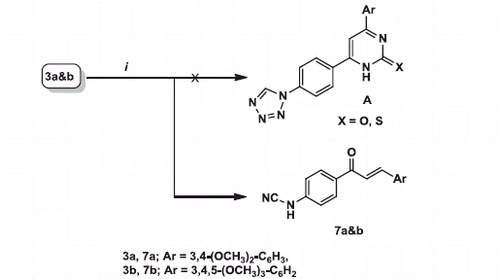
In an attempt to synthesise pyrimidine derivatives A from the reaction of 3a and b with urea or thiourea under reflux temperature in alcoholic KOH, the unexpected chalcone derivatives 7a and b were obtained instead. The deviation of the reaction caused as a result of two factors, heating and presence of a base. Thus, tetrazole ring cleaved with the loss of nitrogen (N2) and forming products bearing cyanamide (-NHCN) group. The same explanation for tetrazole cleavage was published by Vorobiov et al.Citation55 upon using more drastic conditions (NaOH in DMSO), (Scheme 2).
IR spectra of 7a and b showed three sharp peaks at 3210, 3121; 2257, 2203 and 1682, 1674 cm−1 corresponding to NH, C≡N and C = O groups.
1H NMR spectra of 7a and b revealed the presence of two doublet signals at δ 7.66, 7.68 and 7.81, 7.86 due to COCH = CH and COCH = CH protons, respectively with high J value (15.6 Hz). On the other hand, absence of signals for both pyrimidine H-5 and tetrazole H-5 confirmed formation of the unexpected products 7a and b.
13 C NMR spectra of 7a and b showed signals for C≡N and C = O at δ 112.15, 113.61 and 187.48, 187.82, respectively. No evidence for tetrazole C-5 which present in the parent compounds 3a and b and in compound A.
Mass spectra of 7a and b showed molecular ion peak at m/z 308 (11.81%) and 338 (4.68%), sequentially.
Another two groups of triarylheterocycles 8a and b and 9a and b were synthesised using the reaction sequence adopted in Scheme 3. Accordingly, condensation of α,β-unsaturated ketones 3a and b with 4-methanesulphonylphenylhydrazine hydrochloride or 4-benzenesulphonamidehydrazine hydrochloride in absolute ethanol gave the respective 1,3,5-triaryl-pyrazolines 8a and b and 9a and b, in good yields (40–51%).
IR spectra of 8a and b and 9a and b showed two sharp peaks at 1294–1234 and 1151–1125 cm−1 corresponding to SO2. Another broad peak appeared at 3429, 3428 cm−1 observed in case of 9a&b due to NH2.
1H NMR spectra of pyrazolines 8a and b and 9a and b displayed three doublet of doublet (dd) signals each of one-proton intensity at δ 3.29–3.33, 3.99–4.03 and 5.57–5.63 with three different J values corresponding to three protons of pyrazoline ring. The highest J value 17.6–18 Hz was due to germinal coupling of the two protons at C-4 of pyrazoline ring, while the other two J values 12.4 and 5.6–6.4 Hz due to coupling of the two geminal protons at C-4 and the vicinal proton at C-5. Additionally, 1H NMR spectra of 8a and b revealed the presence of a singlet signal at δ 3.09–3.10 due to SO2CH3 protons. Moreover, an exchangeable signal at δ 7.11, 7.64 corresponding to SO2NH2 protons was observed in 1H NMR spectra of 9a and b.
13 C NMR spectra of 8a and b and 9a and b showed two peaks at δ 40.51–43.66 and 63.05–63.73 corresponding to C-4 and C-5 of pyrazoline ring.
Biological evaluation
Anti-inflammatory activity
In vitro cyclooxygenase (COX) inhibition assay
The peroxidase activity of cyclooxygenase enzyme isoforms was measured using the COX activity assay kit. The ability of the test compounds to inhibit both ovine COX-1 and COX-2 enzymes was explored in vitro using a colorimetric enzyme immunoassay (EIA) kit. The appearance of oxidised N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) was monitored at 590 nm. The kit includes isozyme-specific inhibitors for distinguishing COX-2 activity from that of COX-1. The advantages of this COX assay method are screening a vast number of inhibitors and saving much time.
The in vitro test compound concentration required to produce 50% inhibition of COX-1 or COX-2 (IC50%) was measured. Moreover, the COX-2 selectivity index (S.I.) values [IC50 (COX-1)/IC50 (COX-2)] were calculated and compared with that of the standard drug celecoxib (as a selective COX-2 inhibitor), diclofenac (non-selective COX inhibitor) and indomethacin (selective COX-1 inhibitor). All the synthesised compounds were tested; data are listed in .
The results showed that eight compounds (3a, 4c, 4b, 5b, 5c, 5d, 6a, 6b and 8a) could inhibit COX-1 at higher dose range with IC50 (8.67–15.97 μM) which is more than celecoxib and diclofenac (7.31 and 3.9 μM, respectively). While, compounds (3b, 4a, 4b, 5a, 5e, 5f, 9a, 9b and 8b) had closed IC50 to celecoxib or diclofenac in a range of 3.87–7.37 μM and were less potent than indomethacin (IC50 = 0.039 μM). From this point, it was expected that most of the synthesised compounds might be safe with low ulcerogenic effect on gastric mucosa. Regarding to COX-2 inhibitory activity, both trimethoxyphenylpyrazoline derivatives bearing –SO2Me pharmacophore 8b and trimethoxyphenyl pyridine hydroxycarbonitrile analog 5f had the highest COX-2 potency (IC50 = 0.11 and 0.14 μM, respectively), while, IC50 of the reference drug celecoxib was 0.16 μM.
Hydroxypyridine carbonitrile analogs 5a and 5e exhibited appreciable COX-2 inhibitory activity IC50 = 0.18 and 0.19 μM, sequentially, which are closer to celecoxib. Compounds 3a, 3b, 4a, 4b, 4c, 5c, 5d, 6a, 6b, 7a, 7b 8a, 9a and 9b (IC50 = 0.21–0.71 μM, range) were less potent as COX-2 inhibitors than celecoxib and at the same time, more potent if compared to diclofenac. Pyridopyridine hydroxycarbonitrile derivative 5b showed the lowest COX-2 potency (IC50 = 1.14 μM).
Accordingly, the results showed COX-2 selectivity indices in the range 10.88–46.07. Within the synthesised compounds, compound 5f had better COX-2 selectivity index (SI = 46.07) than that of celecoxib (SI = 45.68). Compound 4c had selectivity index (SI = 42.02) near to that of celecoxib. Hydroxypyridine carbonitrile derivatives 5a, 5e and compounds containing pyrazoline scaffold with trimethoxy phenyl ring and SO2Me or SO2NH2 moiety 8b and 9b, respectively, showed good COX-2 selectivity indices ranged from 35.13 to 37.57. While, chalcone derivatives 3a and b, also, 6a and b, 8a, 9a, derivatives and aminopyridine carbontrile derivatives 4b, 5d, showed lower COX-2 selectivity indices (SI = 21.54–28.08) than celecoxib. The smallest COX-2 selectivity index was obtained from 4a and 5b.
Anti-inflammatory activity
In vivo anti-inflammatory activity of the tested compounds was performed using carrageenan-induced rat paw oedema method compared to celecoxib as a reference anti-inflammatory drug.
Mean changes in paw oedema thickness of animals pre-treated with the tested compounds and celecoxib after 1, 3 and 5 h from the induction of inflammation were measured and listed in .
Table 2. Results of in vivo anti-inflammatory activities of tested compounds using carrageenan-induced rat paw oedema assay.
The reference drug, celecoxib showed 76% and 66% inhibitory activity against carrageenan-induced paw oedema after 1 and 3 h, respectively, then the activity decreased to be 19% after 5 h. Most of the tested compounds showed a significant anti-inflammatory activity (p < .001) especially after 1 h from administration.
After 1 h, compound 5f exhibited anti-inflammatory activity (79%) similar to that of celecoxib (76%). This result was in accordance with the in vitro COX-2 assay data.
Compounds containing hydroxypyridine carbonitrile core such as 5d and 5e and those with SO2Me and SO2NH2 pharmacophores like 8a, 8b, 9a, 9b showed strong anti-inflammatory activity of about 71–75%. These compounds also exhibited good COX-2 inhibitory activities.
Moreover, compounds 3b, 4a, 5a, 5c displayed anti-inflammatory activity percentage of about 51–61%.
The lowest anti-inflammatory activity was observed in compounds 3a, 4b, 4c, 5b, 6a, 6b, 7a and 7b in a range 19–49%.
After 3 h and 5 h, the anti-inflammatory activity for most of the synthesised compounds was gradually decreased except for phenyl and N,N-dimethylaminophenyl derivatives of pyridine hydroxycarbonitrile (5a and 5d, respectively) and dimethoxy phenyl pyrazoline derivative 8a showing duration of action extended to 3 h to 4a–c be 54%, 63% and 52%, sequentially.
Ulcerogenic liability
The in vivo ulcerogenic liability was evaluated for celecoxib and the most active anti-inflammatory tested compounds 4c, 5a, 5d, 5e, 5f, 8a, 8b, 9a and 9b relative to indomethacin.
The results of ulcerogenic liability () revealed that indomethacin caused the most ulcerogenic toxicity with ulcer index (UI: 22.50), whereas both celecoxib and tested compounds exhibited much lower UI between (0.50–2.00).
Table 3. Ulcerogenic liability for compounds 4c, 5a, 5d-f, 8a&b and 9a&b compared to reference drugs celecoxib and indomethacin.
Table 4. Molecular modelling data for best poses of the designed compounds 3a&b, 4a-c, 5a-f, 6a&b, 7a&b, 8a&b, 9a&b and SC-558 during docking in COX-2 (PDB: 1CX2) active site.
Compounds 9b and 8b, bearing pyrazoline core, trimethoxyphenyl part and COX-2 pharmacophores (SO2NH2 and SO2Me), showed equal or close ulcerogenic liability to celecoxib (UI: 0.50 and 0.75, respectively).
The rest of the tested compounds 4c, 5a, 5d–f, 8a and 9a exhibited lower toxicity than indomethacin (UI: 22.50), where the UIs were in the range of 1.25–2.00.
Analgesic activity
Hot plate method
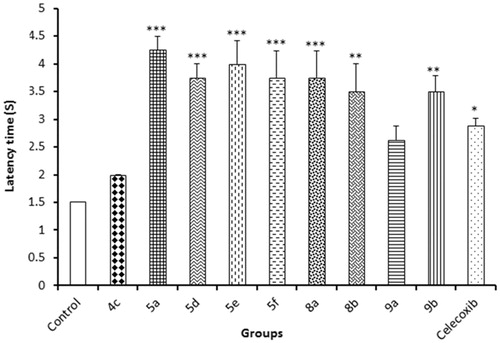
This method was used to evaluate the central analgesic activity of nine compounds from the newly synthesised derivatives by determination of the delay in the latency time of pain response. Celecoxib was used as a reference analgesic drug. The delay in the latency time of pain response of the test compounds compared to vehicle-treated animals was determined. All the tested compounds showed potent analgesic activities specially compounds 5e, 5f, 8a and 8b. These results were consistent with the in vitro data on COX, where compounds 5f, 5e and 8b showed COX-2 inhibitory activity with IC50 equal to 46.07, 37.57 and 35.18 μM, respectively, ().
Figure 3. Results of hot plate assay.
Acetic acid-induced writhing test
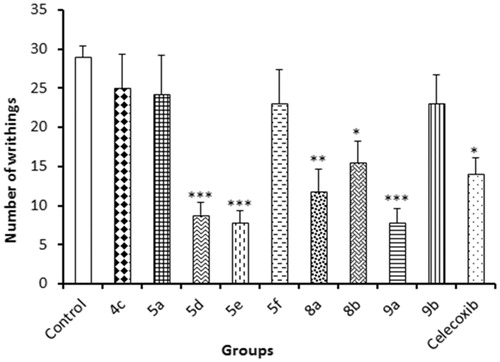
Peripheral analgesic activity for the tested compounds was investigated using intraperitoneal injection of 0.6% acetic acid after oral administration of mice with 10 mg/kg of compounds 4a, 5a, 5d, 5e, 5f, 8a, 8b, 9a, 9b and celecoxib.
The most active compounds were 5d, 5e and 9a compared to normal control. The most potent compound 5e also has a good inhibitory activity against COX-2 as mentioned previously. The data obtained using this method is summarised in .
Figure 4. Results of acetic acid-induced writhing test.
Histopathological studies
The stomach specimen of control treated rats was characterised by normal histological structure of glandular gastric mucosa, submucosa and musculosa (, control)Citation56. Complete disruption of protective mucosal layer was observed in indomethacin treated rats as mucosa showed an erosion formation (starting point of ulcer formation), also some eosinophilic inflammatory cells infiltration in submucosa associated with hyalinosis and coagulative necrosis of muscular layer and this results in accordance with Günnur et al.Citation57 who confirmed that indomethacin induces gastrodoudenal ulcer formation after oral intake (, indomethacin). There were thickening and hyalinosis of basement membrane with lymphocytic infiltration and minimal congested submucosal blood capillaries after celecoxib intake (, celecoxib) that is considered much safer drug than indomethacin.
Figure 5. Haematoxylin and eosin immunohistochemical staining of gastric ulcers after ulcer induction in rats for specimen intact Mucous membrane in control, indomethacin, celecoxib-treated rat and test compounds 4c, 5a, 5d and 5e.
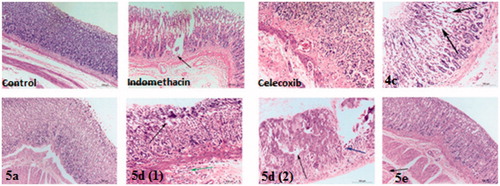
The current tested drug compounds in this study; 4c treated rats has shown that only lesion on the glandular epithelium of gastric mucosa which showed mucous degeneration in gastric glandular epithelium reach to coagulative necrosis (, 4c). For compound 5a, all layers of stomach were more or less normal (, 5a). (Ulcer index = 1.5, the short period of oral intake was not enough to induce the tissue reaction towards the drug compound). Mucous degeneration in gastric glandular epithelium, thickening and hyalinosis of basement membrane, as well as eosinophilic infiltrations in submucosa were observed in 5d-treated rats (, 5d 1, 2). For compounds 5e (, 5e) and 5f (, 5f) affect only muscular layer of the stomach by causing corrugation and hyalinosis in some muscle bundles and reach to coagulative necrosis; this changes suggest presence of nausea and irritability of the stomach reach to vomiting may appear as clinical sign in this two groups.
Figure 6. Haematoxylin and eosin immunohistochemical staining of gastric ulcers after ulcer induction in rats for specimen intact mucous membrane in 5f, 8a, 8 b, 9a and 9 b-treated rats.
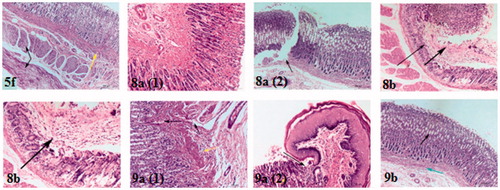
In rats treated with compounds bearing SO2CH3 pharmacophore and dimethoxy or trimethoxy phenyl part, some areas of glandular epithelium were more or less normal associated with sever congested blood capillaries and eosinophilic infiltrations in submucosa of 8a treated rats. Ulcer was found in other areas of glandular epithelium of mucosa, thickening and hyalinosis of basement membrane and associated with congestion of blood capillaries, while massive destruction of localised area of epithelium with formation of an erosion associated with hyalinosis and necrosis of muscular layer in 8b treated rats (, 8a and b).
Scanning of stomach specimens of rats treated with compounds 9a and 9b (have SO2NH2 moiety and di- or trimethoxyphenyl group), showed that sever congestion in blood capillaries and lymphocytic infiltration (, 9a, 1). Area of ulceration between glandular epithelium and keratinised epithelium of gastric mucosa and lymphocytic infiltration were noticed. Coagulative necrosis of some cells of glandular epithelium of gastric mucosa (, 9a, 2) were detected. Mucous degeneration of glandular epithelium of gastric mucosa, and lymphocytic infiltration and mild congestion in submucosa 9b were observed, (, 9b).
Docking study
All new designed compounds were docked using X-ray crystal structure data for COX-2 enzyme obtained from the protein data bank (pdb: ID 1CX2)Citation58.
In docking study, ligand SC-558 and all new designed compounds were docked using Molecular Operating Environment (MOE, Version 2005.06, Chemical Computing Group Inc., Montreal, Quebec, Canada) into the COX-2 receptor.
It was observed that H-bonding interactions between ligand SC-558 (selective COX-2 inhibitor) and COX-2 receptor were achieved via two H-bonds (i) SO2NH2 with His90 (2.42 A°) and (ii) pyrazole N-2 with Tyr355 (2.77 A°). The energy associated with intermolecular interaction was −10.0340 Kcal/mol, and and .
Figure 7. 2D (left image) and 3D (right image) interaction of ligand SC-558 in the active site of COX-2 receptor. It is possible to observe the binding using H-bond to His90 and Tyr355 amino acids.
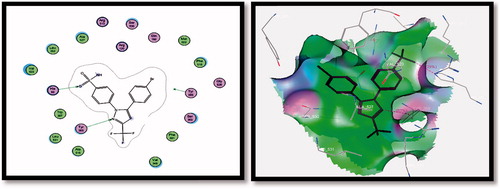
Figure 8. 2D (left image) and 3D (right image) interaction of compound 5f in the active site of COX-2 receptor. It is possible to observe the binding using H-bond to His90, Tyr355, Tyr385 and Ser530 amino acids.
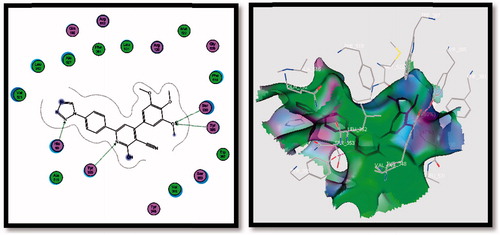
Most of the designed compounds had the same pattern of interactions in the COX-2 receptor through His90, Tyr355, Tyr385, Ser530, Gly354 and Ser353 amino acids, with one to four H-bonds.
Thirteen compounds 3a and b, 4a and c, 5a–f, 6a and b, 8a and b and 9a and b showed appreciable binding interactions (affinity in Kcal/mol ranges from −11.5834 to −22.4323) with one to four H-bonding interactions.
Compounds 4b, 5d, 7a and b exhibited lower binding interactions than ligand and their affinity range was from −3.6634 to −7.0670 Kcal/mol with different numbers of H-bonds from one to four.
Docking compound 5f (with COX-2 S.I.=46.07) in the COX-2 enzyme, four H-bonding interactions with His90, Tyr355, Tyr385 and Ser530 amino acids were observed. Its binding energy was −22.4323 Kcal/mol more than that of SC-558. Compounds 6a and b, 8a and b and 9a and b, containing COX-2 pharmacophores (SO2Me or SO2NH2), were in high response to COX-2.
On the other hand, affinity of some pyridine containing compounds such as 4b and 5d, and the two cyanamide derivatives 7a and b (lack from tetrazole moiety), was lower than that of SC-588.
In most compounds, tetrazole N-2 was important for formation of H-bonding interactions with His90 and Tyr355 amino acids, ().
Conclusions
In summary, the design and synthesis of new tetrazole 3a and b, 4a–c, 5a–f, 8a and b and 9a and b and cyanamide derivatives 7a and b as anti-inflammatory agents were described. In vitro COX-1 and COX-2 assay was evaluated. Compound 5f possessing hydroxypyridine carbonitrile core and bearing trimethoxyphenyl group, was the most potent and selective COX-2 inhibitor with IC50 value of 46.07, more than that of celecoxib (IC50=45.68). Compounds with pyridine ring 4c, 5a, 5e and those with trimethoxyphenyl ring and pyrazoline core-bearing SO2CH3 or SO2NH2 pharmacophores 8b and 9b showed high activity as COX-2 inhibitors with IC50 ranged from 35.13 to 42.02. All test compounds were evaluated for their in vivo anti-inflammatory activities. Analgesic activity (central and peripheral) and ulcerogenic liability were assessed. Compounds 9b and 8b with trimethoxyphenyl part and SO2NH2 or SO2CH3 pharmacophore have the same or near safety profile as the selective COX-2 inhibitor celecoxib (UI = 0.5, 0.75, respectively). The rest of the tested compounds 4c, 5a, 5d–f, 8a and 9a showed remarkable improvement in ulcer index (UI = 1.25–2.00) when compared with indomethacin (UI = 22.5). From histobathological investigations, it was shown that 5a was the most preferable compound with minimal drug effect on tissue. While 9a caused destructive effect on the mucosa and ulceration.
Molecular docking study on COX-2-active site showed that in most compounds, tetrazole N-2 was important for the formation of H-bonding interactions with His90 and Tyr355 amino acids similar to SC-558, the ligand used.
Acknowledgements
Authors would like to thank all members of NMR Lab, Faculty of Pharmacy, Beni-Suef University, Egypt, for their efforts and supports in analyzing samples in this work. We thank Dr. Waleed A. Mohamed, BioChemistry Department, Cairo General Hospital, Egypt, for his kind help in doing COX assay.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Cheng-Xi W, Ming B, Guo-Hua G. Tetrazolium compounds: synthesis and applications in medicine. Molecules 2015; 20:5528–53.
- Demko ZP, Sharpless KB. Preparation of 5-substituted 1H-tetrazoles from nitriles in water. J Org Chem 2001;66:7945–50.
- Vinoth KS, Ranjith KR, Silpa B, et al. The therapeutic journey of tetrazoles: a review. J Compr Pharm 2015;2:42–7.
- Bachar SC, Lahiri SC. Synthesis of chloro and bromo substituted 5-(indan-1′-yl)tetrazoles and 5-(indan-1′-yl)methyltetrazoles as possible analgesic agents. Pharmazie 2004;59:435–8.
- Nazariy TP, Vasyl SM, Mykola DO. New convenient synthesis of 2,3-diaminotheino[2,3-d]pyrimidin-4 (3H)-one derivates from substituted alkyl 2-(1H-tetrazol-1-yl)thiophene-3-carboxylates. Tetrahydron 2008;64:1430–4.
- Bhaskar VH, Mohite PB, Pandhare RB, Khanage SG. In vitro evaluation of tetrazoles as a novel class of Antimycobacterium tuberculosis agents. Acta Pharm Sci 2010;52:502–10.
- Mohite PB, Bhaskar VH. Potential pharmacological activities of tetrazoles in the new millennium. Int J PharmTech Res 2011;3:1557–66.
- Moustafa MA, El-Sherbeny MA, El-Sherbiny DT, El-Sayed SM. Molecular modeling, synthesis and antimicrobial evaluation of new molecular hybrids of tetrazole derivatives. J Am Sci 2012;8:973–86.
- Dhayanithi V, Syed SS, Kumaran K, et al. Synthesis of selected 5-thio-substituted tetrazole derivatives and evaluation of their antibacterial and antifungal activities. J Serb Chem Soc 2011;76:165–75.
- Wu J, Wang Q, Guo J, et al. Characterization of angiotensin II antagonism displayed by Ib, a novel nonpeptide angiotensin AT(1) receptor antagonist. Eur J Pharmacol 2008;589:220–24.
- Yan B, Wang G, Sun J, et al. Identification of the major metabolites of 5-n-butyl-4-{4-[2-(1H-tetrazole-5-yl)-1H-pyrrol-1-yl]phenylmethyl}-2,4-dihydro-2-(2,6-dichloridephenyl)-3H-1,2,4-triazol-3-one, a new angiotensin type 1 receptor antagonist, in rat bile by HPLC-diode array detection-MS and HPLC-MS/MSJ. Biomed Chromatogr 2007;21:912–24.
- Berghmans S, Hunt J, Roach A, Goldsmith P. Zebra fish offer the potential for a primary screen to identify a wide variety of potential anticonvulsants. Epilepsy Res 2007;75:18–28.
- Rostom SA, Ashour HM, El Razik HA, et al. Azole antimicrobial pharmacophore-based tetrazoles: synthesis and biological evaluation as potential antimicrobial and anticonvulsant agents. Bioorg Med Chem 2009;17:2410–22.
- Bhaskar VH, Mohite PB. Synthesis, characterization and evaluation of anticancer activity of some tetrazole derivatives. J Optoelect Biomed Mat 2010;2:249–59.
- Romagnoli R, Baraldi PG, Salvador MK, et al. Synthesis and evaluation of 1,5-disubstituted tetrazoles as rigid analogues of combretastatin A-4 with potent antiproliferative and antitumor activity. J Med Chem 2012;55:475–88.
- Gürsoy A, Demiravak S, Capan G, et al. Synthesis and preliminary evaluation of new 5-pyrazolinone derivatives as analgesic agents. Eur J Med Chem 2000;35:359–64.
- Uchida M, Komatsu M, Morita S, et al. Studies on gastric antiulcer active agents. II.: synthesis of tetrazole alkanamides and related compounds. Chem Pharm Bull 1989;37:322–6.
- Bepary S, Das BK, Bachar SC, et al. Anti-inflammatory activity of indanyltetrazole derivatives. Pak J Pharm Sci 2008;21:295–8.
- Kumar P, Knaus EE. Synthesis and antiinflammatory activity of 5-(1,6-dihydropyridyl)-tetrazol-2-acetic acids, esters and amides. Drug Des Discov 1994;11:15–20.
- Vicini P, Amoretti L, Barocelli E, et al. Synthesis and anti-inflammatory, antipyretic and analgesics properties of 5-(1,2-benzisothiazolyl)tetrazoles. Farmaco Sci 1986;41:111–8.
- Pande K, Tandon M, Bhalla TN, et al. Tetrazoles as potent anti-inflammatory agents. Pharmacology 1987;35:333–8.
- Ikeda T, Kakegawa H, Miyataka H, et al. Anti-allergic and anti-inflammatory actions of 2′-(tetrazole-5-yl)-4-hydroxy-2-methyl-2H-1,2-benzothiazine-3-carboxanilide 1,1-dioxide. Bioorg Med Chem Lett 1992;2:709–14.
- Abou-Ghalia MH, Amr AE, Abdulla MM. Synthesis of some new (Nα-dipicolinoyl)-bis-L-leucyl-DL-norvalyl linear tetra and cyclic octa bridged peptides as new anti-inflammatory Agents. Z Naturforsch 2003;58b:903–10.
- Sondhia SM, Jaina S, Dinodiaa M, Kumarb A. Synthesis of some thiophene, imidazole and pyridine derivatives exhibiting good anti-inflammatory and analgesic activities. Med Chem 2008;4:146–54.
- Thirumurugan P, Mahalaxmi S, Perumal PT. Synthesis and anti-inflammatory activity of 3-indolyl pyridine derivatives through one-pot multi component reaction. J Chem Sci 2010;122:819–32.
- Rathish IG, Javed K, Ahmad S, et al. Synthesis and anti-inflammatory activity of some new 1,3,5-trisubstituted pyrazolines bearing benzene sulfonamide. Bioorg Med Chem Lett 2009;19:255–8.
- Ovais S, Bashir R, Yaseen S, et al. Synthesis and pharmacological evaluation of some novel 2-pyrazolines bearing benzenesulfonamide as anti-inflammatory and blood glucose lowering agents. Med Chem Res 2012; 22:1378–85.
- Ovais S, Yaseen S, Bashir R, et al. Synthesis and anti-inflammatory activity of celecoxib like compounds. J Enzyme Inhib Med Chem 2013;28:1105–12.
- Bashir R, Ovais S, Yaseen S, et al. Synthesis of some new 1,3,5-trisubstituted pyrazolines bearing benzene sulfonamide as anticancer and anti-inflammatory agents. Bioorg Med Chem Lett 2011;21:4301–5.
- Kajal A, Bala S, Sharma N, et al. Therapeutic potential of hydrazones as anti-Inflammatory agents. Int J Med Chem 2014;2014:1–11.
- Won SJ, Liu CT, Tsao LT, et al. Synthetic chalcones as potential anti-inflammatory and cancer chemopreventive agents. Eur J Med Chem 2005;40:103–12.
- Mohite PB, Pandhare RB, Khanage SG. Synthesis, characterization and anti-inflammatory activity of novel N-substituted tetrazoles. Analele UniversităŃii Din Bucureşti – Chimie (Serie Nouă) 2011;20:107–13.
- Ghosh R, Das A. Synthesis and biological activities of chalcones and their heterocyclic derivatives: a review. World J Pharm Pharm Sci 2014;3:578–95.
- Al-Hourania BJ, McDonald R, El-Barghouthi MI, et al. Molecular docking studies and X-ray structure determination of 1-{4-(methylsulfonyl)phenyl}-5-phenyl-1H-tetrazole. Jord J Chem 2015;10:34–40.
- Al-Hourani BJ, El-Barghouthi MI, Mcdonald R, et al. Docking studies and the crystal structure of two tetrazole derivatives:5-(4-chlorophenyl)-1-{4-(methylsulfonyl)phenyl}-1H-tetrazole and4-{5-(4-methoxyphenyl)-1H-tetrazol-1-yl}benzenesul-fonamide. J Mol Struc 2015;1101:21–7.
- Wuest F, Tang X, Kniess T, et al. Synthesis and cyclooxygenase inhibition of various (aryl-1,2,3-triazole-1-yl)-methanesulfonylphenyl derivatives. Bioorg Med Chem 2009;17:1146–51.
- Al-Hourani BJ, Sharma SK, Suresh M, Wuest F. Novel 5-substituted 1H-tetrazoles as cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med. Chem Lett 2012;22:2235–8.
- Al-Hourani BJ, Sharma SK, Kaur J, Wuest F. Synthesis, bioassay studies, and molecular docking of novel 5-substituted 1H tetrazoles as cyclooxygenase-2 (COX-2) Inhibitors. Med Chem Res 2015;24:78–94.
- Al-Hourani BJ, Sharma SK, Mane JY, et al. Synthesis and evaluation of 1,5-diaryl-substituted tetrazoles as novel selective cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem Lett 2011;21:1823–6.
- Eman KA, Phoebe FL, Waleed AM. Cyclooxygenase-2 and 15-lipoxygenase inhibition, synthesis, anti-inflammatory activity and ulcer liability of new celecoxib analogues: determination of region-specific pyrazole ring formation by NOESY. Bioorg Med Chem Lett 2016;26:2893–9.
- Catella-Lawson F, McAdam B, Morrison BW, et al. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther 1999;289:735–41.
- Lamie PF, Phillopes JN, El-Gendy AO, et al. Design, synthesis and evaluation of novel phthalimide derivatives as in vitro anti-microbial, anti-oxidant and anti-inflammatory agents. Molecules 2015;20:16620–42.
- Navidpour L, Amini M, Shafaroodi H, et al. Design and synthesis of new water-soluble tetrazolide derivatives of celecoxib and rofecoxib as selective cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem Lett 2006;16:4483–7.
- Hinz B, Brune K. Cyclooxygenase-2-10 years later. J Pharmacol Exp Ther 2002;300:367–75.
- Khode S, Maddi V, Aragade P, et al. Synthesis and pharmacological evaluation of a novel series of 5-(substituted)aryl-3-(3-coumarinyl)-1-phenyl-2-pyrazolines as novel anti-inflammatory and analgesic agents. Eur J Med Chem 2009;44:1682–8.
- Lamie PF, Ali WAM, Bazgier V, Rarova L. Novel N-substituted indole Schiff bases as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase enzymes: synthesis, biological activities in vitro and docking study. Eur J Med Chem 2016;123:803–13.
- Yamamoto S. Mammalian lipoxygenases: molecular structures and functions. Biochim Biophys Acta 1992;1128:117–31.
- Gaffney BJ. Lipoxygenases: structural principles and spectroscopy. Annu Rev Biophys Biomol Struct 1996;25:431–59.
- Winter CA, Risely EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc Soc Exp Biol Med 1962;111:544–47.
- Cho CH, Ogle CW. Cholinergic-mediated gastric mast cell degranulation with subsequent histamine H 1- and H 2-receptor activation in stress ulceration in rats. Eur J Pharmacol 1979;55:23–33.
- Eddy N, Leimback D. Synthetic analgesics. II. Dithylenylbutenylamines and dithylenylbutyl amines. Pharmacol Exper Therap 1953;3:131–47.
- Koster R, Anderson M,D, Beer E, Acetic acid-induced analgesic screening. In: Federation Proceedings, 1959.
- Bancroft JD, Gamble M, Theory and practice of histological techniques. 6th ed. North Hollywood, Philadelphia (PA): Churchill Livingstone/Elsevier; 2008.
- Vembu S, Parasuraman P, Gopalakrishnan M. Design, in silico molecular docking studies, synthesis, spectral characterization and in vitro antifungal evaluation of 1-(4-(1H-tetrazole-1-yl)phenyl)-3-arylprop-2-en-1-ones. Der Pharma Chemica 2014;6:35–44.
- Vorobiov AN, Gaponik PN, Petrov PT, Ivashkevich OA. One-pot syntheses of 5-amino-1-aryltetrazole derivatives. Synthesis 2006; 1307–12.
- Gartner PL, Hiatt JL. Color atlas and text of histology, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2014.
- Günnur ÖD, Elif ÇY. Histopathologic evaluation of anti-ulcerogenic effect of montelukast in indomethacin-induced experimental ulcer model. Turk J Gastroenterol 2013; 24:88–92.
- Available from: http://www.rcsb.org/pdb/explore/explore.do?structureId = 1CX2