
Abstract
Digoxin and other cardiotonic steroids (CTS) exert their effect by inhibiting Na,K-ATPase (NKA) activity. CTS bind to the various NKA isoforms that are expressed in different cell types, which gives CTS their narrow therapeutic index. We have synthesised a series of digoxin derivatives (γ-Benzylidene digoxin derivatives) with substitutions in the lactone ring (including non-oxygen and ether groups), to obtain CTS with better NKA isoform specificity. Some of these derivatives show some NKA isoform selective effects, with BD-3, BD-8, and BD-13 increasing NKA α2 activity, BD-5 inhibiting NKA α1 and NKA α3, BD-10 reducing NKA α1, but stimulating NKA α2 and α3; and BD-14, BD-15, and BD-16 enhancing NKA α3 activity. A molecular-docking approach favoured NKA isoform specific interactions for the compounds that supported their observed activity. These results show that BD compounds are a new type of CTS with the capacity to target NKA activity in an isoform-specific manner.
Graphical Abstract
Introduction
Na,K-ATPase (NKA) is an enzyme responsible for the transport of Na+ and K+ across the plasma membrane of most animal cells. NKA generates the electrochemical ion gradients that drive many different cell processes, including cell excitability, glucose uptake, and water and salt transport. NKA was discovered by the Danish researcher Jens Skou, who used the cardiotonic steroid (CTS) ouabain, derived from plants of the Strophanthus genus, to specifically define the NKA activityCitation1. According to the most currently accepted mechanism of action of CTS, ouabain binds and locks NKA when in its E2-P conformation, a step in the NKA reaction cycle in which the enzyme has a high affinity for CTSCitation2,Citation3. In E2-P, NKA is phosphorylated, releases Na+ ions to the extracellular side of the cell plasma membrane, and is ready to bind and transfer K+ ions to the cytosol.
NKA is constituted by three different subunits: the α, β, and a smaller polypeptide, which depending on the tissue, consists of one of several members of the FXYD family of polypeptidesCitation4,Citation5. The α subunit is responsible for the catalytic activity of NKA and contains the ATP, Na+, and K+ binding sites. The β subunit is a glycosylated polypeptide responsible for the folding and functional competence of the NKA α subunit. The FXYD peptide functions as a modulator of the catalytic properties of NKA. Four isoforms of the NKA α subunit and three NKA β isoforms have been identified in mammals (α1, α2, α3, α4, β1, β2, and β3). This molecular heterogeneity, in addition to various assemblies of the α and β subunits, provides cells with the versatility of adjusting Na+ and K+ gradients to the needs of each cell typeCitation5.
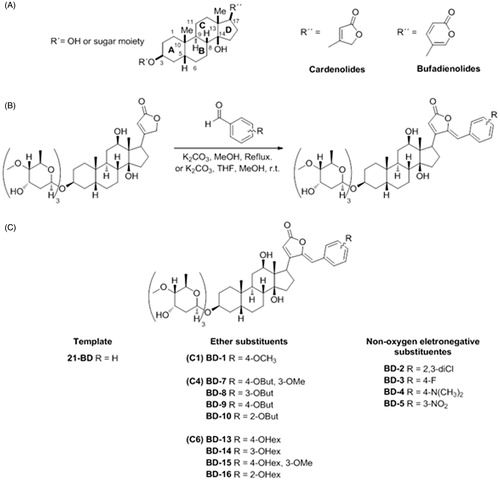
Due to their capacity of inhibiting NKA, CTS secondarily increase intracellular Ca2+ in myocardiocytes and cause higher cardiac contraction force and output. For this reason, CTS have been used in the treatment of congestive heart failureCitation4. These compounds share a common structural characteristic, which is to have a conserved steroidal core with a five-membered lactone ring (in the case of cardenolides) or six-membered lactone ring (for the bufadienolides), attached to C17; and a sugar moiety, composed of one or more hydrocarbons (for the glycosides) or just a hydroxyl group (for the genins), linked to C3 ()Citation4,Citation6.
Figure 1. General scheme of the cardiotonic steroid structure (A), general procedure for the synthesis of digoxin derivatives (B), and group and subgroup split of derivatives according to their structure (C).
Several studies have been conducted to determine the CTS binding site within the NKA. It has been shown that at least part of the ouabain binding site resides on the extracellular side of the NKA α isoform, comprising especially the extracellular loops between the TM1-TM2, TM5-TM6, and TM9-TM10 transmembrane domainsCitation7,Citation8. However, other studies have reported that ouabain can also interact with the transmembrane domains TM5 and TM6, partially unwinding the TM4 helixCitation9,Citation10. The ouabain lactone ring has been shown to accommodate close to Val329 and Ala330 in the TM4 domain, which displaces Gly326, an amino acid residue that is crucial for K+ coordinationCitation9. The steroid core helps the molecule to interact with a hydrophobic pocket at TM4-TM6 domains guided mainly by Phe323, Phe790, and Phe793, whereas the sugar moiety appears to make hydrogen bonds with Glu319 and Arg887 on the TM7-TM8 loop and the TM4 domain, respectivelyCitation9,Citation10. Understanding how ouabain interacts with NKA is of high relevance, since it will help deciphering how other CTS differing on lactone ring, hydroxyl groups attached to the steroidal core, and sugar moieties interact with NKA. This information can then be used to develop compounds with higher binding capacity and potency.
In the 1990s, some researchers observed that ouabain exerted effects on NKA that were independent from inhibition of the enzyme and rather resulted from the activation of a cascade of intracellular events that caused cardiomyocyte hypertrophyCitation11–14. This revealed a new function for the NKA and showed that it can function as a receptor and signal transducer that mediates the effects of ouabain in cells. This role of NKA has been the topic of intense research, with the goal of exploiting NKA signalling for its use in disease and cancer. Several laboratories have designed and generated, particularly via hemi-synthesis, novel CTS derivativesCitation15–17. Our group is interested in developing new CTS derivatives which can exert effects that are more specific. We have generated a series of compounds by modifying the lactone ring of the CTS digoxin. One of these synthetic digoxin analogues, 21-benzylidene digoxin (21-BD), presented cytotoxic and antiproliferative effects when used on HeLa cancer cells, via inducing apoptosis in those cells, but inhibiting NKA activity just at high micromolar levelsCitation18,Citation19. Subsequently, starting with the structure of digoxin, we synthesised a series of γ-benzylidene derivatives and showed that they have cytotoxic effects on HeLa and RKO cancer cell lines that are independent from the modulation of NKA activityCitation20.
One of the disadvantages of the use of CTS is that they exhibit a very narrow safety margin and have toxic effects that are secondary to sustained NKA inhibitionCitation21. In addition, the ubiquitous presence of NKA in all tissues, along with a cell type specific pattern of expression of its various isoforms, all of which being able to respond to CTS, contribute to the widespread action of the CTS. In this manner, it is clear that there is a need for developing compounds that can more specifically target a particular NKA isoform type, which will help to prevent some of their secondary effects.
In this work, we show the synthesis of a series of new CTS compounds, derived from digoxin by introducing non-oxygen and ether substituents into the lactone ring. Analysis of these compounds on NKA activity and studies of their interactions with the NKA α isoforms using a molecular docking approach shows that some of these compounds exhibit isoform specific properties. This shows new data for the structural features underlying the interaction of CTS with NKA and provides new compounds with a better NKA isoform specificity.
Material and methods
General procedure for the synthesis of digoxin derivatives (21-BD), (BD-1), (BD-2), (BD-3), (BD-5), (BD-7), (BD-8), (BD-10), (BD-13), (BD-14), (BD-15), and (BD-16)
Aldehyde (1.8 mmol), digoxin (0.469 g, 0.6mmol), anhydrous K2CO3 (0.249 g, 1.8 mmol) were added to 60 ml of methanol in a round bottom flask. After stirring for 6 h at 70 °C, the solvent was evaporated in a rotary evaporator. The crude product was diluted with 20 ml of water and extracted with hot ethyl acetate (3 × 30 ml). The organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by silica column chromatography (CH2Cl2/MeOH 11:1). After purification, the pure product was diluted in THF, precipitated with hexane and concentrated under reduced pressure to give the benzylidene digoxin derivatives ().
General procedure for the synthesis of digoxin derivatives (BD-4) and (BD-9)
Aldehyde (2 mmol), digoxin (0.781 g, 1.0 mmol), anhydrous K2CO3 (1.1 g, 7.96 mmol), were added to 4 ml of methanol and 2 ml THF in a round bottom flask. After stirring for 6 h at room temperature, the solvent was evaporated in a rotary evaporator. The crude product was diluted with 20 ml of water and extracted with hot ethyl acetate (3 × 30 ml). The organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by silica column chromatography (CH2Cl2/MeOH 11:1). After purification, the pure product was diluted in THF, precipitated with hexane and concentrated under reduced pressure to give the benzylidene digoxin derivatives as a yellow solid ().
Insect cell culture and viral infections
Sf9 insect cells were grown in Grace’s medium with 3.3 g/l lactalbumin hydrolysate, 3.3 g/l yeastolate, and supplemented with 10% (v/v) fetal bovine serum, 100 units/ml penicillin, 100 µg/ml streptomycin, and 0.25 µg/ml fungizone. Cells were grown in suspension cultures and were transferred to 150 mm tissue culture plates before infection. Infections were performed as previously described, using baculoviruses carrying the desired NKA α isoformCitation22.
Membrane preparation
After 72 h of infection, cells were scraped from the culture plates, centrifuged at 1500×g for 10 min and suspended in 250 mM sucrose, 0.1 mM EGTA, and 25 mM imidazole HCl, pH 7.4, using a motor-driven Teflon Potter-Elvehjem homogenizer (Thomas Scientific, Swedesboro, NJ). The sample was then subjected to centrifugation at 4500×g for 10 min. The resulting supernatant was centrifuged at 70,000×g for 1 h. The final pellet was re-suspended in the homogenisation solution and used for NKA activity assays.
NKA activity
The cell membrane preparations were incubated 20 min with digoxin derivatives. The reaction was carried on at 37 °C for 30 min in 120 mM NaCl, 20 mM KCl, 2 mM MgCl2, 3 mM ATPNa2, and 50 mM HEPES (pH 7.5), in the absence and presence of 1 mM ouabain (10 min of incubation). NKA activity was determined by measuring the released inorganic phosphate according to a colorimetric method described previouslyCitation23, and specific activity was considered as the difference between the total and ouabain-treated samples ATPase activities (Table S2).
Molecular docking
Initially, the 3D structures of ligands were generated by Marvin Beans 16.8.1.0 software (ChemAxon, Budapest, Hungary) in PDB formatCitation24, which protonated form and tautomeric conformers were carefully checked using pH 7.4Citation25. Afterwards, all ligands were refined by semi-empirical Parametric Method 7 (PM7)Citation26 implemented on MOPAC2012 using the keyword ef for minimum searchCitation27. Next, refined ligands in pdbqt format were signed to routable bonds, Gasteiger–Marsili net atomic chargesCitation28, and hydrogens from polar atoms were kept by MGLTools softwareCitation29. The visual inspection of ligands geometry was performed by PyMOL applicationCitation30.
In parallel, the amino acid primary sequence of NKA α1 (NP_036636.1), NKA α2 (NP_036637.1), and NKA α3 (NP_036638.1), deposited into National Center for Biotechnology Information (NCBI), was used to generate their 3D structure using Swiss-Model online softwareCitation31 (Swiss Institute of Bioinformatics, Basel, Switzerland). All structures were built by homology modelling using the pig kidney NKA α1 isoform as a model (PDB ID code 4HYT)Citation32. NKA α1 and α2 isoforms were built as precursors, containing the first five amino acids that are not found in mature NKA polypeptide. Then, these targets were prepared for molecular docking using MGLTools softwareCitation29 (The Scripps Research Institute, La Jolla, CA). A grid box was built centred on CTS, covering up all binding sites, with −26.55, −20.197, and 71.573 for x, y, and z axis, respectively, and size of 20, 20, and 36 Å. The exhaustiveness was set to 20 to improve the search for best conformer into the binding site. Next, the ligands in pdbqt format were submitted to molecular docking process using the AutoDock Vina software (The Scripps Research Institute, La Jolla, CA)Citation33. Ramachandran plots were used to validate the models and were generated by Procheck software (European Bioinformatics Institute, Hinxton, Cambridge, UK)Citation34.
Data analysis
The data were analysed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA) and plotted as mean ± SEM. The mean values were compared using Two-way ANOVA, to compare the effects of increasing concentrations of the CTS and different NKA isoform assemblies in the NKA activity, followed by the Bonferroni post hoc test to indicate the source of the observed differences. Dose-response experiments were performed 3–6 times using different membrane preparation samples. NKA activity values at different concentrations of a particular compound and for each NKA isoform were normalised to the activity of the corresponding isoform in the absence of the compound. Statistical significance was set as p < .05.
Results
NKA activity
Sf9 cells were infected with baculoviruses driving the expression of the different NKA α isoforms (α1, α2, and α3) and the β1 isoform. Homogenates from the cells containing the different NKA isoforms were used to test the effects of the digoxin derivatives. As shown in , to facilitate characterizing the effects and interaction of the compounds with different NKA isoforms, these derivatives were divided into three groups.
21-BD, considered the template compound, and the only molecule from this group, did not significantly affect the activity of NKA α2 and α3 isoforms (Figure S1(A)). Previous studies from our group have demonstrated that 21-BD inhibits the NKA activity derived from a mouse kidney membrane preparation, which only contains the NKA α1 isoform; and has no effect on rat brain NKA, which mainly expresses the NKA α2 and α3 isoformsCitation18.
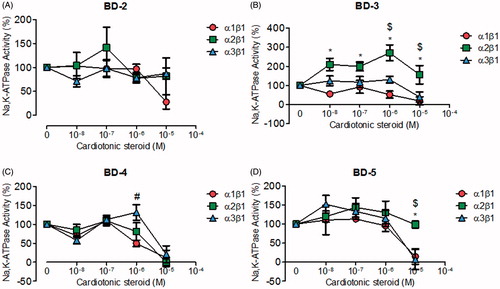
From a second group of compounds (BD-2, BD-3, BD-4, and BD-5), corresponding to chemical scaffolds with non-oxygen electronegative substituents in the aromatic ring level (), BD-2, similar to 21-BD, did not inhibit NKA α2 and α3 isoforms; however, it did reduce NKA α1 isoform activity at micromolar concentrations (). BD-3 exhibited selectivity for the NKA α2 isoform, increasing its activity over 100% at almost all concentrations tested. In contrast, BD-3 inhibited NKA α1 and α3 isoforms at high concentrations (). BD-4 shows no selectivity for the NKA isoforms, but it is able to inhibit all isoforms at micromolar concentrations (). Likewise, BD-5 exhibited no NKA isoform specific effects at relatively low concentrations, but interestingly it inhibited the NKA α1 and α3 isoforms and not NKA α2 at high concentrations ().
Figure 2. NKA activity (%) from Sf9 cell membrane preparations after BD-2 (A), BD-3 (B), BD-4 (C), and BD-5 (D) treatment for 20 min. *p < .05 differences between NKA α1β1 and α2β1 isoforms. $p < .05 differences between NKA α2β1 and α3β1 isoforms. #p < .05 differences between NKA α1β1 and α3β1 isoforms. Each point represents the mean ± SEM of at least three independent experiments performed in triplicate.
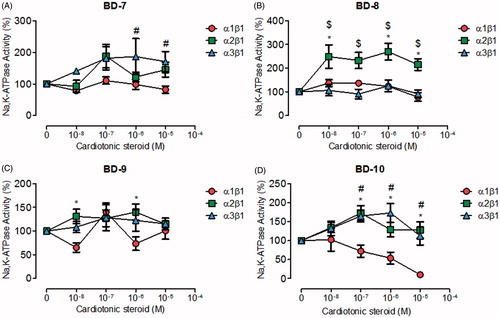
The third group of compounds that we synthesised, consisted in derivatives with ether radicals as functional groups, attached to the aromatic ring of the template 21-BD. These compounds could be further subdivided into three subgroups, according to their carbon chain length, containing one, four, and six-carbons (C1, C4, and C6, respectively). BD-1, the only C1 compound did not exhibit selectivity for any particular NKA α isoforms, nor had any effect on their activity (Figure S1(B)). Concerning the compounds of the C4 subgroup, BD-7 and BD-8 did not affect NKA α1 and α3 isoforms (). However, BD-8 significantly increased NKA α2 activity at all concentrations (). BD-9 showed no effect on NKA α1, α2 and α3 (). However, BD-10 exhibited a strong dose-response inhibitory curve for the NKA α1 isoform, but stimulated the NKA α2 and α3 isoforms at concentrations between 100 nM and 1 µM ().
Figure 3. NKA activity (%) from Sf9 cell membrane preparations after BD-7 (A), BD-8 (B), BD-9 (C), and BD-10 (D) treatment for 20 min. *p < .05 differences between NKA α1β1 and α2β1 isoforms. $p < .05 differences between NKA α2β1 and α3β1 isoforms. #p < .05 differences between NKA α1β1 and α3β1 isoforms. Each point represents the mean ± SEM of at least three independent experiments performed in triplicate.
The compounds of subgroup C6, almost all showed a significant selectivity for NKA α3, stimulating its activity (). BD-13 was the only derivative that demonstrated a different effect, stimulating the NKA α2 isoform in the nanomolar range and inhibiting the NKA α1 and α3 isoforms in the micromolar range (). BD-14, BD-15, and BD-16 significantly stimulated the NKA α3 isoform at all concentrations, whereas they did not affect the NKA α2 isoform (). The effects of these last three compounds on the NKA α1 isoform showed some peculiarities, with BD-14 showing the dual effect observed for classical CTS (), whereas BD-15 also showed a stimulatory effect at nanomolar levels, but no inhibitory effect in the micromolar range (). BD-16 did not significantly affect either the NKA α1 or α2 isoforms ().
Figure 4. NKA activity (%) from Sf9 cell membrane preparations after BD-13 (A), BD-14 (B), BD-15 (C), and BD-16 (D) treatment for 20 min. *p < .05 differences between NKA α1β1 and α2β1 isoforms. $p < .05 differences between NKA α2β1 and α3β1 isoforms. #p < .05 differences between NKA α1β1 and α3β1 isoforms. Each point represents the mean ± SEM of at least three independent experiments performed in triplicate.
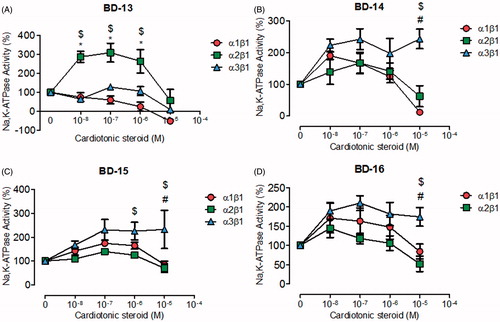
Altogether, these results suggest that changes in the chemical side groups attached to the lactone ring are able to modify the interaction of the resulting CTS with the NKA, in some cases in an isoform specific manner.
Molecular docking
As a correlate to the experimental response of NKA isoforms to each of the synthetic CTS that we generated, we studied their interaction at the molecular level, with the goal of finding the basis for their different NKA effects. The rat NKA α1, α2, and α3 sequences show 96.87, 86.47, and 86.57% of identity, respectively, compared to the pig NKA α1 isoformCitation35–37. Ouabain has been one of the most studied CTS, and therefore, we used it as a model in our molecular docking studies. Our synthetic derivatives, as well as ouabain, were docked on the rat NKA α subunit isoforms. First, the docked compounds were refined through a semi-empirical approach to correct geometric parameters, such as bond lengths. Initially, a re-docking with the refined ouabain ligand and the crystallographic ouabain on the pig NKA α1 isoform was performedCitation18. This provided a root mean square deviation (RMSD) value of 2.24 Å for the best solution, supporting, along with the NKA α isoforms Ramachandran plots (Figures S2–S4), the accuracy of the methodology used. After this, selected compounds were subjected to molecular docking.
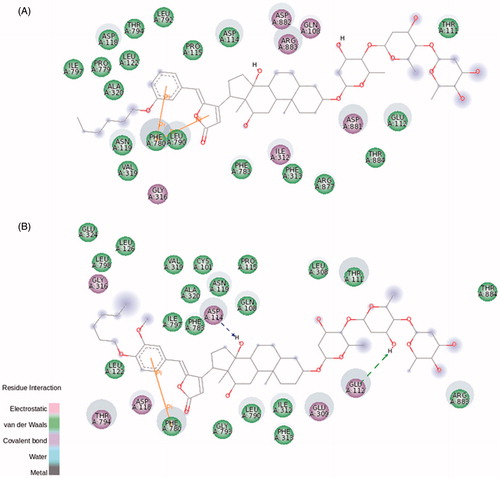
Ouabain docking showed a series of relevant interactions with the NKA α1 isoform (). The lactone ring interacts with Val329, responsible for the coordination of K+ ions, and with Gly803 interacting by electrostatic bonds. Close to the steroidal core and in agreement with previous results obtained with NKA from shark rectal gland, we found some interactions with phenylalanine residues (Phe323, Phe790, and Phe793), through van der Waals interactionsCitation9, as well as with Ala330 and Leu800. Hydrogen bonds are established between the C11α, C19α, and C14β of ouabain and the residues Arg118, Asp128, and Thr804, respectively. Arg118 has been identified as one of the key residues involved in the ouabain resistance of the rodent NKA α1 subunitCitation38. Despite this has been related to an increase in the dissociation constant of the ouabain-rodent NKA α1 complexCitation39,Citation40, the binding energy of the docked ouabain with the three NKA α isoforms shows similar values (Table S1). At the rhamnose level, Glu123 forms hydrogen bonds with the distal hydroxyl groups.
Figure 5. Ouabain molecular docking on rat NKA α1 (A), α2 (B), and α3 (C) isoforms. The green and magenta circles represent residues involved in van der Waals and polar interactions, respectively. The blue halo around the residue is proportional to the solvent accessible surface. Pi interactions are represented by an orange line and symbols indicating the specific interaction. The green dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid main chains. The blue dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid side-chains.
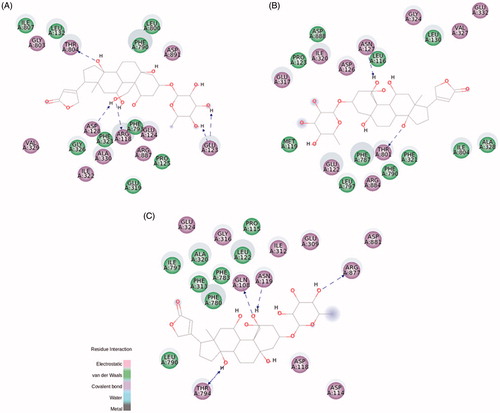
Regarding NKA α2 and α3 isoforms, both of them are slightly smaller than NKA α1 (1020 and 1013 amino acids, respectively, compared to 1023 amino acids for α1 – Figure S5)Citation41. Despite this, most of the interactions with ouabain are conserved, especially for the amino acids interacting with the steroidal core and the sugar moiety (). Some positional correspondences are observed, like the interaction with Arg118 for NKA α1, Leu116 for NKA α2, and Gln108 for NKA α3, but the residue exchange seems to be a key factor to the previously observed effect for the ouabain resistanceCitation38.
21-BD was used as the template compound for docking, due to the simplicity of its structure compared with the other derivatives. The presence of an aromatic ring attached to the lactone ring C21 helps the molecule to interact with a hydrophobic pocket formed by Ala330, Glu786, Phe790, Leu800, and Ile807 at NKA α1 isoform (Figure S6(A)). Thr804 makes hydrogen bond with a different hydroxyl group, attached to C12α, compared to ouabain. The presence of two extra distal sugar units showed new polar interactions with Asp892 and Arg893, as well as hydrogen bonds with Trp894. Indeed, all these different features observed molecularly, compared to ouabain, are evidenced by the absence of any modulatory effect of 21-BD, in nanomolar concentrations, on NKA activityCitation18. Unlike ouabain, not many conserved interactions were found when the three NKA α isoforms were compared, with different residues making polar and nonpolar interactions with all the three CTS moieties (Figure S6(B,C)).
In BD-3, the representative compound of the non-oxygen electronegative substituents group, the fluorine addition at p-position of the aromatic ring rearranges the hydrophobic pocket that accommodates this nonpolar group. Also, some electrostatic contacts are found at this level ( and S7). The electronegative characteristic of fluorine, evidenced by a bulky electronic cloud, keeps the linearity of BD-3 structure into the binding site, compared to a twisted shape of 21-BD docked on NKA α1 and α3 isoforms (Figure S7). BD-3 drastically increased the NKA α2 activity (). The interaction between Thr804 in NKA α1 isoform and C12α is displaced to the lactone ring, compared to the corresponding residue Thr801 of NKA α2 isoform. However, what is really different in this compound compared to 21-BD, and could explain BD-3 ability to increase NKA α2 activity, is the absence of a coordinative sigma–π interaction between Phe787 and the aromatic ring ( and S6(B)). Moreover, some interactions have changed their characteristics, even though the corresponding residues keep the same, as Ala328, Gly324, and Gly800, all of them making polar interactions with BD-3, unlike to nonpolar interactions with 21-BD.
Figure 6. BD-3 (A), BD-8 (B), and BD-13 (C) molecular docking on rat NKA α2 isoform. The green and magenta circles represent residues involved in van der Waals and polar interactions, respectively. The blue halo around the residue is proportional to the solvent accessible surface. Pi interactions are represented by an orange line and symbols indicating the specific interaction. The green dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid main chains. The blue dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid side-chains.
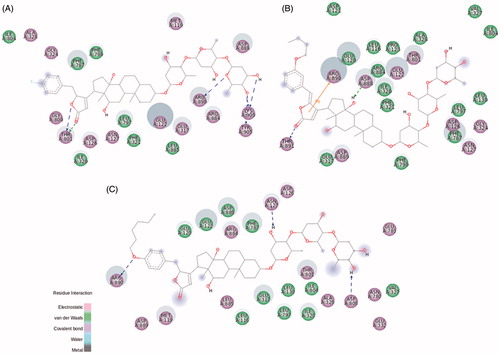
The ether radical derivatives, BD-8 and BD-10 present a butoxy group attached to the aromatic ring at m- and o-position, respectively. Both compounds conserved the interactions between Thr804 and C12α hydroxyl group, and hydrogen bonds between Trp894 and the distal digitoxose unit, when compared to the template 21-BD ( and S8 for BD-8; for BD-10). Interestingly, BD-10 shows the classical inhibitory profile of CTS on NKA α1 activity, whereas this effect is not observed by BD-8. The presence of a butoxy group at o-position clearly drives a distortion at the C17-C20 bond to better fit the butoxy group at a hydrophobic portion of NKA α1 isoform (). Despite both compounds show the same binding energy for the interaction with NKA α1 (Table S1), many hydrophobic interactions are showed for BD-10, especially stabilizing o-butoxy-benzylidene group. Leu132, Val329, Ala330, Glu334, Glu786, Ile787, and additionally, Pro789 and Leu802 are found surrounding this substituent group for BD-10, whereas the possible corresponding residues are spread throughout the BD-8 substituent (Figure S8(A)). These stable hydrophobic contacts between BD-10 and NKA α1, together with a sigma–π interaction between Ile807 and the aromatic ring could explain the inhibitory capacity for this compound.
Figure 7. BD-10 molecular docking on rat NKA α1 (A), α2 (B), and α3 (C) isoforms. The green and magenta circles represent residues involved in van der Waals and polar interactions, respectively. The blue halo around the residue is proportional to the solvent accessible surface. Pi interactions are represented by an orange line and symbols indicating the specific interaction. The green dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid main chains. The blue dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid side-chains.
The addition of a butoxy group to 21-BD drastically changes the interaction with the NKA α2 isoform. Stable polar interactions made by Asp888 and Arg890 at distal digitoxose unit for 21-BD are wondrously found interacting by a cation–π and hydrogen bond at lactone ring and steroidal core level, respectively (). In fact, 21-BD presents a more favourable binding energy compared to BD-8 and BD-10 (Table S1), and this possibly disturbs the binding site for these compounds, favouring an increased flexibility of the enzyme and producing the increased NKA α2 activity that we observed. Looking into interactions of the compounds to NKA α3 isoform, BD-10 seems to displace residues from sugar moiety to lactone ring level, which could explain the slight increase on NKA α3 activity (). BD-8 did not induce this conformational change, and indeed, presents stable interactions at lactone and aromatic ring like 21-BD (Figure S8(B)).
The three derivatives from the C6 subgroup (BD-13, BD-14, and BD-15) present a hexoxy group attached to the aromatic ring: at p-position for BD-13 and BD-15, and at m-position for BD-14. The difference between BD-13 and BD-15 is an additional methoxy group at m-position for BD-15, similar to BD-7 from C4 subgroup. BD-13 was able to inhibit NKA activity reconstituted with NKA α1 and α3 isoforms just at micromolar levels, whereas a huge selectivity to the NKA α2 isoform was observed, increasing NKA activity at sub-micromolar concentrations (). The addition of a six-membered carbonic chain to the aromatic ring helps the molecule to interact with a hydrophobic pocket as stated before, and indeed, the threonine residue (Thr804 for NKA α1 isoform and Thr794 for NKA α3 isoform) forming a hydrogen bond with C12α of 21-BD is otherwise found interacting with the lactone ring and aromatic ring of NKA α1 and α3, respectively, for BD-13 (Figure S9). These changes and other contacts like a cation-π interaction between Arg118 and the lactone ring on NKA α1 isoform, and a sigma–π interaction between Phe780 and the aromatic ring on NKA α3 isoform, could partially stabilise the NKA-CTS complex and explains the ability of BD-13 to inhibit these two isoforms at micromolar range. In contrast, BD-13 did not inhibit, but increased NKA α2 activity at submicromolar levels. Accordingly, the binding energy for BD-13/NKA α2 complex is slightly high (Table S1). In searching for interactions that could explain this activation effect, a completely different interaction pattern for BD-13 and NKA α2 was identified (). Arg890, that was involved in hydrogen bond with the distal sugar unit of 21-BD, is placed at the opposite side of BD-13, making a hydrogen bond with the ether radical oxygen. Any kind of π interaction appears to aid in the stabilisation of the complex. The classical phenylalanine residues covering up the steroidal core (Phe321, Phe787, and Phe790 for NKA α2 isoform) were not observed for BD-13 and some residues, as Glu332, Asn780, Glu783, and Asp808, are involved with polar interactions at sugar moiety level and were not found for any other derivatives. Pedersen et al. have demonstrated that Asn776 from pig kidney NKA α1 is very important in Na+ coordination, and indeed, positional changes in its carboxamide group drastically decreases Na+ bindingCitation42. It is possible that the interaction with the corresponding residue Asn780 in rat NKA α2 could favour Na+ binding and explain the increase in activity that BD-13 causes on NKA α2. Mutations at Asp804 were described to almost completely abolish pig kidney NKA α1 activity by affecting the cation binding siteCitation43. However, Koenderink et al. have reported that some mutations on rat NKA α1 isoform may increase basal NKA activity, and postulated that the presence of Na+ at the cation binding pocket after the dephosphorylation step, quickly shifting the enzyme to E1 conformation is involved in this effectCitation44. We have found a hydrogen bond between the corresponding residue (Asp808) in rat NKA α2 and the distal digitoxose unit of BD-13, suggesting that the lack of K+ coordination could favour a quick conformational shift of the enzyme, which can increase NKA activity. Tepperman et al. have demonstrated that Glu327 residue (corresponding to Glu332 in this study) is deeply involved with the first K+ occlusionCitation45, and we have observed this residue involved with polar interactions at sugar moiety level.
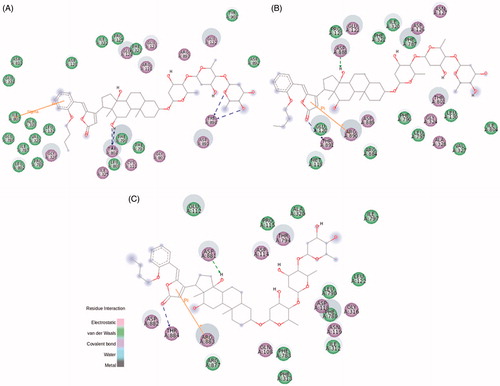
Concerning BD-14 and BD-15, both of them have not presented any effect on NKA α2 activity (). On NKA α1 isoform, BD-14 has demonstrated a well-defined steroidal core attachment on Phe790, Phe793, and Leu800, hydrogen bonds formed between Thr804 and C12α and, these contacts already seen for BD-10 may explain the ability of BD-14 to inhibit this isoform at micromolar range (Figure S10(A)). Instead of that, the large carbon chain keeps barely accommodated, and indeed, it is not possible to observe a well-defined hydrophobic pocket for the BD-13/NKA α1 isoform complex. This statement, and a smaller binding energy compared to BD-13 (Table S1) could explain why BD-14 is able to increase NKA α1 activity at nanomolar concentrations. Regarding BD-15, the hydrogen bond between Thr804 and C12α was not observed, and phenylalanine residues covering up the steroidal core moves together with Thr804, guided by interactions with the lactone ring and, therefore, this molecule is not able to inhibit NKA α1 activity such as BD-14 does (Figure S11(A)).
Finally, BD-14 and BD-15 clearly demonstrated an important selectivity towards the NKA α3 isoform, increasing NKA activity at all concentrations (). The aromatic ring from both compounds and Phe780 are involved in π interactions. Residues involved with steroidal core interactions are totally displaced towards the lactone and aromatic ring, and almost all polar interactions at sugar moiety level are abolished (). Weigand et al. have studied the NKA structural determinants for NKA α isoform selectivity of well-known digitalis-like compounds and, specifically, they have demonstrated a reduction in digoxin affinity for human NKA α2 isoform under pointed mutations at Met119, specifically, Met119Thr, Met119Asp, Met119Lys, Met119Asn, and Met119SerCitation46. Interestingly, the Met119Thr mutation was designed changing methionine residue for the corresponding threonine at NKA α1 isoform that also matches with threonine residue present at the same position of rat NKA α3 isoform. Therefore, a totally different pattern of CTS docking, together with threonine interaction during the pre-binding stateCitation46, could relieve CTS-NKA interaction and support an increased NKA α3 activity.
Figure 8. BD-14 (A) and BD-15 (B) molecular docking on rat NKA α3 isoform. The green and magenta circles represent residues involved in van der Waals and polar interactions, respectively. The blue halo around the residue is proportional to the solvent accessible surface. Pi interactions are represented by an orange line and symbols indicating the specific interaction. The green dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid main chains. The blue dashed arrows are directed towards the electron donor and represent hydrogen bonds with amino acid side-chains.
Discussion
Since the studies of the British physician William Withering on the beneficial and toxic effects of the foxglove (Digitalis purpurea), CTS have been used for the treatment of congestive heart failure. However, because CTS bind to the NKA isoforms that are expressed throughout the body, they show a narrow therapeutic index and need to be carefully used in medicine. This stresses the need for compounds that can specifically target a single type of NKA isoform and encourage the synthesis of new CTS derivatives, with the goal of obtaining compounds with less global effects. Elbaz et al. have demonstrated the ability of a synthetic monosaccharide digitoxin analogue, D6-MA, to inhibit NKA activity and decrease cell viability in lung cancer cellsCitation15. Mijatovic et al. have synthesised a modified version of 2-oxovoruscharin, UNBS1450, with potent anti-cancer activity and also a great inhibitory effect on NKA activityCitation17. Jensen et al. have synthesised mono and bivalent CTS using sulphur linked ethylene glycol moieties of varying length and they have shown their ability to inhibit NKA activity and their cytotoxic effect on the MCF-7 cancer cell lineCitation16. In previous work, we reported the synthesis of 21-BD by the addition of an aromatic ring to C17 of digoxin through a vinylogous aldol reaction. 21-BD was able to reduce the viability of cancer cells inhibiting the NKA activity just at high concentrationsCitation18. This data demonstrated a possible selective antitumor effect overcoming the toxic effects produced by NKA inhibition. Indeed, several authors have reported the ability of CTS to bind to NKA at nanomolar concentrations, without affecting its activity, but triggering signalling pathways inside the cellCitation12,Citation14,Citation47,Citation48. In this work, in an attempt to identify compounds with a selective NKA isoform modulation, we have performed the synthesis of novel digoxin analogues, adding different functional groups to the aromatic ring of 21-BD. We have evaluated their ability to modulate the NKA activity and characterised their interactions with NKA, using a molecular docking approach.
The selectivity of CTS for human NKA α isoforms has been studied and have been assigned to differences in sugar moietyCitation49,Citation50. Recently, it has been shown that some digoxin derivatives, with modifications at the third digitoxose, have a selectivity for NKA α2β1 and α2β3, compared to NKA α1β1, with up to 7.5 and 33.0 fold, respectivelyCitation51. Natural CTS have also been shown to display a NKA α isoform selectivity in rat aorta and cultured cells in the absence and presence of K+Citation50,Citation52,Citation53. Here, we show that changes in the lactone ring also alter the 21-BD selectivity for NKA α isoforms, either stimulating or inhibiting NKA in an isoform specific manner. These findings are important to help understand the specific interactions of CTS with NKA α isoforms and the consequences of their use in cell physiology.
Since 1985, several studies have identified the main amino acids in NKA that interact with CTS and the long-range structural changes that CTS drive in the proteinCitation7,Citation54. These results originally come from random mutagenesis and site-directed mutation analysesCitation38,Citation55,Citation56. More recently, NKA crystals were obtainedCitation10 under various conditions, such as the low-affinity ouabain form E2.2 K+.MgF4Citation2−.ouabainCitation9 and the high-affinity ouabain bound form E2P.nH+.ouabain stateCitation57. It has been shown that the ouabain lactone ring becomes in close proximity with Val329 and Ala330 at the TM4 helix, displacing Gly326 and partially unwinding this helixCitation9. The steroidal core makes nonpolar interactions with three conserved phenylalanine residues (Phe323, Phe790, and Phe793) and the C14β forms a hydrogen bond with Thr804. The rhamnose group can make hydrogen bonds with Glu319 and Arg887Citation9,Citation10. A recent work performed by the Fedosova group has shown that digoxin binding to crystallised NKA from pig kidney involves the formation of a hydrogen bond between Thr804 and the C14β hydroxyl group of the CTSCitation58. Our work shows that the simple addition of an aromatic ring in the digoxin lactone moiety shifts 21-BD in the binding site, stabilizing this hydrogen bond with C12 hydroxyl group on the α surface of the steroidal core (Figure S6(A)). Sugar moiety interactions present a huge difference between digoxinCitation58 and 21-BD. Actually, some residues are changed in ouabain-resistant rat NKA α1 isoform and, in fact, some interactions at sugar moiety level seems to be more conserved compared with ouabain-sensitive rat NKA α2 isoform, such as the corresponding residues Glu122, Glu317, and Arg884 (Figure S6(B)).
Through the use of different mutagenesis approaches, the role of several amino acids in the coordination of Na+ and K+ ions has been demonstrated. Some of these mutations (including substitutions in Asp804) have been even shown to increase the NKA activityCitation42–45. One of the likely mechanisms for this enhancement of the NKA activity was explained by Koenderink et al., who suggested that, in the mutants, Na+ is already present in the enzyme right after the dephosphorylation step, allowing a quick shift in conformation for the enzyme to start a new reaction cycleCitation44. A similar mechanism was proposed by Tepperman et al., who have shown alterations in the kinetic parameters of a mutated NKA α2 isoform derived from ratsCitation45. We have found that some of our compounds interact with amino acids, which have been shown to be involved in NKA ion coordination. Therefore, it is possible that those compounds favour a conformational state similar to that induced by the mutations described in the references mentioned aboveCitation42–45. In this manner, different from the effect of classical CTS, which cause steric blockage of the access of ions from the extracellular aqueous face to the ion binding sites, our NKA activating compounds may allow a rapid access of ions to their enzyme binding sites. It is clear that to assess these possibilities, additional experiments will be needed.
In conclusion, we have made a series of new digoxin derivatives by making substitutions in the lactone ring that exert NKA α isoforms selective effects, either stimulating or inhibiting NKA activity. These findings further advance our understanding of the molecular mechanisms underlying CTS interaction with NKA and open the possibility for the specific targeting of NKA isoforms with CTS. This will aid in further drug design, with the idea of reducing the side and toxic effects that CTS present.
IENZ_1380637_Supplementary_Material.pdf
Download PDF (959.3 KB)Acknowledgements
We are thankful to Marcos Pessoa Conceição for the artwork.
Disclosure statement
The authors report no conflicts of interest. This article does not contain any studies with human or animal subjects.
Additional information
Funding
Related Research Data
References
- Skou JC. Further on a Mg++ + Na+-activated adenosintriphosphatase, possibly related to the active, linked transport of Na + and K + across the nerve membrane. Biochim Biophys Acta 1960;42:6–23.
- Albers RW, Fahn S, Koval GJ. The role of sodium ions in the activation of electrophorus electric organ adenosine triphosphatase. Proc Natl Acad Sci USA 1963;50:474–81.
- Post RL, Sen AK, Rosenthal AS. A phosphorylated intermediate in adenosine triphosphate- dependent sodium and potassium transport across kidney membranes. J Biol Chem 1965;240:1437–45.
- Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev 2009;61:9–38.
- Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol 1998;275:F633–50.
- Mijatovic T, Kiss R. Cardiotonic steroids-mediated Na+/K+-ATPase targeting could circumvent various chemoresistance pathways. Planta Med 2013;79:189–98.
- Keenan SM, DeLisle RK, Welsh WJ, et al. Elucidation of the Na+, K+-ATPase digitalis binding site. J Mol Graph Model 2005;23:465–75.
- Yatime L, Buch-Pedersen MJ, Musgaard M, et al. P-type ATPases as drug targets: tools for medicine and science. Biochim Biophys Acta-Bioenerg 2009;1787:207–20.
- Ogawa H, Shinoda T, Cornelius F, Toyoshima C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc Natl Acad Sci USA 2009;106:13742–7.
- Toyoshima C, Kanai R, Cornelius F. First crystal structures of Na+,K+-ATPase: new light on the oldest ion pump. Cell 2011;19:1732–8.
- Kometiani P. Multiple signal transduction pathways link Na+/K+-ATPase to Growth-related Genes in Cardiac Myocytes. The roles of Ras and mitogen-activated protein kinases. J Biol Chem 1998;273:15249–56.
- Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 2000;275:27832–7.
- Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem 2002;277:18694–702.
- Peng M, Huang L, Xie Z, et al. Partial Inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J Biol Chem 1996;271:10372–8.
- Elbaz HA, Stueckle TA, Wang H-YL, et al. Digitoxin and a synthetic monosaccharide analog inhibit cell viability in lung cancer cells. Toxicol Appl Pharmacol 2012;258:51–60.
- Jensen M, Schmidt S, Fedosova NU, et al. Synthesis and evaluation of cardiac glycoside mimics as potential anticancer drugs. Bioorganic Med Chem 2011;19:2407.
- Mijatovic T, Lefranc F, Van Quaquebeke E, et al. UNBS1450: a new hemi-synthetic cardenolide with promising anti-cancer activity. Drug Dev Res 2007;68:164–73.
- Rocha SC, Pessôa MTC, Neves LDR, et al. 21-benzylidene digoxin: a proapoptotic cardenolide of cancer cells that up-regulates Na,K-ATPase and epithelial tight junctions. PLoS One 2014;9:e108776.
- Silva LND, Pessoa MTC, Alves SLG, et al. Differences of lipid membrane modulation and oxidative stress by digoxin and 21-benzylidene digoxin. Exp Cell Res 2017;359:291–98.
- Alves SLG, Paixão N, Ferreira LGR, et al. γ-Benzylidene digoxin derivatives synthesis and molecular modeling: evaluation of anticancer and the Na,K-ATPase activity effect. Bioorg Med Chem 2015;23:4397–404.
- Melero CP, Medarde M, Feliciano AS. A short review on cardiotonic steroids and their aminoguanidine analogues. Molecules 2000;5:51–81.
- Sanchez G, Nguyen ANT, Timmerberg B, et al. The Na,K-ATPase α4 isoform from humans has distinct enzymatic properties and is important for sperm motility. Mol Hum Reprod 2006;12:565–76.
- Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. J Biol Chem 1925;66:375–400.
- ChemAxon. Marvin Beans [Internet]; 2016. Available from: https://www.chemaxon.com
- Elokely KM, Doerksen RJ. Docking challenge: Protein sampling and molecular docking performance. J Chem Inf Model 2013;53:1934–45.
- Stewart JJP. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J Mol Model 2013;19:1–32.
- Stewart JJP, MOPAC. Colorado Springs: Stewart Computational Chemistry; 2012.
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron 1980;36:3219–28.
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91.
- DeLano WL, The PyMOL molecular graphics system. New York (NY): Schrödinger, Inc; 2002.
- Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42:252–8.
- Laursen M, Yatime L, Nissen P, Fedosova NU. Crystal structure of the high-affinity Na + K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc Natl Acad Sci USA 2013;110:10958–63.
- Trott O, Olson A. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- Laskowski RA, MacArthur MW, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 1993;26:283–91.
- Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011;7:539.
- Li W, Cowley A, Uludag M, et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 2015;43:W580–4.
- McWilliam H, Li W, Uludag M, et al. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res 2013;41:W597–600.
- Price EM, Lingrel JB. Structure-function relationships in the Na,K-ATPase alpha subunit: site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry 1988;27:8400–8.
- Tobin T, Brody TM. Rates of dissociation of enzyme-ouabain complex and K0,5 values in (Na+ + K+) adenosine triphosphatase from different species. Biochem Pharmacol 1972;21:1553–60.
- Wallick ET, Pitts BJR, Lane LK, Schwartz A. A kinetic comparison of cardiac glycoside interactions with Na+,K+-ATPases from skeletal and cardiac muscle and from kidney. Arch Biochem Biophys 1980;202:442–9.
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 1988;16:10881–90.
- Pedersen PA, Nielsen JM, Rasmussen JH, Jorgensen PL. Contribution to Tl+, K+, and Na + binding of Asn776, Ser775, Thr774 Thr772, and Tyr771 in cytoplasmic part of fifth transmembrane segment in alpha-subunit of renal Na,K-ATPase. Biochemistry 1998;37:17818–27.
- Pedersen PA, Rasmussen JH, Nielsen JM, Jorgensen PL. Identification of Asp804 and Asp808 as N + and K + coordinating residues in alpha-subunit of renal Na,K-ATPase. FEBS Lett 1997;400:206–10.
- Koenderink JB, Swarts HGP, Hermsen HPH, et al. Mutation of aspartate 804 of Na+, K+ -ATPase modifies the cation binding pocket and thereby generates a high NA + ATPase activity. Biochemistry 2000;39:9959–66.
- Tepperman K, Millette LA, Johnson CL, et al. Mutational analyses of Glu-327 of Na,K-ATPase reveals stimulation of 86Rb + uptake by external K+. Am J Physiol Physiol 1997;273:2065–79.
- Weigand KM, Laursen M, Swarts HGP, et al. Na+,K+-ATPase isoform selectivity for digitalis-like compounds is determined by two amino acids in the first extracellular loop. Chem Res Toxicol 2014;27:2082–92.
- Xie Z, Askari A. Na(+)/K(+)-ATPase as a signal transducer. Eur J Biochem 2002;269:2434–9.
- Barwe SP, Anilkumar G, Moon SY, et al. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol Biol Cell 2005;16:1082–94.
- Katz A, Lifshitz Y, Bab-Dinitz E, et al. Selectivity of digitalis glycosides for isoforms of human Na,K-ATPase. J Biol Chem 2010;285:19582–92.
- Cherniavsky Lev M, Karlish SJD, Garty H. Cardiac glycosides induced toxicity in human cells expressing α1-, α2-, or α3-isoforms of Na-K-ATPase. Am J Physiol Cell Physiol 2015;309:C126–35.
- Katz A, Tal DM, Heller D, et al. Digoxin derivatives with selectivity for the alpha2beta3 isoform of Na,K-ATPase potently reduce intraocular pressure. Proc Natl Acad Sci USA 2015;112:13723–8.
- Fedorova OV, Bagrov a Y. Inhibition of Na/K ATPase from rat aorta by two Na/K pump inhibitors, ouabain and marinobufagenin: evidence of interaction with different alpha-subunit isoforms. Am J Hypertens 1997;10:929–35.
- Hauck C, Potter T, Bartz M, et al. Isoform specificity of cardiac glycosides binding to human Na+,K+-ATPase alpha1beta1, alpha2beta1 and alpha3beta1. Eur J Pharmacol 2009;622:7–14.
- Repke KRH, Megges R, Weiland J, Schön R. Location and properties of the digitalis receptor site in Na+/K(+)-ATPase. FEBS Lett 1995;359:107–9.
- Croyle ML, Woo a. L, Lingrel JB. Extensive random mutagenesis analysis of the Na+/K+-ATPase alpha subunit identifies known and previously unidentified amino acid residues that alter ouabain sensitivity–implications for ouabain binding. Eur J Biochem 1997;248:488–95.
- Burns EL, Price EM. Random mutagenesis of the sheep Na,K-ATPase α-1 subunit generates a novel T797N mutation that results in a ouabain-resistant enzyme. J Biol Chem 1993;268:25632–5.
- Yatime L, Laursen M, Morth JP, et al. Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J Struct Biol 2011;174:296–306.
- Laursen M, Gregersen JL, Yatime L, et al. Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc Natl Acad Sci USA 2015;112:1755–60.