
Abstract
Histone demethylation is a vital process in epigenetic regulation of gene expression. A number of histone demethylases are present to control the methylated states of histone. Among these enzymes, KDM4s are one subfamily of JmjC KDMs and play important roles in both normal and cancer cells. The discovery of KDM4s inhibitors is a potential therapeutic strategy against different diseases including cancer. Here, we summarize the development of KDM4s inhibitors and some related pharmaceutical information to provide an update of recent progress in KDM4s inhibitors.
Introduction
Post-transcriptional modifications on histones are vital for cells to regulate the expression of genes in epigenetic level. Through affecting micro-environment or recruiting functional proteins, the modifications play different roles in cells. There are several types of histone modifications: acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, etc.Citation1–3. Acetylation and methylation are most significant and well-studied histone modifications among them. Successes on histone deacetylase (HDAC) inhibitors in the pharmaceutical area have stimulated the development of histone methylation regulatory agents, including histone lysine demethylase (KDM) inhibitorsCitation4–7. Like HDAC, some KDMs are thought as new targets for anti-cancer therapy. Comparing with acetylation at lysine residues on histone tails can neutralize the positive charge of histones to promote transcription, the methylation on lysine side chains does not lead to a charge shiftCitation8. The functions of histone methylation are more complex and context-dependentCitation8. The methylated marks (on different positions and states) need to be read by some downstream proteins or protein complexes (such as HDACs), which can regulate the genetic transcription or some other biological processes. Therefore, different histone methylation marks have different functions (transcriptional activation or suppression, and some other functions)Citation9–11. Moreover, the huge number of histone modification proteins also provides some confusion about the regulation of histone modification. So far, more than 30 methyltransferases and 20 demethylases have been identified, and the numbers are still increasingCitation11,Citation12. These enzymes methylate and demethylate different sites and states of histone methylated marks. But, the cooperative or antagonistic relationships among them are hard to be elucidated completely.
So far two families of KDM have been discoveredCitation13,Citation14. One is lysine-specific demethylase (LSD) that belongs to flavin adenine dinucleotide (FAD)-dependent monoamine oxidase. Two members, LSD1 and LSD2, are in this family. Most, if not all, other KDMs belong to Jumonji domain containing lysine demethylases (JmjC KDMs). More than 20 JmjC KDMs have been identified and divided into six subfamilies (KDM2–7) with different demethylated catalysis activities. Although the biological roles of each subfamily are still not elucidated completely, the KDM4s are regarded as promising targets for cancer therapyCitation15–17.
KDM4A (JmjD2A/JHDM3A) and other members (KDM4B/JmjD2B/JHDM3B, KDM4C/JmjD2C/JHDM3C, and KDM4D/JmjD2D/JHDM3D) in KDM4 subfamily are earliest identified as JmjC KDMs which can catalyze the demethylation of histone H3 subunit lysine9 tri-/di-methylated mark (H3K9me3/2)Citation18. High expressions of KDM4s are considered as promote oncogenesis in some types of cancers, including prostateCitation19, breastCitation20,Citation21, colonCitation22, and some othersCitation15. Downregulation of KDM4s via molecular biology methods or inhibition of their catalytic activity by small molecule inhibitors is confirmed as strategy for oncotherapyCitation16. In recent decades, increasing attention is focused on the development of KDM4s inhibitors as antitumor agents. Meanwhile, a number of inhibitors have been disclosed by medicinal chemists. Recently, some potent and selective JmjC KDM inhibitors were reported. Although the good clinical candidates are still undiscovered, many publications have reported various modulators with different chemotypes under the guide of different design strategies or screen assays (especially high-throughput screen, HST). Although some excellent reviews have summarized the development of KDM4s inhibitorsCitation23–29, it is necessary to provide an update for new drug discovery and design works.
In this review, we first introduce the biological roles of KDM4s and some related JmjC KDMs. Second, we demonstrate some structural information of these proteins is summarized based on several reported protein crystal structures with or without co-factor or inhibitor. We mainly emphasize KDM4s inhibitors with different chemotypes and inhibitory mechanisms. Structure-based drug design (SBDD) strategy which is widely applied in the design of KDM4s inhibitors is highlighted. Finally, some prospects about design and development of KDM4s inhibitors are introduced.
Biological function of KDM4s
KDM4s carry a wide range of biological functions including but not limited to removing histone methylated marks (histone code eraser). They are involved in many biological processes such as transcriptional regulation, cell cycle, senescence, DNA damage response, and heterochromatin formation. These roles are vital for cell growth and their over-expressions are crucial for some types of cancers.
All the four members of KDM4s subfamily can catalyze the demethylation H3K9me3/2, furthermore, the KDM4A ∼ C can also demonstrated to demethylate H3K36me3Citation30. KDM4s have also been demonstrated to H1.4K26me3/2Citation31. The positions and states of the methylated marks on histone mostly decide the roles of enzymes in the transcriptional regulation. H3K9me3/2 is a transcriptional suppressive mark that is associated with heterochromatin assemblyCitation32,Citation33. The roles of H3K36me3 are indistinct at some level, but it is generally believed that this histone mark inhibits transcription at start but facilitates transcriptional elongationCitation16,Citation34,Citation35. Thus, KDM4s are thought as transcriptional activators. In fact, overexpression of KDM4A indeed promotes genetic transcription in some types of cancersCitation21,Citation22,Citation36. But the relationship between the activity roles of KDM4s and genetic transcription is not very solid: KDM4A directs the repression of E2F responsive promotersCitation37. Additionally, some studies show that KDM4s catalysis is not only limited to demethylation but also to act on N-alkyl groups other than methylCitation38. Beyond the roles of catalysis, the antagonism between KDM4A and heterochromatin protein 1 gamma (HP1γ) during cell cycle also controls the chromatin accessibility and facilitates transcriptionCitation39.
As a transcription-associated protein, KDM4A, the same as other KDM4 members, is a co-activator of some nuclear receptors mostly including androgen and oestrogen receptors (AR and ER). KDM4A, 4C, and 4D are all co-activators for AR-related genes by binding AR directly and their overexpressions stimulate the AR’s function. KDM4B also can directly bind AR to keep its stability by inhibiting its ubiquitination in prostate cancer cellsCitation21,Citation40–43. The co-factor roles of KDM4s may be vital for the growth of cancer cells because siRNA-blocking of KDM4A indicates the inhibition of cancer cell growth in some cell-based experimentsCitation44,Citation45. Some small molecule inhibitors can also suppress cancer growth and it indicates that the functions of KDM4A are depended on its catalytic activity. But KDM4A-siRNA-induced gene silence inhibited not only the proliferation of the ER-positive but also the ER-negative breast cancer cell lines (MCF-7 and MDA-MB-231)Citation44,Citation45. These results implied that KDM4A may also benefit the proliferation of breast cancer cells via some other mechanisms.
KDM4s are involved in some protein complexes. KDM4A directly binds HDAC and retinoblastoma (pRb) to form a complex. In this complex, KDM4A associated with pRb and class I HDACs and mediated repression (but not promotion as a transcriptional activator) of E2F-regulated promoters in vivoCitation37. One of the E2F-regulated genes aplasia Ras homolog member I (ARHI), which is a tumour suppressive gene, is repressed by this complexCitation46. The downregulation of ARHI may lead to the silence of specificity protein1 (SP1) in advanced breast cancer cells and promotes the metastasis of cancer. The transcription factor SP1 has been considered as an oncoprotein which is highly expressed in early-stage breast cancer. But in advanced breast cancer cells, SP1 is downregulated due to its inhibitory roles for migratory and invasive abilities. Moreover, SP1 also interact with KDM4A strongly which may be a reason for the SP1-inactivity. Taken together, KDM4A seems to play an important role in SP1 downregulation in advanced breast cancer cells. However, in some advanced human bladder cancers, downregulation of KDM4A may promote lymphovascular invasionCitation47.
p53 is a well-known tumour suppressor. It is also a potential non-histone substrate of KDM4s. The function of p53 in senescence process is biphasic and related to KDM4s: p53 is essential in the early phase of senescence but must be removed in the late phase. In this process, the SCFFbxo22–KDM4A–p53 complex is an indispensable element which functions as an E3 ubiquitin ligase complex targeting methylated p53 for its degradation at the late senescent stage. The overexpression of a catalytic mutant KDM4AH188A results in p53 stabilityCitation48. Thus, blocking the interactions between KDM4A and some other proteins (such as pRb and SP1) may be a therapeutic strategy for some types of cancers. However, SCFFbxo22 complex was also identified as an E3 ubiquitin ligase for KDM4s themselvesCitation49. The FIST-C domains of the Fbxo22 subunit interact with JmjC and JmjN domains of KDM4A to mediate the ubiquitination of KDM4A. SCF–FbxL4 complex is another ubiquitin ligase for KDM4ACitation50. A previous study has revealed that siRNA-interference of KDM4A was sufficient to trigger a p53-dependent senescence response via transcriptional negative regulating CHD5 (Chromodomain-helicase-DNA-binding protein 5), a tumour suppressor, in A549 lung cancer cellsCitation51.
In DNA damage response process, 53BP1 plays a crucial role and binds to methylated H4K20 at DNA damage sitesCitation52. KDM4A can bind to methylated H4K20 via its tudor domains. The degradation of KDM4A is necessary for the recruitment of 53BP1 to DNA damage sites. Two E3 ubiquitin ligases, RNF8 (ring finger protein 8) and RNF168, degrade KDM4A via ubiquitin–proteasome pathway to trigger DNA damage repair processCitation53. Thus, the overexpression of KDM4A may interfere with the DNA damage repair.
The protein level of KDM4A changes in a cell-cycle-dependent manner and is high in G1/S phase. The overexpression of KDM4A, but not catalytical inactive KDM4AH118A, resulted in a faster S phaseCitation39. In contrast, silencing KDM4A with KDM4A–siRNA induced G2/M and S phase arrests in a LPS-stimulated p53-null neuroectodermal stem cell modelCitation54.
The overexpression of KDM4A also plays significant roles in some other types of cancersCitation15. In colon cancer HCT116 cells, KDM4A interacted with p53 directlyCitation22. The downregulation of KDM4A resulted in increased expression of p21 and Puma protein, whereas the level of the anti-apoptotic Bcl-2 protein decreased. Knockdown of KDM4A led to reduction of the proliferation in three different colon cancer cell lines (HCT116, DLD-1, and HT-29). Similar role of KDM4A is also found in gastric cancerCitation36. Overexpression of KDM4A in Kaposi’s sarcoma-associated herpesvirus (KSHV) increases its reactivationCitation55. KDM4B is involved in neural stem cells (NSCs) inflammation. In an LSP-stimulated in vitro model, knockdown of KDM4B downregulated the expression levels of p65, iNOS, Bcl2, and TGF-betaCitation56. This function was dependent on its H3K9me3/2 demethylated activity. For embryonic stem cells (ESCs), the reduction of H3K9me3/2 level by KDM4B improved the reprograming of ESCs into cloned embryosCitation57. The same as KDM4A, KDM4B plays a significant role in cancer. Beside its ER-related functions in breast cancerCitation20,Citation58, KMD4B is highlighted in gastric cancer. KDM4B was also regulated by hypoxia and radiation to promote the proliferation in gastric cancer cellCitation59. It was expressed in gastric cancer cells and was required for the proliferation and survival of tumour. Silence of KDM4B gene by siRNA inhibited cell growth and elevated the level of p53 and p21 proteinCitation60. Cooperating with β-catenin, KDM4B could enhance gastric cancer metastasis and colon cancer cell proliferationCitation61,Citation62. Depletion of KDM4C (which is regarded as oncogene) from cancer cells resulted in inhibition of proliferation of cancer cellsCitation41,Citation63.
KDM4C is a hypoxia-inducible factor 1α (HIF-1α) co-activator that enhances the expression of GLUT1, LDHA, and other HIF-1α-related genes. It is also required for breast cancer progressionCitation64. The oncoprotein Mouse Double Minute 2 (MDM2) was up-regulated by high level of KDM4C, tooCitation65.
Compared with KDM4A-C, KDM4D encodes only a shorter protein consisting of JmjN and JmjC domain. Methylation of H3K79 has been linked to actively transcribed genes and is involved in the regulation of telomeric silencing, cellular development, cell-cycle checkpoint, DNA repair, and regulation of transcriptionCitation66. In a recent researchCitation67, KDM4D was validated to affect H3K79me3 level in vivo: upregulation of KDM4D led to a severe reduction in H3K79me3 level in cells, despite the result in vitro assay revealed that KDM4D showed no reproducible reduction in H3K79me3. Even though, KDM4D lysine demethylase might be a potential regulator for trimethylated H3 at Lys79.
Protein structure and demethylation mechanism of KDM4A
Protein structure of KDM4A
The structural information of KDM4s is vital for rational drug design of inhibitors. KDM4A, 4B, and 4C share several domains: JmjN domain, JmjC domain, tandem PHD domain, tudor domain, and F-box domain. The interaction between JmjN and JmjC domain is important for the catalytic function and stability of KDM4sCitation68. Both PHD domain and tudor domain can recognize the methylated lysine mark on H3/H4 tail of histone, but the function of PHD domain seems not dependent on H3 N-terminalCitation69. F-box domain is involved in interaction between KDM4A and the SKP1-Cul1-F-box ubiquitin ligaseCitation50. KDM4C is much shorter than KDM4A and 4B: the PHD and tudor domains are not involved in its gene sequence.
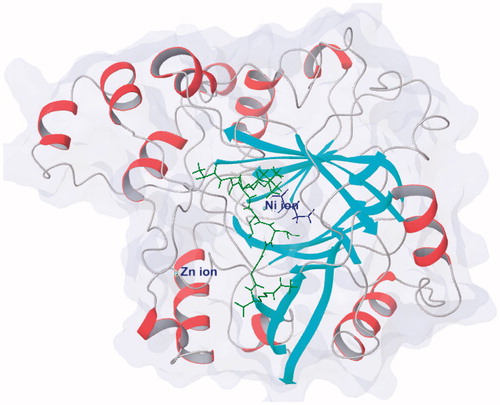
KDM4A and other JmjC KDMs contain a JmjC domain as catalytic regionCitation18,Citation70,Citation71. This JmjC domain is conserved in different species and belongs to the superfamily of Fe(II) ion and 2-OG dependent oxygenase ()Citation63,Citation72. In this domain, a Zinc ion is bound by residues Cys234, His240, Cys306, and Cys308Citation71. The zinc-finger motif is much close to histone substrate peptide recognition region. Two adjacent residues, Lys241 and Arg309, are involved in substrates reorganization. It indicates that the zinc-finger motif plays an important role for keeping the substrate-binding conformation. This result is in accordance with the study of KDM4A Zn-ejection inhibitorsCitation73.
Figure 1. Protein structure of KDM4A (PDB code: 2OQ6) shows as white/red rainbow; H3K9me3 peptide shows in green; 2-OG shows in blue.
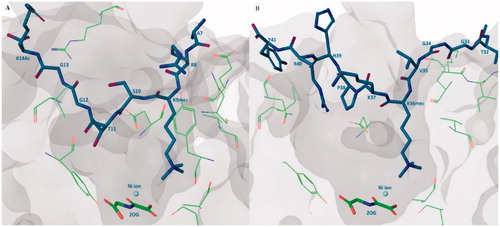
KDM4s can demethylate the histone substrates in different ratesCitation30. H3K9me3 is the most preferable substrate and H3K9me2 along with H3K36me3 can also be demethylated by KDM4A but with lower catalytic rate. Additionally, some studies show that the KDM4s can also catalyze the demethylation of H3K27me3/2 substrateCitation74. Crystal structures revealed that the binding positions of H3K9me3 peptide and H3K36me3 peptide are very superimposable. In catalytic region, H3K9me3 substrate binds approximately in a broad “W” shape while H3K36me3 substrate is in a “U” shape. The H3Ser10 side chain forms a hydrogen bond with the main-chain H3Gly12 and this intra-interaction is vital for keeping the binding conformation. Ser10Ala mutant leads to a strongly reduced activity. This result is in accordance with a previous study that the phosphorylation at H3Ser10 is a “switch” for the regulation of genetic transcription. Phosphorylation at H3Thr11 also plays a similar role. In the same way, mutants of H3Gly12 and H3Gly13 also reduce the affinity. H3Pro30Gly mutant makes H3K27me3 peptide substrate gain activity. This “W” or “U” conformation may be important for the design of substrate-mimic inhibitorsCitation73 ().
Figure 2. Substrate binding pose. (A) KDM4A-H3K9me3 peptide-2-OG structure (PDB code: 2OQ6), H3K9me3 peptide shows in purple stick, 2-OG shows in green stick, some important residues of KDM4A shows in green line; (B) KDM4A-H3K36me3 peptide-2-OG structure (PDB code: 2OS2), H3K36me3 peptide shows in purple stick, 2-OG shows in green stick, some important residues of KDM4A shows in green line.
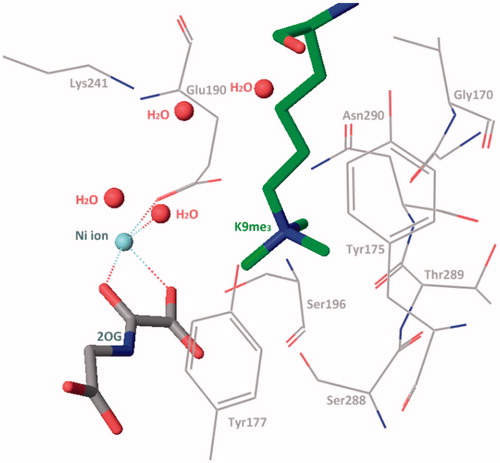
In the methylated lysine side chain binding sub-pocket, the positively charged nitrogen of methylated lysine side chain is surrounded by some nucleophilic side chains of Try177, Glu190, Ser288, and Asn290 (). These three methyl groups project toward Asn290, Fe(II) ion (replace by Ni(II) ion in crystal structure), and Try177. At the one or two methylated state, the methyl group is preferentially pointing to Asn290 and then Try177. The rest positions are occupied by water molecules. This structural information elucidates the methylated state selectivity of KDM4ACitation73.
Figure 3. Trimethylated lysine side chain in catalytic region (PDB code: 2OQ6).
KDM4A, together with 4B and 4C, can catalyze both H3K9me3 and H3K36me3 substrates, whereas the KDM4D could only recognize H3K9me3 motif. The main reason of the difference between KDM4s is a different recognition mode between H3His39 and Arg40 which is indicated by the crystal structure and some docking studies. The His90 and Leu75 in KDM4D present a steric clash with H3Arg40 compared with the corresponding residues Asn86 and Ile71 in KDM4A. The arginine in -1 position also plays an important role for both KDM4A and 4D: its side chain forms multiple hydrogen bonds with Asp135, Tyr175, and Glu169 in KDM4A, corresponding to Asp139, Tyr179, and Glu173 in KDM4DCitation75.
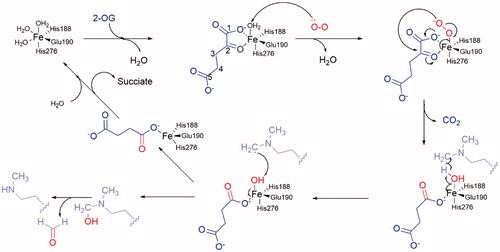
Figure 4. Mechanism of demethylation. 2-OG shows in blue; O2 shows in red; substrate lysine side chain shows in pink.
Demethylation mechanism
Crystal-structural studies reveal the catalysis region of KDM4A and take insight of the catalysis process. Unlike the LSDs, JmjC KDMs can demethylate tri-, di-, and monomethylated modification in some specific sites. Furthermore, most JmjC KDMs are more inclined of tri- or di-methylated lysine. Their catalytic activity is dependent on Fe(II) ion and 2-OG. Its molecular mechanism has been well revealedCitation14,Citation63 (). In the catalytic region of KDM4A, Fe(II) ion (replaced by Ni(II) ion) is chelated by residues His188, Glu190, and His276. Two water molecules are also involved in this site. In the co-crystal structure, 2-OG replaces the water molecules and interacts with the Fe(II) ion by its C1 carboxylate oxygen and the C2 ketone oxygen. The other carboxylate moiety of C5 forms hydrogen bonds with Tys132, Asn198, and Lys206 to stabilize the binding pose of 2-OGCitation71. At the start, the water molecules are replaced by 2-OG (in blue) and then one molecule of negative oxygen ion (in red) attacks the Fe(II) positive ion. Then the negatively charged oxygen of the Fe(III)-O2 complex attacks the C2-position of 2-OG, resulting in decarboxylation to carbon dioxide, succinate, and Fe(IV) = O group. The Fe(IV) = O group’s oxygen forms a hydrogen bond with the hydrogen of the aminomethyl group of lysine side chain (in pink), which is nearby. The C–H bond of aminomethyl group breaks and then forms a carbon free radical. In the next step, the hydroxyl group transfers from the Fe(III) ion to the carbon free radical. The aminomethyl group is hydroxylated and then takes off one molecule of formaldehyde. At last, the succinate is replaced by water molecules or 2-OG. The 2-OG and succinate are also intermediate products in Krebs’s Cycle. So, it is presumed that this reaction could be influenced by energy metabolism.
KDM4s inhibitors
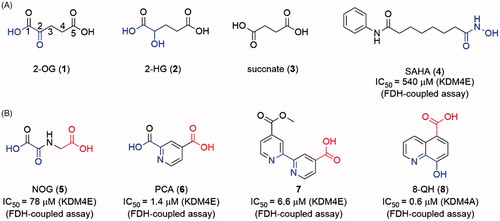
Like many other 2-OG-dependent enzymes, KDM4s are naturally inhibited by some 2-OG-like endogenous substances, such as alpha-hydroxygluarate (2-HG, 2, the reduzate of 2-OG) and succinate (3, the by-product of histone demethylation)Citation76,Citation77 (). The 2-HG could be converted from 2OG and isocitrate by mutant isocitrate dehydrogenase (IDH) in some types of brain tumorsCitation78. The up-regulation of 2-HG increases the level of HIF1α by inhibiting PHD2 (a 2-OG-dependent oxygenase).
Figure 5. (A) Structures of endogenous substances and SAHA; (B) scaffolds of “generic” 2-OG-dependent inhibitor.
The development of JmjC KDM inhibitors was initiated after the identification of the enzymes. The primal drug discovery works focused on natural product and 2-OG mimicsCitation79. SAHA (4, an HDAC inhibitor with a metal-chelated moiety) also showed mild inhibitory activity. With a growing number of structural biology works being reported, SBDD strategy was used in the discovery of JmjC KDMs inhibitors. Several different chemotypes of inhibitors have been reported. Most of the inhibitors are 2-OG mimics with metal-chelated moiety which can bind to the Fe(II) ion in the catalytic core. Many natural inhibitors, such as catechol, also contain metal-binding moieties. Some other types of inhibitors (including Zn(II) ion ejection inhibitors, peptide based inhibitors, etc.) were also reported with limited quantity. So far, the 2-OG competitive inhibitors are still the major strategy for JmjC KDMs.
It should be indicated that it is still a challenge to improve selection among JmjC KDMs subfamilies. Inside KDM4s subfamily, no member-selective inhibitor has been reported as far as we know. Thus, the inhibitory activity on KDM4E (a pseudogene) or 4 C was sometimes used to represent the inhibitory activity of compounds on KDM4s. Additionally, the IC50 values of the various inhibitors were obtained by different methods, such as FDH-coupled assay, MS related assay, LANCE assay, and AlphaScreen. Although some reference compounds were involved in the experiments, a proper comparison of the activities of different compounds could be correctly done only within the context of the same assay. Thus, we label the types of test assay of the inhibitors’ IC50 values on the figures for reference.
Metal-chelated inhibitors
As members of the 2-OG-dependent oxygenase family, JmjC KDMs can be inhibited by some 2-OG dependent enzymes inhibitors (5–8) which can coordinate the Fe(II) ion like 2-OG. These inhibitors are also called as “generic” inhibitors for 2-OG dependent oxygenasesCitation24,Citation79 (). The common features of metal-chelated inhibitors are a 1,4-bidentate moiety (represented in blue in structures) and a carboxyl group (represented in red in structures). Based on different metal-chelated moieties, the inhibitors could be divided into (1) oxalyl acid scaffold; (2) hydroxamic acid scaffold; (3) hydrazide scaffold; (4) 8-hydroxygenquinoline (8-HQ) scaffold and benzimidazole pyrazolone scaffold inhibitors; (5) pyridine-based scaffold.
Oxalyl acid, hydroxamic acid, and hydrazide scaffold inhibitors
N-Oxalylglycine (NOG, 5) is a human HIF prolyl hydroxylase PHD2 inhibitor with Ki = 8 μM. As a 2-OG mimic, NOG showed a 24 μM IC50 value for KDM4A. Although the inhibitory activity of 5 is medium, its small molecular weight (MW) makes 5 a good hit for the development of KDM4s inhibitorsCitation79(). Co-crystal structure indicates that the oxalyl group of NOG binds to the Fe(II) ion and the carboxyl group of glycine is positioned to form hydrogen bonds with Lys241 and Tyr177 of KDM4ACitation73. The structural studies and modelling results imply that a subpocket is adjacent to the activity site of KDM4s but not exists in PHD2. Thus, one usual approach to provide selectivity toward KDM4A is to occupy this hydrophobic pocket by introducing a big side chain on NOG. By synthesizing and evaluating many N-substituted derivatives and amino acid derivatives of NOG, compound 9, an N-oxalyl tyrosine derivate, was identified as a potent and selective KDM4A inhibitor. Its side chain occupies the large pocket beside the 2-OGCitation80. Additionally, the different side chains of NOG can provide selectivity toward the subfamily of 2-OG dependent oxygenaseCitation24.
Figure 6. Structures of oxalyl acid, hydroxamic acid and hydrazide scaffold inhibitors.
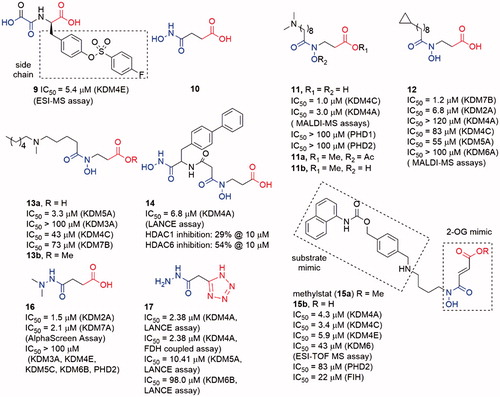
The metal-chelated moiety of SAHA, hydroxamic acid group (10), could replace the oxalyl group to bind the Fe(II) ion in KDM4s. Compound 11 was a hydroxamic acid derivative with a long carbon chain. The introduction of the long chain provided 11 good selectivity toward KDM4s over PHD2Citation81. A combination of NCL-2 (a LSD1 selective inhibitor) with 11a or 11b, two prodrugs of 11, displayed synergistic cell growth inhibitory activity on LNCaP, PC3, and HCT116 cell lines. However, all the three compounds did not show obvious inhibitory activity alone. Interestingly, replacing the dimethylamine moiety of 11 by a cyclopropyl moiety (12) led to good selectivity toward KDM2/7 subfamiliesCitation82. Docking study indicated that this selectivity was possibly due to the different environments around the subpocket of KDM4s and KDM2/7s: the pocket in KDM4A was composed of some polar residues such as Tyr179 and Asp137, while in KDM7A the residues belonged to some hydrophobic amino acid (Phe250, Phe359, and Tyr234). Moreover, 12 showed anti-proliferative activity in N2a, KYSE150, and Hela cell lines (GI50 value is 86, 16, and 40 µM, respectively) via downregulating E2F1 gene expressionCitation83.
Another hydroxamic acid derivative compound 13a shared similar scaffold with 11 and 12Citation84. However, 13a displayed selectivity toward KDM5A (IC50 = 3.3 μM) than KDM3A (IC50 > 100 μM), KDM4C (IC50 = 43 μM), and KDM7B (IC50 = 73 μM)Citation84. The docking study showed that the interaction between the NH group and Asp412 in KDM5A might play an important role for the selectivity. Its methyl ester prodrug 13b could increase the accumulated amount of H3K4me3 in a dose-dependent manner in A549 cell. In contrast, it had minor effect on the levels of H3K9me3 and H3K27me2. 13b could also inhibit the proliferation of A549 cells.
Similar to SAHA, another HDAC inhibitor SW55 also showed weak inhibitory activity to JmjC KDMs with IC50 = 25.4 μM. Based on the scaffold of SW55, Jung et al.Citation85 synthesized a series of 4-biphenylaline and 3-phenyltyrosine derived hydroxamic acids as KDM4A inhibitors. The best compound 14 contained two hydroxamic acid moieties and exhibited potent inhibitory activity to KDM4A with IC50 = 1.7 μM (LANCE assay). 14 also had potency for HDAC6 and HDAC1. In the proliferation experiments, 14 displayed antiproliferative effect against two human cancer cell lines: KYSE-150 and HL-60 with GI50 = 44 μM and 53 μM, respectively. An immunofluorescence microscopy assay ascertained that 14 could significantly increase the H3K9me3 in KYSE-150 cells.
The Methylstat (15a) is a selective inhibitor that links a 2-OG mimic moiety with a substrate mimic moiety from a HDAC inhibitor MS-275Citation86. Compound 15a showed good selectivity toward KDM4s than PHDs and HDACs, but it did not show potent inhibitory activity on KDM4s (IC50 = 4.3 μM). Compound 15a inhibited KYSE150 cell growth with GI50 = 5.1 μM, but the unmethylated compound 15b showed almost no effect on the cell growth.
The hydrazide group is not regarded as a good metal-chelated group because its amino nitrogen atom is potential to be protonated and lose its lone pair electrons. However, some simple hydrazide derivatives (16 and 17) displayed good JmjC KDMs inhibitory activity. Similar to 12, the two amino methyl groups of 16 occupied the hydrophobic pocket of KDM7s to provide selectivity. In contrast, 17 is a selective KDM4s inhibitor with medium activity. The docking result indicated that the tetrazole group instead of the carboxylate interacted with KDM4s. Structure–activity relationship (SAR) studies indicated that the unsubstituted hydrazide group might be the reason for its selectivity.
8-Hydroxygenquinoline (8-HQ) scaffold inhibitors
8-HQ possesses potent coordinating ability and good metal-recognition property. A derivative of 8-HQ, 5-carboxy-8-hydroxyquinoline (8), is a potent KDMs inhibitorCitation87. Notably, 4-carboxy-8-hydrogenquinoline (18) displays a better superimposition with 2-OG scaffold, but its activity is lower than the 5-carboxy derivative (). Crystallographic studies demonstrated that 8 could cause translocation of the Fe(II) ion in activity siteCitation88.
Figure 7. Structures of 8-hydroxygenquinoline core inhibitors.
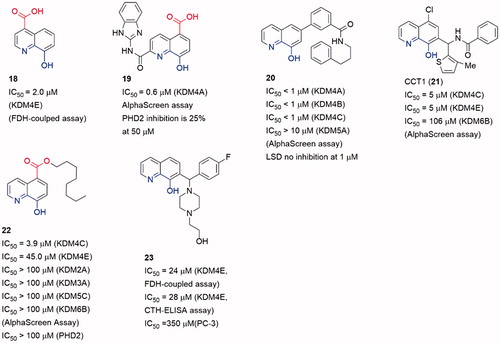
Several 8-HQ derivatives were discovered from HTS assays or SBDD. Some derivatives were designed to enhance the selectivity towards KDM4s or the physicochemical property (19–23)Citation89–91 (). Such optimization enhances the selectivity of inhibitors on KDMs and antiproliferative activity on cancer cells. 19 displayed promising antiproliferative activities against three cell lines (HCT116, MCF-7, and A549) with IC50 = 1.5 μM, 27.6 μM and 21.7 μM. It also exhibited good aqueous solubility and appropriate LogD7.4, indicating a better physicochemical property than 8Citation89. Compound 20 was a selective KDM4s inhibitor and had high selectivity for a variety of cancer cells including PC3 cells that lack AR. It also inhibited the in vivo growth of tumours derived from PC3 cells and ex vivo human PCa explants. Additionally, 20 could block the binding of KDM4B to the promoterCitation90.
Similar to the SAR of compounds 11 and 12, the length of the carbon chain of compound 22 offered selectivity among subfamilies of JmjC KDMsCitation92. The carbon chain also provided a better cell permeability. Treatment with 8 or its ester derivative 22 caused a dose-dependent increase in H3K9me3 fluorescence intensity, implying KDM4A inhibition in Hela cells.
Compound 23 (as known as CBN207192) indicated modest inhibitory activity and promising selectivity toward KDM4s by employing the formaldehyde dehydrogenase (FDH)-coupled fluorescence assay and the CTH-ELISA (IC50 = 24 μM, FDH-coupled fluorescence assay and IC50 = 28 μM, CTH-ELISA). All members of the KDM4(A-E) subfamily were inhibited with similar potenciesCitation93. Cell-based activities assays demonstrated its anti-cancer effect at high inhibition concentrations by reducing the proliferation of PCa cell lines.
Pyridine-based inhibitors
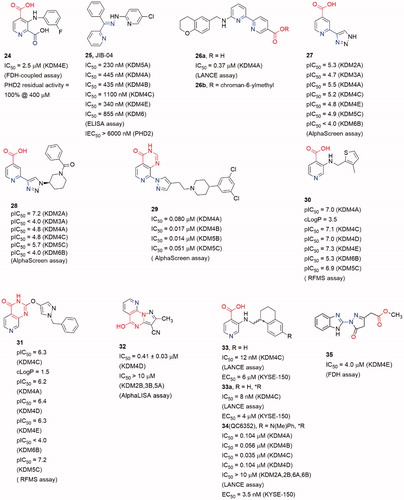
Pyridine-based inhibitor 2,4-pyridinedicarboxylic acid (6, PCA) is one of the most potent inhibitors for KDMs, although its physicochemical properties and selectivity are not satisfactory. However, the pyridine moiety as metal ion binding group is often involved in KDM4A inhibitor scaffoldsCitation79 (). The pyridine nitrogen and the 2-position carboxylate of PCA bind to the Fe(II) ion in a bidentate manner as the oxalyl acid group of NOG. And the 4-position carboxylate forms hydrogen bonds with Lys241 and Tyr177. Introducing a phenyl ring on 3-position to occupy the pocket beside activity site, 24 displayed selectivity toward KDMs than PHD2Citation94.
Figure 8. Structures of pyridine-based inhibitors.
Based on a cell-based assay (named locus derepression), JIB-04 (25) was identified as a JmjC KDMs inhibitorCitation95. Compound 25 showed a pan inhibitory activity on JmjC KDMs but weak affection on PHD2. It was noteworthy that the Z-isomer which lacked a pyridine-based chelated moiety showed almost no activity. It indicated that 25 may inhibit KDM4A in a Fe(II) ion binding manner. Compound 25 inhibited KDM4A activity in vivo and its action was modulated by KDM4A enzyme level. Moreover, in an immune-competent tumour mouse model, 25 diminished tumour growth compared with vehicle-treated mice.
Compound 26a was identified by Jung et al. through virtual screening and structural optimizing as a potent KDM4A inhibitor with IC50 = 0.94 μMCitation96. This inhibitor also showed good potency towards KDM5A (IC50 = 0.44 μM) and moderate potency towards KDM6B (IC50 = 36.5 μM). The co-crystal structure of KDM4A and 26a indicated that the nitrogen atoms of pyridine and pyrimidine chelated the Fe(II) ion while the carboxylate group interacted with Lys206 and Tyr132. The secondary amine formed a hydrogen bond with the carboxylate of Glu190. And the terminal pyridine ring protruded into a large pocket formed by Tyr175, Val171 and Asp191. The combination of optimal side chain in 26a with this ester led to 26b, which was indeed soluble at much higher concentrations and exhibited obvious inhibition on proliferation of KYSE-150 cells.
Brennan et al. replaced carboxylate group by a triazole moiety to gain compound 27. Compound 27 showed moderate inhibitory activity toward KDM4s (pIC50 = 5.5 ± 0.33)Citation97. Co-crystal structure of KDM4A and 27 indicates that some five-member sp2-hybridized nitrogen containing heterocyclic could play the same role of pyridine in 7 or carboxylate in 6. Moreover, introducing some moieties on the heterocyclic can provide more selective and potent inhibitors. A number of analogous of 27 were synthesized leading to selective KDM inhibitors. One of the analogous, compound 28, was a potent and selective KDM2A inhibitors with pIC50 = 7.2 (pIC50 = 4.8 for KDM4).
Recently, another co-2-OG and -substrate mimic 29 was reported. Although the design of the inhibitors started from a 2-OG mimic from an HTS assay, the docking result showed that the chlorobenzyl piperidine group inserted into the substrate methylated lysine side subpocketCitation98. This may be the reason for its good potency on KDM4s (IC50 value is 17 nM for KDM4B, and 80 nM for KDM4A). In this scaffold, the 4-carboxylate group was replaced by pyrido[3,4-d]pyrimidin-4(3H)-one group (in red). This replacement led to an obvious enhancement of cellular permeability.
The pyrido[3,4-d]pyrimidin-4(3H)-one group was developed by Westaway et al.Citation99,Citation100. In Westaway’s work, some 3-amino-4-pyridine carboxylate derivatives have been identified as hits for KDM4 inhibitors. Compounds 30 and 31 were developed as the potent cell penetrant inhibitors with IC50 ≤ 100 nMCitation99. These inhibitors bound Fe(II) ion only via the nitrogen of pyridine ring (single pyridine binding JmjC KDMs inhibitors was common in patent literatures). In a subsequent work, the 4-carboylate group and the amino group were linked to form a bicyclic scaffold which kept potencyCitation100. This replacement abandoned the carboxylate group which frequently occurred in KDM4s inhibitors and it may be able to be introduced into some other types of inhibitors to optimize the physicochemical properties.
In Zhen Fang’s workCitation101, a novel selective KDM4D inhibitor (32), which had a pyrazolo-[1,5-a]-pyrimidine-3-carbonitrile scaffold similar to 31, was reported. A molecular docking-based virtual screening against various chemical libraries include Specs and ChemDiv was performed to retrieve two compounds with modest inhibition rate against KDM4D (> 50% at 10 μM). The one with better selectivity against KDM4D became the starting point of structural optimization which led to compound 32 and analogs. Compound 32 was the most potent one with an IC50 of 0.41 ± 0.03 μM and displayed good selectivity for KDM4D over other tested KDM family members (> 10 μM against KDM2B, 3B and 5A).
Chen et al. developed a novel series of potent KDM4 inhibitors with promising selectivity from a small fragment lead using structure-based design. The SAR study led to the most potent and efficacious inhibitor compound 34 (as known as QC6352)Citation102. They explored the substitutions of the 3-aminoisonicotinic acid nitrogen then identified the tetrahydronaphthalene compound 33 as a potent inhibitor of KDM4C (IC50 = 12 nM). Compound 33 also demonstrated a measurable inhibition activity to cell proliferation in the KYSE-150 cell line (EC50 = 6 μM). The R enantiomer, 33a, was determined to be the active enantiomer (KDM4C IC50 = 8 nM, KYSE-150 cell EC50 = 4 μM). After Molecular modelling of 33a, researchers developed a series of derivatives with 6-position substitutions which were designed to impart favourable hydrophobic interactions and improve cellular permeability. Among them all, compound 34 showed favourable in vivo efficacy (KYSE-150 cell EC50 = 3.5 nM, tumour growth inhibitions =61% at dose level:50 mg/kg) in a breast cancer PDX model and reduced the tumour initiating cell populations, which were associated with resistance to chemotherapy treatments.
In a recent work, Metzger et al. established a method to isolate and propagate breast cancer stem-like cells (BCSC) from individual triple-negative tumours stemmed from patients after neoadjuvant chemotherapy. This method is intended to assess the antitumor properties of 34 (QC6352)Citation103. After they validated KDM4A is directly related to proliferation and xenogaraft tumour growth of BCSC, 34 was tested if it is qualified for the treatment of BCSC-originating tumours. Results showed that 34 revealed excellent potency and selectivity against the KDM4A-D subfamily (hundredfold selectivity over KDM2 and KDM6 and weak inhibition of KDM5B), with some favourable pharmacokinetic properties. Moreover, it’s corroborated that 34 blocked proliferation and self-renewal of BCSCs through EGFR regulation in vitro, and xenograft tumour growth of BCSCs in vivo.
David et al. who identified the 23 also validated compound 35 (CBN209350) as a novel benzimidazole byrazolone scaffold KDM4 inhibitor by HTS. Compound 35 represented an undescribed scaffold in KDM inhibitors, it exhibited some specificity toward KDM4 enzymes. Compound 35 also displayed cytostatic activity in PCa cell lines at a higher concentration. By Rosen et al.Citation94, they described that 35 had key structural similarities to other privileged scaffolds that inhibited Fe2+/α-KG-dependent hypoxia inducible factor prolyl hydroxylase (PHD) enzymes such as the Molidustat.
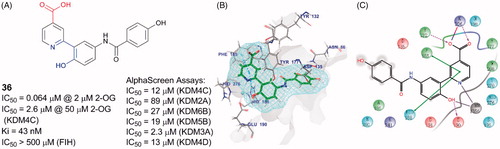
A docking and linking of fragment strategy was applied to discover novel, potent JmjC KDM inhibitorsCitation104. After analyzing the docking models of two fragments, 5-carboxypyridine and 5-aminosalicylate, a new fusional structure type was designed and optimized to get 36 (). This combined approach improved affinity by ∼3 log-orders (Ki = 43 nM). This compound showed great potency with an IC50 value of 64 nM (at 2 μM of 2-OG) and good selectivity towards KDM4s than PHD2. Unlike most of Fe(II) ion coordinating inhibitors, 36 was a 1,5-bidentate ligand instead of a 1,4-bidentate ligand. Co-crystal structure demonstrates that the pyridine ring and benzene ring form dihedral angle of 36°. By testing its KDM4C inhibitory activity in various concentrations of 2-OG or substrate peptide, 36 showed competition with 2-OG but no competition with the substrate peptide (IC50 = 4.8 μM at 50 μM of 2-OG and 50 μM peptide, IC50 = 5.5 μM at 50 μM of 2-OG and 250 μM peptide, IC50 = 1.1 μM at 5 μM of 2-OG and 50 μM peptide).
Figure 9. Compound 36 and its binding pose. (A) Structure of 36; (B) and (C) 36 binding pose (PDB code: 5A7W).
Non-chelated inhibitors
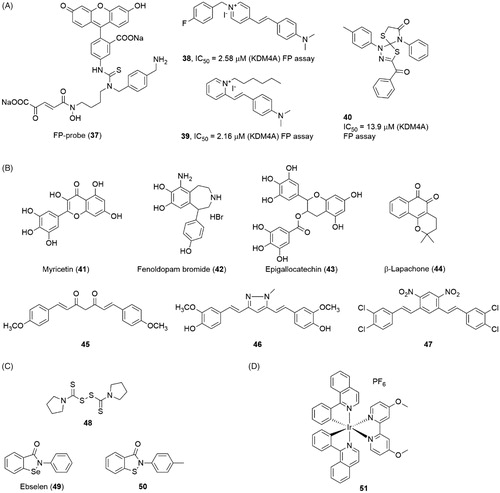
The metal-chelated agents reveal good potency on KDM4s. But considering the large number of metal-containing proteins in cells, this feature also rises the potential off-target risk of the inhibitors. The non-chelated inhibitors are also necessary for the KDM4s. Linking a fluorescent group FITC (fluoresceinisothiocyanate) with a KDM4s inhibitor resulted in a FP-Probe(37) which was used in fluorescent polarization (FP) assay to screen novel JmjC KDMs inhibitorsCitation105 (). Using this fluorescent probe, several non-chelated inhibitors, compounds 38, 39 and 40, were identified through a FP-based HTS assayCitation106. Compounds 38 and 39 shared a pyridinium fragment and a dimethylamino-styrene fragment. All three compounds had moderate inhibitory activities in FP assay with IC50 = 2.58 μM, 2.16 μM, and 13.9 μM.
Figure 10. Structures of (A) FP-probe and screen inhibitors; (B), natural product inhibitors; (C) Zinc ion ejection inhibitors; and (D) metal-containing inhibitor.
Natural originated inhibitors
Many natural products show KDM4s inhibitory activity (compounds 41, 43 and 44) (). Although a large part of them contain metal-chelated groups (such as catechol scaffold 41, 42 and 43), we prefer to discuss the natural products separately. Many catechols (including some synthesis compounds such as Fenoldopam 42) showed inhibitory activity to KDM4E with moderate IC50 value (1–10 μM)Citation107. However, a shape and electrostatic similarity search indicated that the simple catechol moiety could inhibit KDM4A alone. The docking model and SAR study both indicated that the two adjacent hydroxide groups chelated the Fe(II) ion and the electrical property of the phenyl ring greatly affected the affinityCitation108. This study implied that natural catechol may be too complex for KDM4s. And on the other side, the ultra-small molecule inhibitors might be an opportunity for KDM4s. Curcumin and its derivatives (45, 46, and 47) also showed good activity on inhibiting histone demethylation in cell levelCitation109,Citation19. However, the curcumin is a well-known pan-targets inhibitor and is regarded as one of the pan-assay interference compounds (PAINS)Citation110.
Zinc ion ejection inhibitors
Ejection of structural Zn(II) ion is another possible strategy for inhibiting KDM4A. Structural study revealed that the Zn(II) ion binding site is very close to the catalysis region in KDM4ACitation73. The Zn(II) ion binding motif (His220, Cys234, Cys306 and Cys308 in KDM4A) was next to the substrate-recognition residues, Lys241 and Arg309. So, this motif may play an important role for keeping substrate-recognition conformation. Several Zn-ejecting compounds, including disulphiram (48) and ebselen (49), could inhibit KDM4A with IC50 = 3.3 and 10.6 μM, respectivelyCitation111(). However, considering Zinc ion is contained in a large number of proteins, the selectivity of Zinc ion ejection inhibitors should be further studied. An ebselen-structure similar compound 50 was identified as a KDM5B inhibitor through HTS assay. Compound 50 also showed a moderate activity for KDM4E with IC50 = 28 µM).
Metal-contain inhibitors
Nickel ion, together with some other divalent ion like Co(II), could inhibit JmjC KDMs activityCitation112. The IC50 value of nickel ion is 25 μM for KDM3A. These ions could replace the Fe(II) ion in the catalytic site to block the hydroxylation process. Recently, an Iridium (III) complex 51 was reported as a KDM4 inhibitor with IC50 = 15 μMCitation113(). Absorbance spectrum and ESI mass spectrometry showed that 51 did not inhibit KDM4A via ion-sequestration method. In the A549 cell line, 51 led to the increase of H3K9me3 and the amplification of the p21 gene promoter. Moreover, 51 displayed weaker potencies on KDM5A and KDM3 than KDM4 in an ELISA assay which was based on H3K4me3 and H3K27me3 antibody separately.
Peptide-based inhibitors
Some attempts for developing substrate mimic inhibitors were truncating or modifying substrate peptides (). By truncating the H3K9me3 substrate, H3(7–14)Kme3 peptide was identified and displayed the best KDM4A affinity (kcat = 0.01 min−1, KM = 121 μM), even better than some longer peptidesCitation114. In accordance with previous structural study, the absences of Gly–Gly led to a rapid decreasing in enzyme kinetic activity (kcat value) for KDM4C. Introducing a metal-chelated moiety (uracil) or a carboxylate group compounds 52 and 53 which showed moderate inhibition activities to KDM4ACitation114. Compounds 54 and 55 were obtained by linking NOG (N-oxalylcysteine) with substrate peptidesCitation115. The two inhibitors showed better inhibitory activities and provided selectivity between KDM4A and 4 D. However, 54 and 55 did not link NOG to the lys9 side chain but the 10 position of H3K9me3 peptide or the Pro38 of H3K36me3 peptide.
Figure 11. Peptide based inhibitors.
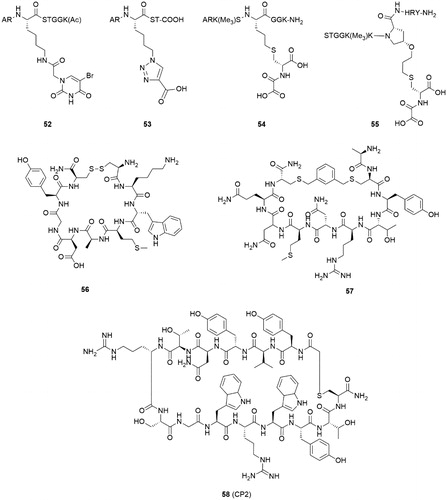
Two peptide inhibitors were developed through a hydrogen/deuterium exchange coupled to mass spectrometry (HDX-MS) methodCitation116. After amino acid replacement, truncation, and chemical modification, two cyclic peptides (56 and 57) showed KDM4C inhibitory activities. It was important that the two peptides inhibited the KDM4C in a substrate- and 2-OG-independent manner. Additionally, the two peptide inhibitors bound to KDM4C in different regions. This study may provide some novel binding sites for KDM4C inhibitors.
Some highly potent and selective cyclic peptide inhibitors of KDM4A-C were developed by Random nonstandard Peptides Integrated Discovery (RaPID) system using two thioether-macrocycle librariesCitation117. Compound 58 (CP2) demonstrated potent IC50 against KDM4A-C (IC50 = 42 nM, 33 nM, and 39 nM against KDM4A, B, and C, respectively) in a luminescence-based AlphaScreen assay. Moreover, 58 represented an exceptional selectivity in intra-subfamily: it displayed high potency against KDM4A-C and much less active against KDM4D-E (IC50 = 6270 nM, 9200 nM against KDM4D, KDM4E, respectively). Arg6 of 58 showed a significant function for KDM4A binding which indicated different binding modes between histone substrates (H3K9me3/K36me3) and 58. Compound 58 derivatives based on structure-guided and mass spectrometry (MS)-guided modifications revealed meliorative stability in intracellular target. Although further studies of cyclic peptides at cellular level are necessary for deeper investigation.
Other JmjC KDM inhibitors
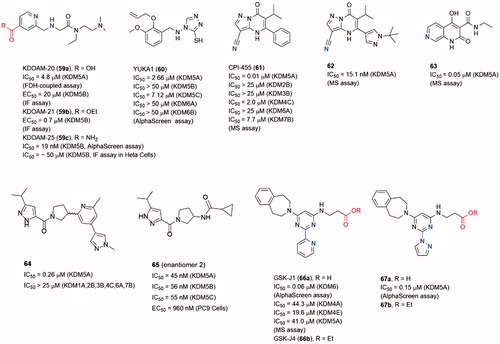
Selective inhibitors for JmjC KDMs subfamilies are important as chemical biological tools to explore the biological roles of the JmjC KDMsCitation17,Citation28,Citation118. Although discovery of the selective inhibitors is difficult, several selective inhibitors have been reported as mentioned before (such as 12 and 16 as KDM7 inhibitors, 28 as KDM2 inhibitor). To make an integrated comprehension for JmjC KDM inhibitors, several other subfamily selective inhibitors will be introduced in this section ().
Figure 12. Structures of some other subfamily selective inhibitor.
KDM5 subfamily inhibitors
After KDM4 subfamily, the roles of KDM5 subfamily in cancer have attracted increasingly attention recentlyCitation119. Especially, KDM5A and 5B could promote the development of drug tolerance and maintain tumour-initiating cells. Many evidences indicate that KDM5s are potential targets for cancer therapy. Meanwhile, many KDM5 selective inhibitors have been disclosed as anticancer agents.
Compound 59a (KDOAM-20), and its cell-permeable ethyl ester derivative 59b (KDOAM-21), were potent and selective KDM5s inhibitorsCitation120, Citation121. Several structural studies based on 59a and its derivatives revealed the details of its binding pose in KDM5A. Treatment of MCF7 and MDA-MB-231 breast cancer cells with 59b significantly increased the global levels of H3K4me3 while had little impact on H3K4me2/1 or other histone methylated marks. In colony-formation experiment, the growth of MCF7, BT474, and ZR-75–1 cells was inhibited at 5 μM of 59b. Treatment of Hela cells transiently overexpressing WT KDM5B with 59a and 59b, assessed by immunofluorescence (IF) assay, showed potent inhibition of H3K4me3 demethylation, with EC50 = 20 μM and 0.7 μM, respectively. Unexpectedly, 59b still showed modest activity against KDM5B in cells despite it has low cell permeabilityCitation122.
In Tumber’s workCitation123, researchers replaced the ester of KDOAM-21 with a less labile group to improve KDM5 potency and selectivity. Some amide derivatives were synthesized, among them 59c (KDOAMA-25) showed the highest potency, with a KDM5B IC50 of 19 nM. Through AlphaScreen or MALDI-TOF enzymatic assays, 59c was proved to be selective for KDM5 sub-family, because the IC50 values for other 2-OG oxygenases were over 4.8 μM. The cellular activity of 59c was assessed in an IF assay by using an overexpression system in HeLa cells, it can increase H34Kme3 levels in a two-digit micromolar range. Compound 59c also showed an inverse relationship between KDM5B expression and overall survival from three clinical multiple myeloma trials’ data, suggesting a potential application for KDM5B inhibitors in myeloma therapy.
Yan et al.Citation124 identified some small molecule inhibitors through a HTS assay of full-length KDM5ACitation125. YUKA1 (60) was one of the potent inhibitors with IC50 = 2.66 μM for KDM5A and was a cell permeable compound. But it showed little to no activity against KDM5B, KDM6A, and KDM6B. Treatment with 60 led to increased H3K4me3 levels and inhibited growth of Hela and ZR-75–1 cells which depended on KDM5A expression for their proliferation. In contrast, it exhibited no significant effect to MCF7, MCF10A, and PC9 cells. As KDM5A was showed to mediate drug tolerance, YUKA1 displayed the ability to prevent drug tolerance in EGFR-mutant lung cancer cells treated with gefitinib and HER2+ breast cancer cell treated with trastuzumab.
CPI-455 (61) was identified as an inhibitor of KDM5 through an HTS assay against KDM4C JmjC domain and subsequent structural modification workCitation124,Citation125. It exhibited good potency toward KDM5A with IC50 = 0.02 μM and KDM4C with IC50 = 0.62 μM. 61 could specifically increase H3K4me3/2 in a dose-dependent manner in PC9, M14 and SKBR3 cells. In the co-crystal structure, the nitrile N atom of 61 interacted with the active Fe(II) ion; the carbonyl O atom formed hydrogen bonds with Lys501 and Asn575 (PDB code 5CEH). Treatment with 61 reduced the number of surviving cells after lethal drug exposures in some cell culture models. Further lead optimization work led to a potent, orally bioavailable KDM5 inhibitors 62Citation126. Compound 62 did not display improved potency against 61 (IC50 = 15.1 versus 10.0 nM in parallel experiment), but it possessed a more “balanced” human plasma protein binding (hPPB) and cell permeability which led its superior cell potency. This inhibitor also exhibited an excellent pharmacokinetic (PK) profile in mice. A hybrid naphthyridone inhibitor 63 that combined 61 and another pyrido[3,4-d]pyrimidin-4(3H)-one KDM5 inhibitor also displayed good potency (IC50 = 0.05 μM for KDM5A) in vitroCitation127.
In Liang’s workCitation128, some KDM5 inhibitors with potent, selective and orally bioavailable characteristics were identified through a high-throughput screening (HTS) of the Genentech/Roche library. From the HTS campaign, 64 (as a racemic mixture) had reasonable physiochemical properties, excellent KDM5 potency and selectivity (IC50 = 0.26 μM against KDM5A). After comprehensive SAR studies and optimizations, a more potent compound 65 (IC50 of 45, 56, and 55 nM against KDM5A, KDM5B and KDM5C, respectively; EC50 = 960 nM in PC9 cells), was identified with promising in vivo PK profiles.
KDM6 selective inhibitor
Members of KDM6 (JmjD3) subfamily can demethylated the H3K27me3 mark. KDM6s are involved in several physiological functions, including the inflammatory response. GSK-J1 (66a) was a selective KDM6 inhibitor with IC50 = 60 nM in AlphaScreen assayCitation129. In the co-crystal structure, the pyridyl-pyrimidine biaryl made a bidentate interaction with the catalytic metal; the propanoic acid of 66a mimicked 2-OG binding by Lys1381, Thr1387 and Asn1480; the aromatic ring of the tetrahydrobenzazepine moiety sat in a narrow cleft between Arg1246 and Pro1338. The ethyl ester prodrug 66b was examined for the efficacy at inhibiting the LPS-induced response of human primary macrophages derived from healthy volunteers. Administration of 66b significantly reduced the expression of 16 in 34 LPS-driven cytokines by PCR array. Xiong et al. designed and synthesized a series of derivatives of 66a to study the detail of SAR of this scaffold of selective KDM6 inhibitorsCitation130. The representative compound 67a had an equivalent potency against 66a (IC50 = 0.15 μM versus 0.15 μM). However, its ethyl ester prodrug 67b had a higher activity for inhibition of TNF-α production in LPS-induced murine macrophage cell line Raw264.7 cells.
Future preview
Histone modifications are basic and vital post-transcription regulations for normal and tumor cells in epigenetic process. Histone lysine methylation seems a “massage-contain” step in the whole epigenetic regulation. A big number of methyltransferases and demethylases work together and elaborately control the behaviour of DNA and chromatin. It seems very hard to figure out the relationship among the histone modifications and even just among the KDMs. Some scholars raised the questionsCitation131: was histone methylation a “cause or cog” in epigenetics? Would the affection of blocking single KDMs be drowned in total epigenetic regulatory flood? Further research works need to carry on for these questions.
However, overexpression of some histone modification enzymes, such as G9a, LSD1, MLL1, is certain to contribute to cancersCitation2,Citation3. KDM4 is involved in tumourigenesis in different types of cancers. Biological and physiological studies demonstrate that KDM4 is a promising target for cancer, especially breast and prostate cancer. Thus, the KDM4 inhibitors are still worth being expected as a strategy for cancerCitation21.
Moreover, targeted indication of JmjC KDMs inhibitors should not be limited in cancer treatment. As a milder method to affect gene transcription, histone methylation and demethylation could be involved in many therapeutic strategies for some other conditions. For example, some selective H3K27 demethylase KDM6 inhibitors showed anti-inflammation activityCitation129.
The development of KDM4s inhibitors is a tough but attractive work for improvements of potency and selectivity. Recently, some breakthroughs were reportedCitation98–100,Citation104,Citation120. We note that both academia and industry are participating in this workCitation132. Several future directions of KDM4s inhibitors may be focused on (a) more potent KDM4s inhibitors are still in urgent need, very limited number of nanomole level inhibitors have been presented now; (b) JmjC KDMs subfamily-selective inhibitors should be designed and synthesized for clinical application and providing biological chemistry tools; (c) some other non-2-OG mimic inhibitors are very desirable while a number of 2-OG-dependent enzymes, such as PHD2, are potential off-targets; (d) some further breakthroughs of the inhibitors may focus on physicochemical and pharmacokinetics property.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Andreoli F, Barbosa AJ, Parenti MD, et al. Modulation of epigenetic targets for anticancer therapy: clinicopathological relevance, structural data and drug discovery perspectives. Curr Pharm Des 2013;19:578–613.
- Liu Y, Liu K, Qin S, et al. Epigenetic targets and drug discovery: Part 1: histone methylation. Pharmacol Ther 2014;143:275–94.
- Liu K, Liu Y, Lau JL, et al. Epigenetic targets and drug discovery. Part 2: histone demethylation and DNA methylation. Pharmacol Ther 2015;151:121–40.
- New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code?. Mol Oncol 2012;6:637–56.
- Lee J, Huang RS. Cancer epigenetics: mechanisms and crosstalk of a HDAC inhibitor, vorinostat. Chemotherapy 2013;2:111. doi: 10.4172/2167-7700.1000111
- Srinivas NR. Clinical pharmacokinetics of panobinostat, a novel histone deacetylase (HDAC) inhibitor: review and perspectives. Xenobiotica 2016;1–15:354–368.
- Yoon S, Eom GH. HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J 2016;52:1–11.
- Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol 2004;14:R546–51.
- Jenuwein T, Allis CD. Translating the histone code. Science 2001;293:1074–80.
- Kouzarides T. Chromatin modifications and their function. Cell 2007;128:693–705.
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012;13:343–57.
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 2005;74:481–514.
- Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119:941–53.
- Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006;439:811–6.
- Guerra-Calderas L, Gonzalez-Barrios R, Herrera LA, et al. The role of the histone demethylase KDM4A in cancer. Cancer Genet 2015;208:215–24.
- Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res 2013;73:2936–42.
- Rotili D, Mai A. Targeting histone demethylases: a new avenue for the fight against cancer. Genes Cancer 2011;2:663–79.
- Katoh M, Katoh M. Identification and characterization of JMJD2 family genes in silico. Int J Oncol 2004;24:1623–8.
- Chu CH, Wang LY, Hsu KC, et al. KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor. J Med Chem 2014;57:5975–85.
- Kawazu M, Saso K, Tong KI, et al. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One 2011;6:e17830.
- Berry WL, Shin S, Lightfoot SA, et al. Oncogenic features of the JMJD2A histone demethylase in breast cancer. Int J Oncol 2012;41:1701–6.
- Kim TD, Shin S, Berry WL, et al. The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J Cell Biochem 2012;113:1368–76.
- Lohse B, Kristensen JL, Kristensen LH, et al. Inhibitors of histone demethylases. Bioorg Med Chem 2011;19:3625–36.
- Rose NR, Mcdonough MA, King ON, et al. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev 2011;40:4364–97.
- Suzuki T, Miyata N. Lysine demethylases inhibitors. J Med Chem 2011;54:8236–50.
- Lillico R, Stesco N, Khorshid Amhad T, et al. Inhibitors of enzymes catalyzing modifications to histone lysine residues: structure, function and activity. Future Med Chem 2016;8:879–97.
- Mcallister TE, England KS, Hopkinson RJ, et al. Recent progress in histone demethylase inhibitors. J Med Chem 2016;59:1308–29.
- Nowak RP, Tumber A, Johansson C, et al. Advances and challenges in understanding histone demethylase biology. Curr Opin Chem Biol 2016;33:151–9.
- Maes T, Carceller E, Salas J, et al. Advances in the development of histone lysine demethylase inhibitors. Curr Opin Pharmacol 2015;23:52–60.
- Whetstine JR, Nottke A, Lan F, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006;125:467–81.
- Trojer P, Zhang J, Yonezawa M, et al. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem 2009;284:8395–405.
- Nakayama J, Rice JC, Strahl BD, et al. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001;292:110–3.
- Peters AH, Mermoud JE, O’Carroll D, et al. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet 2002;30:77–80.
- Gilbert TM, Mcdaniel SL, Byrum SD, et al. A PWWP domain-containing protein targets the NuA3 acetyltransferase complex via histone H3 lysine 36 trimethylation to coordinate transcriptional elongation at coding regions. Mol Cell Proteomics 2014;13:2883–95.
- Kizer KO, Phatnani HP, Shibata Y, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 2005;25:3305–16.
- Hu CE, Liu YC, Zhang HD, et al. JMJD2A predicts prognosis and regulates cell growth in human gastric cancer. Biochem Biophys Res Commun 2014;449:1–7.
- Gray SG, Iglesias AH, Lizcano F, et al. Functional characterization of JMJD2A, a histone deacetylase- and retinoblastoma-binding protein. J Biol Chem 2005;280:28507–18.
- Hopkinson RJ, Walport LJ, Munzel M, et al. Is JmjC oxygenase catalysis limited to demethylation?. Angew Chem Int Ed Engl 2013;52:7709–13.
- Black JC, Allen A, Van Rechem C, et al. Conserved antagonism between JMJD2A/KDM4A and HP1γ during cell cycle progression. Mol Cell 2010;40:736–48.
- Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun 2007;359:742–6.
- Wissmann M, Yin N, Muller JM, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 2007;9:347–53.
- Coffey K, Rogerson L, Ryan-Munden C, et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res 2013;41:4433–46.
- Qiu MT, Fan Q, Zhu Z, et al. KDM4B and KDM4A promote endometrial cancer progression by regulating androgen receptor, c-myc, and p27kip1. Oncotarget 2015;6:31702–20.
- Li BX, Zhang MC, Luo CL, et al. Effects of RNA interference-mediated gene silencing of JMJD2A on human breast cancer cell line MDA-MB-231 in vitro. J Exp Clin Cancer Res 2011;30:90.
- Li BX, Luo CL, Li H, et al. Effects of siRNA-mediated knockdown of jumonji domain containing 2A on proliferation, migration and invasion of the human breast cancer cell line MCF-7. Exp Ther Med 2012;4:755–61.
- Li L, Gao P, Li Y, et al. JMJD2A-dependent silencing of Sp1 in advanced breast cancer promotes metastasis by downregulation of DIRAS3. Breast Cancer Res Treat 2014;147:487–500.
- Kauffman EC, Robinson BD, Downes MJ, et al. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol Carcinog 2011;50:931–44.
- Johmura Y, Sun J, Kitagawa K, et al. SCF(Fbxo22)-KDM4A targets methylated p53 for degradation and regulates senescence. Nat Commun 2016;7:10574.
- Tan MK, Lim HJ, Harper JW. SCF(FBXO22) regulates histone H3 lysine 9 and 36 methylation levels by targeting histone demethylase KDM4A for ubiquitin-mediated proteasomal degradation. Mol Cell Biol 2011;31:3687–99.
- Van Rechem C, Black JC, Abbas T, et al. The SKP1-Cul1-F-box and leucine-rich repeat protein 4 (SCF-FbxL4) ubiquitin ligase regulates lysine demethylase 4A (KDM4A)/Jumonji domain-containing 2A (JMJD2A) protein. J Biol Chem 2011;286:30462–70.
- Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Rep 2012;2:1233–43.
- Zimmermann M, De Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol 2014;24:108–17.
- Mallette FA, Mattiroli F, Cui G, et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J 2012;31:1865–78.
- Das A, Chai JC, Jung KH, et al. JMJD2A attenuation affects cell cycle and tumourigenic inflammatory gene regulation in lipopolysaccharide stimulated neuroectodermal stem cells. Exp Cell Res 2014;328:361–78.
- Chang PC, Fitzgerald LD, Hsia DA, et al. Histone demethylase JMJD2A regulates Kaposi’s sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J Virol 2011;85:3283–93.
- Das ND, Choi MR, Jung KH, et al. Functional analysis of histone demethylase Jmjd2b on lipopolysaccharide-treated murine neural stem cells (NSCs). Neurotox Res 2013;23:154–65.
- Antony J, Oback F, Chamley LW, et al. Transient JMJD2B-mediated reduction of H3K9me3 levels improves reprogramming of embryonic stem cells into cloned embryos. Mol Cell Biol 2013;33:974–83.
- Shi L, Sun L, Li Q, et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci USA 2011;108:7541–6.
- Kim JG, Yi JM, Park SJ, et al. Histone demethylase JMJD2B-mediated cell proliferation regulated by hypoxia and radiation in gastric cancer cell. Biochim Biophys Acta 2012;1819:1200–7.
- Li W, Zhao L, Zang W, et al. Histone demethylase JMJD2B is required for tumor cell proliferation and survival and is overexpressed in gastric cancer. Biochem Biophys Res Commun 2011;416:372–8.
- Zhao L, Li W, Zang W, et al. JMJD2B promotes epithelial-mesenchymal transition by cooperating with beta-catenin and enhances gastric cancer metastasis. Clin Cancer Res 2013;19:6419–29.
- Berry WL, Kim TD, Janknecht R. Stimulation of β-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int J Oncol 2014;44:1341–8.
- Cloos PA, Christensen J, Agger K, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006;442:307–11.
- Luo W, Chang R, Zhong J, et al. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci USA 2012;109:E3367–76.
- Ishimura A, Terashima M, Kimura H, et al. Jmjd2c histone demethylase enhances the expression of Mdm2 oncogene. Biochem Biophys Res Commun 2009;389:366–71.
- Farooq Z, Banday S, Pandita TK, et al. The many faces of histone H3K79 methylation. Mutat Res Rev Mutat Res 2016;768:46–52.
- Jbara M, Guttmann-Raviv N, Maity SK, et al. Total chemical synthesis of methylated analogues of histone 3 revealed KDM4D as a potential regulator of H3K79me3. Bioorg Med Chem 2017;25:4966–70.
- Huang F, Chandrasekharan MB, Chen YC, et al. The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J Biol Chem 2010;285:24548–61.
- Ozboyaci M, Gursoy A, Erman B, et al. Molecular recognition of H3/H4 histone tails by the tudor domains of JMJD2A: a comparative molecular dynamics simulations study. PLoS One 2011;6:e14765.
- Schofield CJ, Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr Opin Struct Biol 1999;9:722–31.
- Chen Z, Zang J, Whetstine J, et al. Structural insights into histone demethylation by JMJD2 family members. Cell 2006;125:691–702.
- Clifton IJ, Mcdonough MA, Ehrismann D, et al. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J Inorg Biochem 2006;100:644–69.
- Ng SS, Kavanagh KL, Mcdonough MA, et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature 2007;448:87–91.
- Williams ST, Walport LJ, Hopkinson RJ, et al. Studies on the catalytic domains of multiple JmjC oxygenases using peptide substrates. Epigenetics 2014;9:1596–603.
- Krishnan S, Trievel RC. Structural and functional analysis of JMJD2D reveals molecular basis for site-specific demethylation among JMJD2 demethylases. Structure 2013;21:98–108.
- Smith EH, Janknecht R, Maher LJ 3rd. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet 2007;16:3136–48.
- Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011;12:463–9.
- Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009;324:261–5.
- Rose NR, Ng SS, Mecinovic J, et al. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J Med Chem 2008;51:7053–6.
- Rose NR, Woon EC, Kingham GL, et al. Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches. J Med Chem 2010;53:1810–8.
- Hamada S, Suzuki T, Mino K, et al. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of Jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem 2010;53:5629–38.
- Rose NR, Woon ECY, Tumber A, et al. Plant growth regulator daminozide is a selective inhibitor of human KDM2/7 histone demethylases. J Med Chem 2012;55:6639–43.
- Suzuki T, Ozasa H, Itoh Y, et al. Identification of the KDM2/7 histone lysine demethylase subfamily inhibitor and its antiproliferative activity. J Med Chem 2013;56:7222–31.
- Itoh Y, Sawada H, Suzuki M, et al. Identification of Jumonji AT-rich interactive domain 1A inhibitors and their effect on cancer cells. ACS Med Chem Lett 2015;6:665–70.
- Morera L, Roatsch M, Furst MC, et al. 4-Biphenylalanine- and 3-phenyltyrosine-derived hydroxamic acids as inhibitors of the JumonjiC-domain-containing histone demethylase KDM4A. ChemMedChem 2016;11:2063–83.
- Luo XL, Liu YX, Kubicek S, et al. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J Am Chem Soc 2011;133:9451–6.
- King ON, Li XS, Sakurai M, et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One 2010;5:e15535.
- Hopkinson RJ, Tumber A, Yapp C, et al. 5-Carboxy-8-hydroxyquinoline is a broad spectrum 2-oxoglutarate oxygenase inhibitor which causes iron translocation. Chem Sci 2013;4:3110–7.
- Feng T, Li D, Wang H, et al. Novel 5-carboxy-8-HQ based histone demethylase JMJD2A inhibitors: introduction of an additional carboxyl group at the C-2 position of quinoline. Eur J Med Chem 2015;105:145–55.
- Duan L, Rai G, Roggero C, et al. KDM4/JMJD2 histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-regulated genes. Chem Biol 2015;22:1185–96.
- Thinnes CC, Tumber A, Yapp C, et al. Betti reaction enables efficient synthesis of 8-hydroxyquinoline inhibitors of 2-oxoglutarate oxygenases. Chem Commun (Camb) 2015;51:15458–61.
- Schiller R, Scozzafava G, Tumber A, et al. A cell-permeable ester derivative of the JmjC histone demethylase inhibitor IOX1. ChemMedChem 2014;9:566–71.
- Carter DM, Specker E, Przygodda J, et al. Identification of a novel benzimidazole pyrazolone scaffold that inhibits KDM4 lysine demethylases and reduces proliferation of prostate cancer cells. SLAS Discov 2017;22:801–12.
- Thalhammer A, Mecinovic J, Loenarz C, et al. Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Org Biomol Chem 2011;9:127–35.
- Wang L, Chang J, Varghese D, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun 2013;4:2035.
- Roatsch M, Robaa D, Pippel M, et al. Substituted 2-(2-aminopyrimidin-4-yl)pyridine-4-carboxylates as potent inhibitors of JumonjiC domain-containing histone demethylases. Future Med Chem 2016;8:1553–71.
- England KS, Tumber A, Krojer T, et al. Optimisation of a triazolopyridine based histone demethylase inhibitor yields a potent and selective KDM2A (FBXL11) inhibitor. Medchemcomm 2014;5:1879–86.
- Bavetsias V, Lanigan RM, Ruda GF, et al. 8-Substituted pyrido[3,4-d]pyrimidin-4(3H)-one derivatives as potent, cell permeable, KDM4 (JMJD2) and KDM5 (JARID1) histone lysine demethylase inhibitors. J Med Chem 2016;59:1388–409.
- Westaway SM, Preston AG, Barker MD, et al. Cell penetrant inhibitors of the KDM4 and KDM5 families of histone lysine demethylases. 1. 3-Amino-4-pyridine carboxylate derivatives. J Med Chem 2016;59:1357–69.
- Westaway SM, Preston AG, Barker MD, et al. Cell penetrant inhibitors of the KDM4 and KDM5 families of histone lysine demethylases. 2. Pyrido[3,4-d]pyrimidin-4(3H)-one derivatives. J Med Chem 2016;59:1370–87.
- Fang Z, Wang TQ, Li H, et al. Discovery of pyrazolo[1,5-a]pyrimidine-3-carbonitrile derivatives as a new class of histone lysine demethylase 4D (KDM4D) inhibitors. Bioorg Med Chem Lett 2017;27:3201–4.
- Chen YK, Bonaldi T, Cuomo A, et al. Design of KDM4 inhibitors with antiproliferative effects in cancer models. ACS Med Chem Lett 2017;8:869–74.
- Metzger E, Stepputtis SS, Strietz J, et al. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res 2017;77:5900–12.
- Korczynska M, Le DD, Younger N, et al. Docking and linking of fragments to discover Jumonji histone demethylase inhibitors. J Med Chem 2016;59:1580–98.
- Xu W, Podoll JD, Dong X, et al. Quantitative analysis of histone demethylase probes using fluorescence polarization. J Med Chem 2013;56:5198–202.
- Wang W, Marholz LJ, Wang X. Novel scaffolds of cell-active histone demethylase inhibitors identified from high-throughput screening. J Biomol Screen 2015;20:821–7.
- Sakurai M, Rose NR, Schultz L, et al. A miniaturized screen for inhibitors of Jumonji histone demethylases. Mol BioSyst 2010;6:357.
- Feng T, Chen W, Li D, et al. Identification of novel JMJD2A inhibitor scaffold using shape and electrostatic similarity search combined with docking method and MM-GBSA approach. RSC Adv 2015;5(101):82936–46.
- Kim TD, Fuchs JR, Schwartz E, et al. Pro-growth role of the JMJD2C histone demethylase in HCT-116 colon cancer cells and identification of curcuminoids as JMJD2 inhibitors. Am J Transl Res 2014;6:236–47.
- Baell J, Walters MA. Chemistry: chemical con artists foil drug discovery. Nature 2014;513:481–3.
- Sekirnik R, Rose NR, Thalhammer A, et al. Inhibition of the histone lysine demethylase JMJD2A by ejection of structural Zn(II). Chem Commun (Camb) 2009;6376–8.
- Chen HB, Kluz T, Zhang RH, et al. Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells. Carcinogenesis 2010;31:2136–44.
- Liu LJ, Lu L, Zhong HJ, et al. An iridium(III) complex inhibits JMJD2 activities and acts as a potential epigenetic modulator. J Med Chem 2015;58:6697–703.
- Lohse B, Nielsen AL, Kristensen JB, et al. Targeting histone lysine demethylases by truncating the histone 3 tail to obtain selective substrate-based inhibitors. Angew Chem Int Ed Engl 2011;50:9100–3.
- Woon EC, Tumber A, Kawamura A, et al. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew Chem Int Ed Engl 2012;51:1631–4.
- Leurs U, Lohse B, Rand KD, et al. Substrate- and cofactor-independent inhibition of histone demethylase KDM4C. ACS Chem Biol 2014;9:2131–8.
- Kawamura A, Munzel M, Kojima T, et al. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat Commun 2017;8:14773.
- Pilka ES, James T, Lisztwan JH. Structural definitions of Jumonji family demethylase selectivity. Drug Discov Today 2015;20:743–9.
- Rasmussen PB, Staller P. The KDM5 family of histone demethylases as targets in oncology drug discovery. Epigenomics 2014;6:277–86.
- Horton JR, Liu X, Gale M, et al. Structural basis for KDM5A histone lysine demethylase inhibition by diverse compounds. Cell Chem Biol 2016;23:769–81.
- Rotili D, Mattevi A. At long last potent and selective KDM5 inhibitors. Cell Chem Biol 2016;23:749–51.
- Hatch SB, Yapp C, Montenegro RC, et al. Assessing histone demethylase inhibitors in cells: lessons learned. Epigenet Chromatin 2017;10:9.
- Tumber A, Nuzzi A, Hookway ES, et al. Potent and selective KDM5 inhibitor stops cellular demethylation of H3K4me3 at transcription start sites and proliferation of MM1S myeloma cells. Cell Chem Biol 2017;24:371–80.
- Gale M, Sayegh J, Cao J, et al. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 2016;7:39931–44.
- Gehling VS, Bellon SF, Harmange JC, et al. Identification of potent, selective KDM5 inhibitors. Bioorg Med Chem Lett 2016;26:4350–4.
- Liang J, Zhang B, Labadie S, et al. Lead optimization of a pyrazolo[1,5-a]pyrimidin-7(4H)-one scaffold to identify potent, selective and orally bioavailable KDM5 inhibitors suitable for in vivo biological studies. Bioorg Med Chem Lett 2016;26:4036–41.
- Labadie SS, Dragovich PS, Cummings RT, et al. Design and evaluation of 1,7-naphthyridones as novel KDM5 inhibitors. Bioorg Med Chem Lett 2016;26:4492–6.
- Liang J, Labadie S, Zhang B, et al. From a novel HTS hit to potent, selective, and orally bioavailable KDM5 inhibitors. Bioorg Med Chem Lett 2017;27:2974–81.
- Kruidenier L, Chung CW, Cheng Z, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012;488:404–8.
- Hu J, Wang X, Chen L, et al. Design and discovery of new pyrimidine coupled nitrogen aromatic rings as chelating groups of JMJD3 inhibitors. Bioorg Med Chem Lett 2016;26:721–5.
- Henikoff S, Shilatifard A. Histone modification: cause or cog?. Trends Genet 2011;27:389–96.
- Chin YW, Han SY. KDM4 histone demethylase inhibitors for anti-cancer agents: a patent review. Expert Opin Ther Pat 2015;25:135–44.