
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The inhibition of the enzyme acetylcholinesterase (AChE) increases the levels of the neurotransmitter acetylcholine and symptomatically improves the affected cognitive function. In the present study, we searched for novel AChE inhibitors by docking-based virtual screening of the standard lead-like set of ZINC database containing more than 6 million small molecules using GOLD software. The top 10 best-scored hits were tested in vitro for AChE affinity, neurotoxicity, GIT and BBB permeability. The main pharmacokinetic parameters like volume of distribution, free fraction in plasma, total clearance, and half-life were predicted by previously derived models. Nine of the compounds bind to the enzyme with affinities from 0.517 to 0.735 µM, eight of them are non-toxic. All hits permeate GIT and BBB and bind extensively to plasma proteins. Most of them are low-clearance compounds. In total, seven of the 10 hits are promising for further lead optimisation. These are structures with ZINC IDs: 00220177, 44455618, 66142300, 71804814, 72065926, 96007907, and 97159977.
Introduction
The enzyme acetylcholinesterase (AChE) is a serine protease (EC 3.1.1.7) catalysing the hydrolysis of the neurotransmitter acetylcholine (ACh) to choline and acetic acid. Low levels of ACh lead to cognitive impairment and dementiaCitation1. The inhibition of AChE increases the ACh levels and symptomatically improves the affected cognitive functionCitation2. AChE inhibitors (AChEIs) are the main drugs currently in use for treatment of Alzheimer’s disease (AD), the most common form of dementiaCitation3. Donepezil, galantamine, and rivastigmine are AChEIs. They have moderate affinity to AChE and provide delay in AD progression.
The binding site on AChE is deep and narrow gorge and consists of several domains: catalytic, anioinic, acylic, oxyanionic, and peripheral anionicCitation4–7. The most important of them are the catalytic active site (CAS) where ACh hydrolysis happens and the peripheral anionic site (PAS) placed near the entrance of the gorge and associated with the formation of amyloid plaquesCitation8. Thus, AChE is a target with dual functionality: ACh hydrolysis and amyloid beta (Aβ) peptide aggregation. Because of its importance, AChE is a focus of many intensive and extensive drug discovery studies during the last two decadesCitation9–11. These studies could be grouped into three directions: lead optimisation of known AChEIsCitation12–16, hybrids between themCitation17–24 and search for new scaffoldsCitation25–28. A wide range of computational tools are involved in these studiesCitation29.
One of the most useful structure-based computational methods in the discovery of novel hits binding to a specific target is the molecular docking used solely or in combination with 2D- and 3D-QSAR, high-throughput screening, and/or machine learning methodsCitation30. Recently Chen et al. performed virtual screening on 263,146 entries from Specs database using a structure-based pharmacophore (SBP) derived on two tacrine hybrids with nanomolar affinity to AChECitation31. The top 50 compounds were docked in the AChE and the best 15 of them were tested in vitro by Ellman’s methodCitation32. Ten hits showed IC50 values below 10 µM. Dhanja et al. derived a ligand-based pharmacophore using the 3D structures of 16 known AChE inhibitorsCitation33. The pharmacophore was used to screen 50,000 small molecule natural compounds from ZINC database, followed by docking studies. The best two binders were analysed for molecular interactions with AChE, but have not been tested experimentally. Chen et al. conducted a virtual screening on Chemdiv compound collection, which contains 1,293,896 moleculesCitation34. Initially, the collection was screened by rapid overlay of chemical structures using the AChEI donepezil as a template. Then, the top 1% of the most similar structures was screened by SBP generated on the basis of donepezil-AChE X-ray complex. Finally, 24 compounds of the best hits were tested for AChE and BuChE activity in vitro by Ellman’s method. Among them, five new inhibitors were discovered.
In the present study, we conducted a docking-based virtual screening on ZINC database containing over 6 million biologically active small molecules in order to identify novel hits binding to AChE. The top 10 best-scored binders to AChE were tested in vitro for affinity to AChE, neurotoxicity, blood-brain barrier (BBB), and gastrointestinal (GI) permeability. Predictions of their physicochemical and ADME properties also were performed.
Materials and methods
Database and docking protocol
ZINC (www.zinc.docking.org) contains several databases of biologically active structures. We selected the Standard Lead-like database which consists of 6,053,287 small molecules with molecular weights between 250 and 350 g/mol, log P up to 3,5 and up to 7 rotatable bonds. The set was downloaded in March 2015. The molecules were docked into the X-ray structure of human recombinant acetylcholinesterase (rhAChE, pdb id: 4EY6, R = 2.40 Å)Citation35. The docking simulations were performed by GOLD v. 5.1. [CCDC Ltd., Cambridge, UK] using a protocol previously optimised in terms of scoring function, rigid/flexible ligand and binding site, radius of the binding site, the presence/absence of a water molecule (HOH846) within the binding site, number of genetic algorithm (GA) runsCitation36–39. The docking simulations in the present study were performed at the following settings: scoring function ChemPLP, flexible ligand, rigid protein, radius of the binding site 6 Å, no water molecule, 10 GA runs. The top 20 best-scored compounds were selected and docked five times with 100 GA runs each. The top 10 best-scored compounds from all runs were selected for tests.
Compounds
N-Methyl-3-(2-oxo-1-pyridyl)-N-[(2-phenylphenyl)methyl]propanamide (1, ZINC72065926) and N-[3-(benzimidazol-1-yl)propyl]-3-indol-1-yl-propanamide (2, ZINC71804814) were purchased from AKos GmbH, Germany. 3-(6-Oxo-3-phenylpyridazin-1(6H)-yl)-N-phenethylpropanamide (3, ZINC00220177), 1-[3-(2-fluorophenyl)-1-methyl-pyrazol-4-yl]-N-(6-quinolylmethyl) methanamine (4, ZINC23159164), N1-((1-methyl-3-phenyl-1H-pyrazol-4-yl)methyl)piperidine-1,4-dicarboxamide (6, ZINC96007907), N-((1-methyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl)methyl)-2-(quinolin-8-yl)ethanamine (7, ZINC97159977), 2–(2,4-dioxoquinazolin-1-yl)-N-[2-(3-fluorophenyl)ethyl]acetamide (9, ZINC08993868) and N-[2-(4-hydroxyphenyl)ethyl]-2-(1-isopropylindol-3-yl)acetamide (10, ZINC96116182) were obtained from MolPort SIA, Latvia. The purchased compounds arrived with analytical data for identity and purity. Compounds 2-indol-1-yl-N-[2-(8-quinolyloxy)ethyl]acetamide (5, ZINC44455618) and N-((1S,2S)-1-(4-aminophenyl)-1,3-dihydroxypropan-2-yl)-2-(4-methoxyphenyl)acetamide (8, ZINC66142300) were synthesised.
Isothermal titration calorimetry (ITC) protocol
The ITC measurementsCitation40 were performed on NanoITC tool (TA Instruments, Lindon, UT) with 190 µL sample cell and 50 µL syringe. The lyophilised AChE from Electrophorus electricus (electric eel) (Sigma Aldrich, St. Luis, MO) was reconstructed in 50 mM TRIS-HCl pH 7.4 buffer to obtain ca. 1140 U/mL with the addition of 0.1% BSA as an enzyme stabilising factor, according to the manufacturer’s instructions. The tested compounds were prepared in 5 mM stock solutions in ethanol and diluted to 0.5 mM in 50 mM Tris-HCl pH 7.4 buffer. All samples were degassed prior the experiments. The AChE solution was placed into the sample cell and titrated by the tested compounds in 25 steps of 2 µL at 5 min intervals at 25 °C. The blank samples (buffer lacking AChE) were titrated at the same conditions. The corresponding Kd values were calculated using NanoAnalyze software (TA Instruments, Lindon, UT).
Parallel artificial membrane permeability assay (PAMPA)
The intestinal and BBB permeabilities were measured by PAMPA Permeability Analyzer (pION Inc.) at the following settings: wavelength analysed 250–500 nm, pH 7.4, temperature 25 °C, permeation time 4 h, lipid formulation GIT-0 and stirring 60 rpm and BBB-1 and no stirring, respectively. The intestinal permeability was tested at three pH values: 5.0, 6.2, and 7.4, while the BBB permeability was tested at pH 7.4. The permeability was presented as −logPe, where Pe is the permeability coefficient (10−6 cm/s). Compounds with −logPe below 5 are considered as highly permeable, with −logPe between 5 and 6 – as medium permeable and with above 6 – as low permeableCitation41. Carbamazepine, ketoprofen, and ranitidine were used as controls for intestinal permeability. Theophylline, progesterone, and propranolol were used as controls for BBB permeabilityCitation41.
Neurotoxicity test
Murine neuroblastoma NEURO-2 A cells (German collection DSMZ, Braunschweig, Germany) were cultivated under standard conditions: complete medium (90% DMEM, 10% heat-inactivated FBS, and 1 × non-essential amino acids); 37 °C and5% CO2 in fully humidified atmosphere. The cell line was kept in the logarithmic growth phase by splitting 1:4 once a week using trypsin/EDTA. About 30% of the cells grow like neuronal cells. For the experimental evaluation of the cytotoxicity NEURO-2A, cells were plated in 96-well flat-bottomed cell culture plates at the recommended density of 1 × 106 cells/25 cm2. After 24 h, the cells were treated with various concentrations of the investigational compounds and after 72-h incubation, a MTT-dye reduction assay was performedCitation42. Briefly, at the end of incubation, a MTT stock solution (10 mg/ml in PBS) was added (10 µL/well). Plates were further incubated at 37 °C for 4 h. Next, the formazan crystals were dissolved by the addition of 110 µL/well 5% formic acid in 2-propanol (v/v). Absorption was measured at 580 nm wavelength on an automated ELISA reader Labexim LMR1. At least six wells per concentration were used, and data were processed using the GraphPad Prism 5.0 software 2.
Calculation of physicochemical properties and prediction of pharmacokinetic (PK) parameters
The main physicochemical properties pKa, logP, logD7.4, polar surface area (PSA), number of hydrogen-bond donors (HBD), and hydrogen-bond acceptors (HBA) in the molecules of the tested compounds were calculated using ACD/logD v. 9.08 (ACD Inc., Canada). The fraction ionised as a base at pH = 7.4 (fB) was calculated according to the equation:
The key PK parameters were predicted by quantitative structure–activity relationships (QSPkRs) models, derived previouslyCitation43–46. Briefly, the PK parameters of 145 neutral and/or 262 basic drugsCitation47 were used to derive QSPkR models by multiple linear regression (MLR) with MDL QSAR v. 2.2 (MDL Information Systems Inc., San Leandro, CA). Three PK parameters were modelled: the steady-state volume of distribution (VDss), free fraction of drug in plasma (fu), and unbound clearance (CLu). The total clearance (CL) and half-life (t1/2) were calculated following the equations.
The models are statistically significant and meet the criteria for good performing QSPkRsCitation48,Citation49. The AChEI galantamine (GAL) is given as a reference compound.
Results
Docking-based screening of ZINC database on rhAChE
The top 10 best-scored hits by ChemPLP from the docking of 6,053,287 molecules from ZINC database on rhAChE are given in . The dockings were performed with flexible ligands and rigid binding site lacking the water molecule necessary for the binding of GAL. The ChemPLP scores are given in .
Figure 1. Structures of the top 10 best-scored hits. The fragments binding in CAS are given by ellipse.
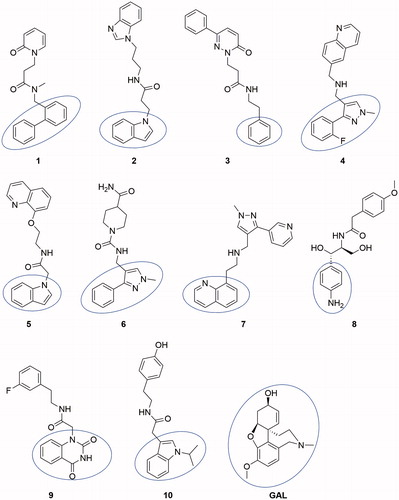
Table 1. Docking score, eeAChE affinity, neurotoxicity, BBB, and GIT permeability of the tested compounds.
All compounds consisted of two aromatic moieties connected by a linker of 3–7 carbon chain containing NH or NHCO group. An exception is made by compound 6 which contain one non-aromatic piperidine ring in the linker. The docking poses showed that the first aromatic moiety binds in CAS, the aliphatic chain is stretching along the binding gorge and the second aromatic ring binds in PAS ().
Compounds 1–4, 6, 7, 9, and 10 were purchased; compounds 5 and 8 were synthesised.
Synthesis of compounds 5 and 8
The synthetic strategy towards the synthesis of compound 5 was based on the formation of amide bond between 2-indoleacetic acid 11 and quinoline derived amine 12 (Scheme 1).
Scheme 1. Synthesis of compound 5.
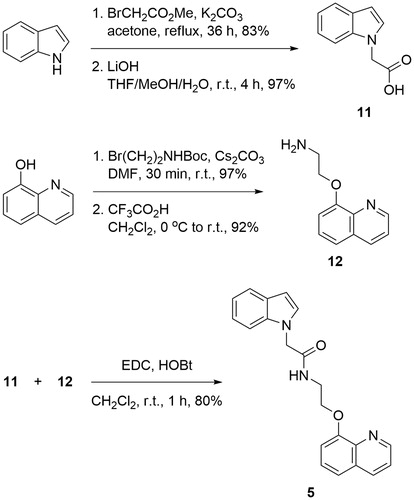
The desired acid 11 was synthesised via nucleophilic substitution of methyl 2-bromoacetate with deprotonated indole, followed by hydrolysis of the resulting methyl esterCitation50. Reaction of 8-hydroxyquinoline with N-Boc protected 2-bromoethanamine in the presence of Cs2CO3 as a base, and subsequent deprotection of the amine afforded 2-(quinolin-8-yloxy)ethanamine 12 in excellent yield. The coupling between the two building blocks was achieved applying 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 1-Hydroxybenzotriazole hydrate (HOBT) in dichloromethane (Scheme 1).
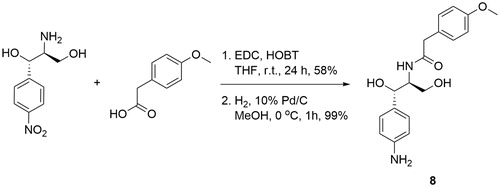
Compound 8 was synthesised by coupling of the commercially available (1S,2S)-2-amino-1-(4-nitrophenyl)propane-1,3-diol with 4-methoxyphenylacetic acid and subsequent catalytic hydrogenation of the nitro group to the corresponding amine (Scheme 2).
Scheme 2. Synthesis of compound 8.
The detailed synthesis, analytical data (incl. LC-MS and elemental analyses) and copies of 1H and Citation13C NMR spectra for compounds 5 and 8 are given in the Supplementary File.
Estimation of binding affinity by ITC
The binding affinity of the best-scored compounds was tested in vitro by ITC as described in “Materials and methods” section. AChE from electric eel (eeAChE) was used in the measurements. The UniProt alignment of rhAChE (UniProt: P22303) and еeAChЕ (UniProt: O42275) have showed that all 17 residues forming the binding gorges are identicalCitation39. Our previous experience has shown that the docking scores predicted for rhAChE correlate well with the experimental binding affinity measured on eeAChECitation36–39. The Kd values of the tested compounds are given in . Compound 9 does not interact with eeAChE. For the rest, the Kds are in the range of 0.517–0.735 µM.
Estimation of GIT and BBB permeability by PAMPA
The permeability of the compounds tested by PAMPA shows that all of them are highly permeable through the GIT and BBB (). Only compound 6 has intermediate BBB permeability with −logPe = 5.008. The −logPe for the rest compounds ranges from 4.146 to 4.731 for the BBB permeability and from 4.202 to 4.469 for the GIT permeability at pH = 6.2. For comparison, GAL has −logPe 5.060 and 4.268 for BBB and GIT permeability, respectivelyCitation39, which corresponds well to its low ability to penetrate BBB by passive diffusionCitation51 and to 90% oral bioavailabilityCitation52. Moderate correlations exist between logD7.4 and BBB permeability (r = 0.693) and between logD7.4 and GIT permeability (r = 0.637) of the tested compounds.
Estimation of neurotoxicity on neuro-2A cells
The neurotoxicity of the compounds was tested on NEURO-2A cells as described in “Materials and methods” section. All of them are non-toxic at concentrations up to 100 µM, apart from compounds 4 and 10. Compound 4 has IC50 = 35 µM and compound 10 has IC50 = 53 µM.
Physicochemical properties and PK parameters
The molecular weights of the tested compounds vary in a short range from 330.38 to 347.41 g/mol. The pKa values are from −0.69 to 8.14, logPs are between 0.13 and 3.65, logD7.4 s – between 0.13 and 3.42 (). According to fB, compounds 4 and 7 are moderate bases, partially ionised at physiological pH, and the rest are neutral molecules. Most of the compounds are lipophilic, only compounds 6 and 8 are rather hydrophilic with PSAs above 80 Å2. The number of HBDs in the molecules is up to 5 and the number of HBAs – up to 10.
Table 2. Calculated physicochemical properties and predicted PK parameters of the tested compounds. VDss is the volume of distribution in steady state, fu – the fraction of free (unbound) compound in plasma, CL – total plasma clearance, t1/2 – half-life.
The predicted VDss values of the bases 4 and 7 are 3.17 L/kg and 3.67 L/kg, respectively. They are considerably higher than the VDsss of the neutral molecules, which range from 0.73 to 1.61 L/kg. This is in a good agreement with the distribution of VDss of the drugs from the training set of bases used to derive the modelCitation41. The VDsss in the training set range between 0.073 and 140 L/kg, with mean 5.90 L/kg and median 2.45 L/kg. The lower VDsss of the neutral molecules also is in accordance with the VDsss of the training set of neutral molecules, which vary between 0.16 and 25 L/kg, with mean 1.94 L/kg and median 1 L/kgCitation44. As logP is one of the descriptors with high positive effect on VDssCitation43, the hydrophilic compounds 6 and 8 have reasonably the lowest VDsss.
The tested compounds bind extensively to plasma proteins (PP). The predicted fus are between 0.01 and 0.27, i.e. 73–99% of the compounds are bound to plasma proteins. Again, lipophilicity is the major governing factor for high PP bindingCitation44. The hydrophilic compounds 6 and 8 have the highest fus.
The predicted CL values vary between 0.59 and 9.82 mL/min/kg. The CL of compound 2 is overpredicted, as it exceeds the maximum total body blood flow (80 mL/min/kg). Most of the tested compounds are low-clearance compounds with CLs below 30% of hepatic blood flow (QH = 21 mL/min/kg)Citation53,Citation54. Only compounds 5 and 10 are moderate-clearance drugs with CLs around 50% of QH.
The half-lives t1/2 were predicted from the corresponding VDss and CL values. Expectedly, the t1/2 of compound 2 is underpredicted. Compounds 5 and 10 have very short t1/2 due to the low VDss and relatively high CL. The rest of compounds have moderate t1/2 in the range of 6–21 h, allowing convenient multiple-dose regimens.
Discussion
The standard lead-like set of ZINC database was virtually screened by molecular docking on rhAChE and the 10 best-scored structures were tested in vitro for binding affinity to the enzyme, neurotoxicity, permeability across GIT and BBB. ADME properties were predicted. Nine of the compounds bind well the enzyme with Kd in nanomolar range, eight of them are non-toxic at concentrations up to 100 µM. All of the tested compounds are highly permeable across the GIT and BBB, have MWs up to 350 g/mol, logD7.4 up to 3.5 and bind extensively to plasma proteins. Most of them are low-clearance compounds.
Compound 1 is a neutral molecule with logD7.4 of 2.69 and high affinity to AChE (Kd = 525 nM). It is non-toxic on Neuro-2 A cells, permeates easily the gastric mucosa and the BBB, 96% of the molecules are bound to plasma proteins. Although the VDss is 1.09 L/kg, compound 1 is cleared very slowly (CL = 0.59 mL/min/kg) and has long half-life (t1/2 = 21.23 h). The docked pose of 1 into rhAChE shows that the biphenyl fragment binds in CAS, while the oxo-pyridyl moiety is placed in PAS (). Hydrogen bonds are formed between Tyr337 and carbonyl oxygen atom from the linker and between His447 and the carbonyl oxygen atom from the oxo-pyridyl moiety. Hydrogen–π interactions exist between Trp86, Gly121, and biphenyl, and between Phe295 and oxo-pyridyl. Trp286, Val294, Phe297, Phe338, and His447 take part in a network of hydrophobic interactions with the linker and the oxo-pyridyl fragment.
Figure 2. Interactions between rhAChE residues and (1) compound 1, (2) compound 2, (3) compound 3, (4) compound 5, (5) compound 6, (6) compound 7, (7) compound 8, and (8) GAL in the docked complex.
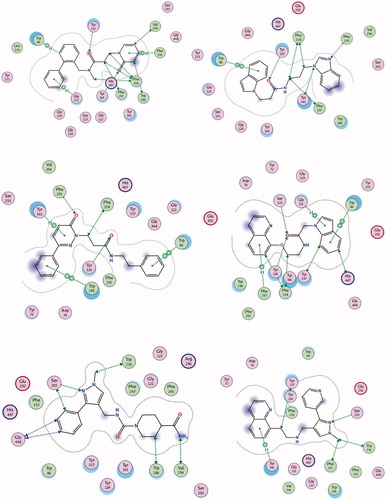
Figure 2. Continued.
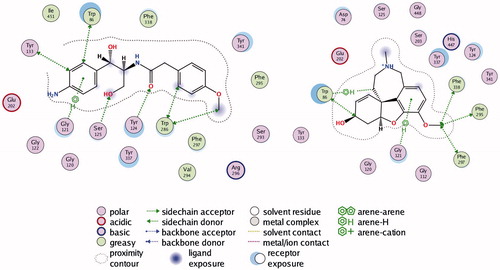
Compound 2 is a weak base with logD7.4 of 3.17, high affinity to AChE (Kd = 618 nM), good permeability and absence of toxicity. The predicted ADME properties show that 98% of compound 2 are bound to plasma proteins and the VDss is 1.34 L/kg. Compound 2 has very intensive probably overpredicted clearance (CL =143 mL/min/kg) and ultra-short half-life (t1/2 = 0.11 h). The indole moiety stacks with Trp86 in CAS, while the benzimidazol fragment is positioned in PAS making a hydrogen bond with Phe295 (). Another hydrogen bond is formed between Tyr337 and the carbonyl oxygen atom from the linker. Phe297, Tyr341, and Phe338 are involved in hydrophobic interactions with the linker.
Compound 3 is a high binder to AChE with Kd = 555 nM, logD7.4 of 1.96, VDss of 1.29 L/kg, moderate clearance (CL =15.48 mL/min/kg) and half-life of 6 h. It is permeable and non-toxic. The single phenyl ring of 3 binds in CAS stacking with Trp86, while the phenylpiperazine moiety is placed in PAS interacting with Trp286 and Tyr341 and forming a hydrogen bond with Phe295 (). Compound 4 binds well to the enzyme with Kd = 618 nM but because of its toxicity (IC50 = 35 µM), it is not considered as a perspective hit for further optimisation.
Compound 5 is a neutral molecule with logD7.4 of 2.82 and Kd of 517 nM. It is non-toxic, easily permeable with VDss = 0.96 L/kg, binds extensively (99%) with plasma proteins, is cleared with moderate rate (CL =9.82 mL/min/kg) and has short half-life (t1/2 = 1.13 h). The indolyl fragment inhibits CAS making π–π interaction with Trp86 and hydrogen–π bond with Gly121, while the quinolyl moiety binds in PAS forming hydrogen–π bond with Phe297 (). The carbonyl oxygen from the linker makes hydrogen bonds with Tyr124 and Ser125.
Compound 6 binds tightly to eeAChE with Kd of 613 nM. It is a neutral molecule with logD7.4 of 0.15, VDss = 0.84 L/kg, 73% is bound to plasma proteins, has low clearance (CL =1.28 mL/min/kg) and half-life of 7.57 h. The phenyl-pyrazolyl moiety is bound in CAS making hydrogen bond with Ser203, while the piperidine fragment is oriented towards the PAS and binds hydrophobically to Trp286 and Val294 ().
Compound 7 is a weak base partially protonated at pH 7.4 with logD7.4 of 1.21. Binds to AChE with Kd of 577 nM. It absorbs easily through the intestinal mucosa, crosses the BBB, distributes extensively with VDss of 3.67 L/kg, is cleared moderately (CL = 5.45 mL/min/kg) and has half-life of 7.78 h. The quinolyl fragment enters CAS interacting with Tyr341, while the pyridyl-pyrazolyl moiety makes hydrogen bond with Ser203 near PAS ().
Compound 8 binds to eeAChE with Kd = 682 nM. It is a neutral molecule with small VDss, slow clearance (CL = 0.59 mL/min/kg) and half-life of 14.27 h. The aminophenyl binds in CAS, while the methoxy-benzene is placed in PAS. Hydrogen bonds are formed between Ser125 and oxygen from the CH2OH group and between Tyr124 and the carbonyl oxygen from the linker (). Compound 9 is a non-binder and compound 10 is toxic on Neuro-2A (IC50 = 53 nM). Both of them are not considered as prospective hits for AChE inhibition.
GAL is a well-known AChE inhibitor. The Kd found by ITC is 388.2 µM. It is a weak base with pKa = 7.92, partially protonated at pH 7.4. Most of it (83%) exists as a free, non-bound fraction in plasma, distributed extensively with VDss of 2.30 L/kg, has moderate clearance (CL = 5.60 mL/min/kg) and half-life of 5.30 h47. GAL easily crosses the intestinal mucosa (pPe GIT =5.060) which corresponds well to its oral bioavailability of 90%Citation52. The value of 5.060 for pPe BBB, found in our previous studyCitation39, indicates for moderate ability to cross the BBB by passive diffusion but mediation by choline transport system has been suggestedCitation55. GAL binds mainly in CAS; only the methoxy group interacts with Phe295, Phe297 and Phe338 from PAS. The pose presented in has RMSD of 1.0396 Å from the X-ray data of the complex GAL-AChECitation35 which is a good validation of the docking protocol used in the present study.
Conclusions
The virtual screening on ZINC database and the following in vitro tests and PK predictions featured seven new hits for acetylcholinesterase inhibition (compounds 1, 2, 3, 5, 6, 7, and 8). The hits are non-toxic, GIT and BBB permeable and bind the enzyme AChE with nanomolar affinity. They could be considered for further lead optimisation.
IENZ_1458031_Supplementary_Materials.pdf
Download PDF (343.2 KB)Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
Related Research Data
References
- Bartus RT, Dean RLIII, Beer B, et al. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982;217:408–14.
- Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol 2000;163:495–529.
- Kumar A, Kumar A, Alzheimer’s disease therapy: present and future molecules. In: Kunal R, ed. Computational modeling of drugs against Alzheimer’s disease. New York: Humana Press; 2018:3–22.
- Ordentlich A, Barak D, Kronman C, et al. Dissection of the human acetylcholinesterase active center determinants of substrate specificity. Identification of residues constituting the anionic site, the hydrophobic site, and the acyl pocket. J Biol Chem 1993;268:17083–95.
- Ordentlich A, Barak D, Kronman C, et al. Functional characteristics of the oxyanion hole in human acetylcholinesterase. J Biol Chem 1998;273:19509–17.
- Radic Z, Pickering NA, Vellom DC, et al. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 1993;32:12074–84.
- Sussman JL, Harel M, Frolow F, et al. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 1991;253:872–9.
- Carvajal FJ, Inestrosa NC. Interactions of AChE with Aβ aggregates in Alzheimer's brain: therapeutic relevance of IDN 5706. Front Mol Neurosci 2011;4:19.
- Geldenhuys WJ, Van der Schyf CJ. Rationally designed multi-targeted agents against neurodegenerative diseases. Curr Med Chem 2013;20:1662–72.
- Viayna E, Sabate R, Muñoz-Torrero D. Dual inhibitors of β-amyloid aggregation and acetylcholinesterase as multi-target anti-Alzheimer drug candidates. Curr Top Med Chem 2013;13:1820–42.
- Rosini M, Simoni E, Minarini A, et al. Multi-target design strategies in the context of Alzheimer’s disease: acetylcholinesterase inhibition and NMDA receptor antagonism as the driving forces. Neurochem Res 2014;39:1914–23.
- Greenblatt HM, Guillou C, Guénard D, et al. The complex of a bivalent derivative of galanthamine with torpedo acetylcholinesterase displays drastic deformation of the active-site gorge: implications for structure-based drug design. J Am Chem Soc 2004;126:15405–11.
- Camps P, Formosa X, Galdeano C, et al. Novel donepezil-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation. J Med Chem 2008;51:3588–98.
- Jia P, Sheng R, Zhang J, et al. Design, synthesis and evaluation of galanthamine derivatives as acetylcholinesterase inhibitors. Eur J Med Chem 2009;44:772–84.
- Bartolucci C, Haller LA, Jordis U, et al. Probing torpedo californica acetylcholinesterase catalytic gorge with two novel bis-functional galanthamine derivatives. J Med Chem 2010;53:745–51.
- Kozurkova M, Hamulakova S, Gazova Z, et al. Neuroactive multifunctional tacrine congeners with cholinesterase, anti-amyloid aggregation and neuroprotective properties. Pharmaceuticals 2011;4:382–418.
- Scherzer-Attali R, Pellarin R, Convertino M, et al. Complete phenotypic recovery of an Alzheimer’s disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS One 2010;5:e11101.
- Bolea I, Juárez-Jiménez J, de Los Ríos C, et al. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer's disease. J Med Chem 2011;54:8251–70.
- Simoni E, Daniele S, Bottegoni G, et al. Combining galantamine and memantine in multitargeted, new chemical entities potentially useful in Alzheimer's disease. J Med Chem 2012;55:9708–21.
- Bautista-Aguilera OM, Esteban G, Chioua M, et al. Multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer's disease: design, synthesis, biochemical evaluation, ADMET, molecular modeling, and QSAR analysis of novel donepezil-pyridyl hybrids. Drug Des Devel Ther 2014;8:1893–910.
- Wang L, Esteban G, Ojima M, et al. Donepezil + propagylamine +8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer's disease. Eur J Med Chem 2014;80:543–61.
- Nepovimova E, Uliassi E, Korabecny J, et al. Multitarget drug design strategy: quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J Med Chem 2014;57:8576–89.
- Korabecny J, Andrs M, Nepovimova E, et al. 7-Methoxytacrine-p-anisidine hybrids as novel dual binding site acetylcholinesterase inhibitors for Alzheimer's disease treatment. Molecules 2015;20:22084–101.
- Singh M, Kaur M, Chadha N, et al. Hybrids: a new paradigm to treat Alzheimer's disease. Mol Divers 2016;20:271–97.
- Piazzi L, Rampa A, Bisi A, et al. 3-(4-{[Benzyl(methyl)amino] methyl}phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation: a dual function lead for Alzheimer's disease therapy. J Med Chem 2003;46:2279–82.
- Hernández-Rodríguez M, Correa-Basurto J, Martínez-Ramos F, et al. Design of multi-target compounds as AChE, BACE1, and amyloid-β(1-42) oligomerization inhibitors: in silico and in vitro studies. J Alzheimers Dis 2014;41:1073–85.
- Meena P, Nemaysh V, Khatri M, et al. Synthesis, biological evaluation and molecular docking study of novel piperidine and piperazine derivatives as multi-targeted agents to treat Alzheimer’s disease. Bioorg Med Chem 2015;23:1135–48.
- Guzior N, Bajda M, Skrok M, et al. Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and β-amyloid aggregation inhibitors with neuroprotective properties. Eur J Med Chem 2015;92:738–49.
- Basile L, Virtual screening in the search of new and potent anti-Alzheimer agents. In: Kunal R, ed. Computational modeling of drugs against Alzheimer’s disease. New York: Humana Press; 2018:107–137.
- Fradera X, Babaoglu K. Overview of methods and strategies for conducting virtual small molecule screening. Curr Protoc Chem Biol 2017;9:196–212.
- Chen Y, Fang L, Peng S, et al. Discovery of a novel acetylcholinesterase inhibitor by structure-based virtual screening techniques. Bioorg Med Chem Lett 2012;22:3181–7.
- Ellman GL, Courtney KD, Andreas V Jr, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Dhanjal JK, Sharma S, Grover A, et al. Use of ligand-based pharmacophore modeling and docking approach to find novel acetylcholinesterase inhibitors for treating Alzheimer's. Biomed Pharmacother 2015;71:146–52.
- Chen Y, Lin H, Yang H, et al. Discovery of new acetylcholinesterase and butyrylcholinesterase inhibitors through structure-based virtual screening. RSC Adv 2017;7:3429–38.
- Cheung J, Rudolph MJ, Burshteun F, et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem 2012;55:10282–6.
- Atanasova M, Yordanov N, Dimitrov I, et al. Molecular docking study on galantamine derivatives as cholinesterase inhibitors. Mol Inf 2015;34:394–403.
- Atanasova M, Stavrakov G, Philipova I, et al. Galantamine derivatives with indole moiety: docking, design, synthesis and acetylcholinesterase activity. Bioorg Med Chem 2015;23:5382–9.
- Stavrakov G, Philipova I, Zheleva D, et al. Docking-based design of galantamine derivatives with dual-site binding to acetylcholinesterase. Mol Inform 2016;35:278–85.
- Stavrakov G, Philipova I, Zheleva-Dimitrova D, et al. Docking-based design and synthesis of galantamine-camphane hybrids as inhibitors of acetylcholinesterase. Chem Biol Drug Des 2017;90:709–18.
- Freyer MW, Lewis EA. Isothermal titration calorimery: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. In: Biophysical Tools for Biologists, volume one: In Vitro Techniques. Methods Cell Biol 2008;84:79–113.
- Di L, Kerns EH, Fan K, et al. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem 2003;38:223–32.
- Momekov G, Ferdinandov D, Bakalova A, et al. In vitro toxicological evaluation of a dinuclear platinum(II) complex with acetate ligands. Arch Toxicol 2006;80:555–60.
- Zhivkova Z, Doytchinova I. Quantitative structure – pharmacokinetics relationships analysis of basic drugs: volume of distribution. J Pharm Pharm Sci 2015;18:515–27.
- Zhivkova Z. Quantitative structure – pharmacokinetics relationships for plasma protein binding of basic drugs. J Pharm Pharm Sci 2017;20:349–59.
- Zhivkova Z, Doytchinova I. Quantitative structure – pharmacokinetic relationships for plasma clearance of basic drugs with consideration of the major elimination pathway. J Pharm Pharm Sci 2017;20:135–47.
- Zhivkova Z. Quantitative structure – pharmacokinetics relationship for the steady state volume of distribution of basic and neutral drugs. World J Pharm Pharm Sci 2018;7:94–105.
- Obach RS, Lombardo F, Waters NJ. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab Dispos 2008;36:1385–405.
- Berellini G, Waters NJ, Lombardo F. In silico prediction of total human plasma clearance. J Chem Inf Model 2012;52:2069–78.
- Roy K, Kar S, Das RN, Statistical methods in QSAR/QSPR. In: Roy K, Kar S, Das RN, eds. A primer on QSAR/QSPR modeling. Fundamental concepts, Springer Briefs in Molecular Science. Cham: Springer; 2015:37–59.
- Zou Y, Li L, Chen W, et al. Virtual screening and structure-based discovery of indole acylguanidines as potent β-secretase (BACE1) inhibitors. Molecules 2013;18:5706–22.
- Karasova JZ, Korabecny J, Zemek F, et al. Acetylcholinesterase inhibitors used or tested in Alzheimer’s disease therapy; their passive diffusion through blood brain barrier: in vitro study. Afr J Pharm Pharmacol 2013;7:1471–80.
- Farlow MR. Clinical pharmacokinetics of galantamine. Clin Pharmacokinet 2003;42:1383–92.
- Boxenbaum H. Interspecies variations in liver weight, hepatic blood flow, and antipyrine intrinsic clearance: extrapolation of data to benzodiazepines and phenytoin. J Pharmacokinet Biopharm 1980;8:165–76.
- Peters SA. Physiologically-Based Pharmacokinetic (PBPK) modeling and simulations: principles, methods, and applications in the pharmaceutical industry. Hoboken (NJ): John Wiley & Sons Inc; 2012:407–421.
- Lee NY, Kang YS. The inhibitory effect of rivastigmine and galantamine on choline transport in brain capillary endothelian cells. Biomol Ther 2010;18:65–70.