
Abstract
Cathepsin K (Cat K), highly expressed in osteoclasts, is a cysteine protease member of the cathepsin lysosomal protease family and has been of increasing interest as a target of medicinal chemistry efforts for its role in bone matrix degradation. Inhibition of the Cat K enzyme reduces bone resorption and thus, has rendered the enzyme as an attractive target for anti-resorptive osteoporosis therapy. Over the past decades, considerable efforts have been made to design and develop highly potent, excellently selective and orally applicable Cat K inhibitors. These inhibitors are derived from synthetic compounds or natural products, some of which have passed preclinical studies and are presently in clinical trials at different stages of advancement. In this review, we briefly summarised the historic development of Cat K inhibitors and discussed the relationship between structures of inhibitors and active sites in Cat K for the purpose of guiding future development of inhibitors.
Introduction
Cathepsin K (Cat K), a member of cysteine proteases, is predominantly expressed in osteoclasts and plays crucial roles in degradation of bone matrix composited of hydroxyapatite and protein, especially type I collagenCitation1. Both decalcification of hydroxyapatite in an acidic microenvironment and degradation of the protein matrix are inevitable during the bone resorption process. Furthermore, the imbalance between bone resorption (influenced by osteoclasts) and bone formation (influenced by osteoblasts) ultimately results in bone loss, which is significantly associated with osteoporosisCitation2,Citation3. Cat K with a relatively restricted expression pattern exhibits high activity against elastin and type I collagen and is obviously responsible for the relation of osteoclastic bone resorption, which leads to the development of Cat K inhibitors for the treatment of diseases characterised by excessive bone, loss such as osteoporosisCitation4,Citation5.
Moreover, with the clear understanding of the structure of Cat K () and wide consideration of Cat K as a novel target for osteoclast-related maladies, many efforts have been devoted to screening the natural products and developing the therapeutically useful inhibitors for treatment of diseases, such as osteoporosis and other bone disorders displaying excessive levels of resorptionCitation6–9. At present, research results demonstrated the strong antiresorptive activity and high selectivity of some inhibitorsCitation10. Furthermore, several Cat K inhibitors under preclinical or clinical investigation for indications such as bone metastases, rheumatoid arthritis or osteoporosis have made optimistic and positive progressesCitation11–13.
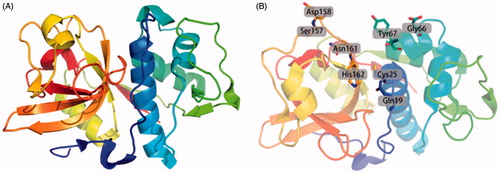
Figure 1. Ribbon drawing of human Cat K and the active sites of Cat K. (A) The overall ribbon structure of human Cat K. The structure is from Protein Data Bank (PDB ID: 5TDI). (B) The residues in active sites of human Cat K.
To date, more than 200 papers on the development of Cat K inhibitors have been published, yielding a large number of compounds with different skeletons, in particular encompassing biologically partial structure of L-leucine. However, only a few reviews on structure–activity relationships of Cat K inhibitors have been publishedCitation14,Citation15. Considering the importance and attractiveness of Cat K inhibitors in pharmaceutical chemistry and preclinical medicine, we would therefore like to deliver a survey on the recent development in structure–function relationship of Cat K inhibitors.
Cat K inhibitors
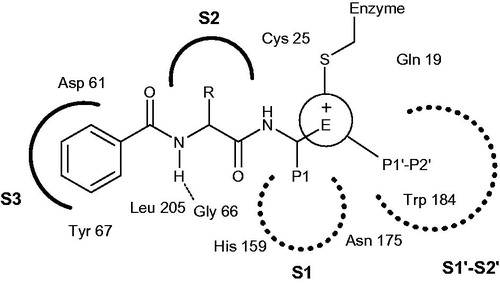
The search and design of potent and selective active-site Cat K inhibitors for human use have been a highly intense and competitive area over the past 20 yearsCitation16–18. By summing up the experimental results, these inhibitors typically consist of an electrophilic group for covalent binding with active sites in cysteine and some addressed regions for enzyme recognition ()Citation7,Citation15. Therefore, according to the available compound sources, the following sections are intended to provide a broad introduction to Cat K inhibitors from literature reports that were generally divided into two classes, synthetic compounds and natural products.
Figure 2. Key binding features of active sites in Cat K.
Cat K inhibitors based on designed synthesis
A large number of competitive efforts have been made, in terms of the disclosure of Cat K as a key role in osteoclast-mediated degradation of bone matrix, in order to design the structures of Cat K inhibitors and evaluate the biological activities and selectivity followed by modulating the chemical moieties for covalent reversible or non-covalent reversible inhibition.
Cat K inhibitors based on ketone warhead
In 1997, Veber et al. reported a series of selective and reversible Cat K inhibitors based on a poorly electrophilic 1,3-bis(acylamino)-2-propanone scaffoldCitation19. Through modelling the interaction of active-sites and simplifying the structure of inhibitors, they developed an accessible symmetrical ketone 1 with a Ki,app value of 22 nM against Cat K (). It was interesting to note that 1 exhibited excellent selectivity over other members of cathepsin family (Ki,app cathepsin L (Cat L), 0.34 μM; cathepsin B (Cat B), 1.3 μM; cathepsin S (Cat S), 0.89 μM)Citation20. N-methyl analog of 1 examined effects of methylation, ketone 2, was 4-fold less active than 1. Whereas, in order to span the distance of both sides of its active site (picking up the Trp184 aromatic interaction) 3, the chemical moiety of Cbz-Leu in 1 substituted by 4-phenoxyphenyl sulfonamide, showed 10-fold more active than their original peptide-based lead.
Figure 3. Molecular structures of Cat K inhibitors based on ketone warhead.
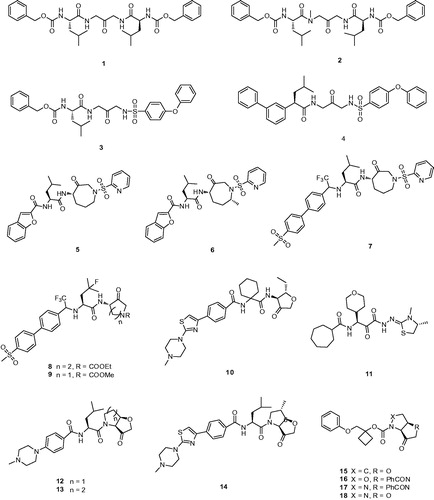
On the other hand, extension of aromatic moiety interacted with Try67, DesJarlais et al.Citation21 developed a variety of sulfonyl inhibitors, among which 4 with the biphenyl group replacing Cbz showed greater than 500-fold selectivity over Cat B, S, L (Ki,app Cat K, 1.4 nM; Cat B, >10000 nM; Cat S, 910 nM; Cat L, >1000 nM) (). The biphenyl group that best matched the conformation of prime side is more rigid and bulky than the benzyl carbamate. From the analysis of X-ray co-crystal structure, the biphenyl system in 4 occupied the S3 site rather than the substrate backbone binding site and formed an aromatic–aromatic interaction with Tyr67.
Marquis et al.Citation22 designed an azepanone-basediInhibitor of Cat K 5, which possessed some special structures including a C-4 chiral center as S and an azepanone ring in a pseudo-boat conformation (). The C-4 S stereochemistry was critical for potent inhibition that predicted the higher energy axial orientation bound within the active site of Cat K by molecular modelling. Compound 5, which incorporated the replacement of the carbonylbenzyloxy group with the benzofuran-2-carboxyamide showed a potently reversible inhibitor of human Cat K with a Ki = 0.16 nM and a relatively acceptable selectivity against Cat B, S, L (Ki,app Cat B, 500 nM; Cat S, 4 nM; Cat L, 2.2 nM). Comparison of the transport of cyclic and acyclic analogs, the results from pharmacokinetic analysis revealed inhibitor 5 with cyclic has good oral bioavailability in the rat of 42% with a T1/2 of 30 min.
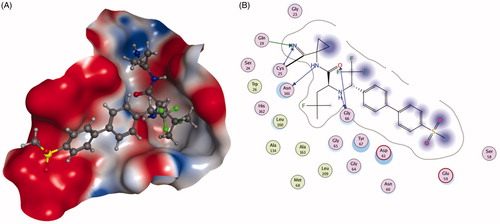
The ketone inhibitors of Cat K pioneered by GSK scientists have been taken a huge number of efforts to realise the desired inhibition and selectivity. The discovery of 6 embodying extremely potent inhibition with picomolar affinity, known as relacatib or SB-462795 (Developed by GSK), was considered as an important milestone ()Citation23. Compound 6 in a chair conformation has an axial methyl group at C-7 position, which contacts with the S1′ hydrophobic pocket, while the sulfonylpyridine interacts with the S2′ hydrophobic pocket. The interactions between compound 6 and Cat K are shown in . Furthermore, conformational analysis revealed that the methyl group at C-4 increased the configurational stability. The 7-methyl substituted azepanone analog shows favorable pharmacokinetic characteristics, good oral bioavailability (89%), and an in vivo clearance rate of 19.5 ml/min/kg. However, in spite of those advantages, compound 6 exhibits a rather low or no selectivity over other off-target cathepsins (Ki,app Cat K, 0.041 nM; Cat L, 0.068 nM; cathepsin V (Cat V), 0.053 nM; Cat B, 15 nM; Cat S, 1.6 nM)Citation24.
Figure 4. The interactions between 6 and Cat K from molecular modelling. (A) The pocket is shown in electrostatics representation. (B) The detailed interactions between 6 and Cat K. The molecular docking is calculated by AutoDock Vina. Green line: Sidechain hydrogen bond; Blue line: Backbone hydrogen bond; Black line: Covalent bond.
During the systemic research of odanacatib, Boyd et al. investigated that the replacement of nitrile with cyclic ketone warheads was based on the experience of ketones as reversible Cat K inhibitorsCitation8,Citation25. Substitution of the benzofuran moiety of compound 5 with an odanacatib-like 4-methylsulfonylphenyl backbone, to provide compound 7, conceivably allowed interactions on the prime sideCitation26. The biphenyl of the substrate intensively participates a ring-ring interaction between Tyr67 at the S3 pocket. In spite of this, compound 7 only furnished improved partial selectivity as well as more than 10-fold reduced inhibition.
Merck Frost also provided a series of ketone inhibitors of various cyclic aminoketone ring size and nitrogen substitution, such as 8, 9 ()Citation27. The IC50 values against humanised rabbit Cat K and selectivity against human Cat L, B, and S exhibited no significant difference compared with inhibitor 7Citation28. Moreover, from the rat bile cannulation study, these inhibitors were rapidly cleared in rats due to cleavage between the amide nitrogen and α-keto stereocenter, as well as oxidation on the ketone heterocyclic.
Medivir UK Ltd. (Little Chesterford, Essex, UK) invented MIV-711, a highly selective Cat K inhibitor; however, the structure of MIV-711 still remains unknown. This company revealed a potent and selective Cat K inhibitor MV061194 (10), analog of MIV-711, which featured a reversible ketone electrophile and a piperazine S3 substitute ()Citation29. With high selectivity, compound 10 was selected as a candidate to be active in reducing bone degradation in preclinical studies (Ki,app Cat K, 2.5 nM; Cat L, >40,000 nM; cathepsin H (Cat H), >4000 nM; Cat B, >4000 nM; Cat S, >40,000 nM)Citation30. Moreover, through degradation of other crucial matrix-embedded growth factors and restricting collagen degradation, compound 10 augmented bone formation probably by preventing Cat K activity.
ONO-5334 (11) (Ono Pharmaceutical, Tokyo, Japan) as a potent inhibitor of Cat K with Ki value of 0.1 nM is undergoing II clinical trials, which showed its inhibitory activity for other cathepsins was 8 to 320-fold lower than that for Cat K (Ki,app Cat K, 0.1 nM; Cat B, 32 nM; Cat L, 17 nM; Cat S, 0.83 nM) ()Citation12,Citation31. Furthermore, 11 has been shown to prevent the decrease in bone mineral density (BMD) in the ovariectomised cynomolgus monkey osteoporosis model and showed a significant increase in BMD compared with placebo and a similar magnitude of suppression on bone resorption compared with the current well known anti-resorptive agents in a 12-month clinical study with postmenopausal osteopenia or osteoporosisCitation32.
Quibell et al.Citation33 stereo-selectively synthesised a series of novel tetrahydrofuro[3,2-b] pyrrol-3-one and hexahydrofuro[3,2-b]pyridine-3-one Cat K inhibitors and demonstrated the cis-fused geometry with better inherent stability compared with the corresponding trans-fused, such as 12 and 13 (). Although the bicyclic structure was in direct contrast with all other scaffolds based upon substrate-like binding sites, the thiolate addition to 12 was predicted to exhibit the same interaction with P1′–P2′ pocket. The compound 12 is one of the successful applications of cis-fused bicycle exhibited as a very potent and selective Cat K inhibitor (Ki,app Cat K, 8.7 nM; Cat L, >10,000 nM; Cat S, >40,000 nM). Some of preferred compounds similar to 12 have been filed by Amura and presented an improved potency and activity in osteoclast resorption assayCitation34,Citation35. Unfortunately, those compounds showed no or little selectivity against other cathepsin enzymes.
Subsequent patents published a series of several novel nonbasic cis-fused 5,5-bicyclic ketone Cat K inhibitors, which possessed a small substituent at the 6-position of the tetrahydrofuro[3,2-b]pyrrol-3-one warheadCitation36,Citation37. By virtue of configurationally stable cis-fusion, these compounds were able to access both S1 and S1′–S2′ binding sites. With differently preferred C-6 substituents including methoxyl, amino, heteroatom, hydroxyl or ethyl group, these inhibitors showed considerable affinityCitation35–37. Several exemplified compounds exhibited sub-nanomolar inhibition for Cat K, such as 14 (Ki app, 0.6 nM). However, from researching the comparable data, the stereochemistry at C-6 showed no obvious influence on potency.
With the embodied advantages of cis-fused bicycle, Amura claimed a series of 5,5-bicyclic ketone Cat K inhibitors, which were based on the replacement of S2 and S3 residues from the aldehyde Cat K inhibitorsCitation38, for example, 15, 16, 17, and 18 (). However, there was no further data provided for describing their inhibition and selectivity.
Cat K inhibitors based on nitrile warhead
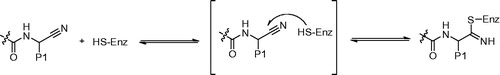
The electrophilic nitrile warhead has been reported to be an important component for inhibitors of Cat B, K, and SCitation39–42. It was well known that the nitrile inhibitors could undergo slow nucleophilic attack of the nitrile moiety to form the reversible thioimidate ester bond between the inhibitor and enzyme (). The nitrile warhead as an effect region interacted with active sites of cysteine protease has received much attention during the last decadesCitation43,Citation44. The achievement for Cat K inhibitors between potency and selectivity is partly depended on controlling the reactivity of the nitrile warhead.
Figure 5. Activity of a nitrile-based Cat K inhibitor is the result of a reversible covalent bond with the active site cysteine of the enzyme.
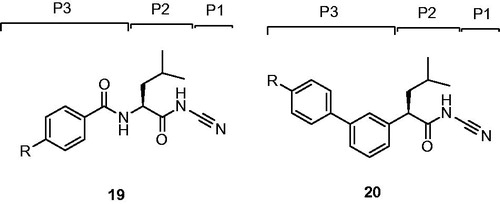
Robichaud et al. Citation46 replaced P3 amide bond of dipeptide inhibitors 19 with a phenyl ring and gave rise to a series of nonpeptidic biaryl compounds 20 ()Citation45.
Figure 6. Schematic representation of the assumed binding mode of Cat K inhibitors.
Replaced compound 21 as a potent inhibitor retained activity against Cat K and showed a significantly improved selectivity profile against the other cathepsins (IC50 Cat K, 3 nM; Cat B, 3950 nM; Cat L, 3725 nM; Cat S, 2010 nM)Citation28. From more experimental data, other alkylated peperazines 22 had similar Cat K potencies but did not show any substantial advantages over the lead unsubstituted piperazine 21 (). Moreover, compound 21, which possessed the inhibition without observably time-dependent, had good pharmacokinetic properties in rhesus monkeys and showed excellent in vivo efficacy in the rhesus monkey model for inhibition of bone resorption.
Figure 7. Molecular structures of Cat K inhibitors based on nitrile warhead.
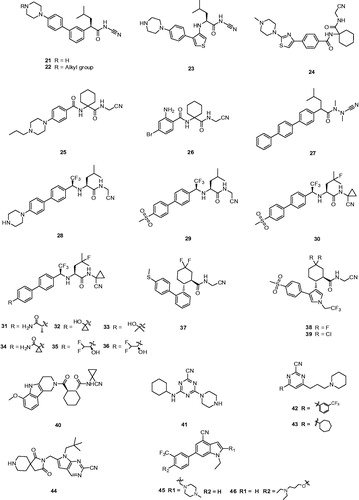
Then, a novel structure of Cat K inhibitor showed that the introduction of a NH linker between P3 aryl and P2 leucinamide moiety formed a H-bond with the Gly66 residue and hopefully enhanced the interaction between inhibitor and Cat K. Preferred compound 23 () showed more than 10-fold improvement in potency over its predecessor 21 and a good selectivity profile against other cathepsins (IC50 Cat K, 0.2 nM; Cat B, 123 nM; Cat L, 352 nM; Cat S, 102 nM)Citation46. From the model deduction, five-membered heterocycle adequately placed the P3 moiety of 23 into the S3 pocket of Cat K.
Meanwhile, a great deal of efforts from researchers has been made to optimise α-amidoacetonitrile – containing Cat K inhibitors by using a dipeptide templateCitation47. A series of inhibitors based on a novel tri-ring benzamide moiety and an aminocyclohexanecarboxylate have been provided by Palmer groupCitation48. For example, the preferred compound 24 (L-006235) (), in which 1,1′-cyclohexyl ring occupying P2 position gave excellent binding affinity combined with high selectivity and reduced hydrolysis of the reversible bond, displayed excellent inhibition of cathepsin as well as good selectivity against other cathepsins (IC50 Cat K, 0.2 nM; Cat B, 1000 nM; Cat L, 6000 nM; Cat S, 47,000 nM). In addition, the pharmacokinetic study with 24 at 300 mg/kg daily treatment demonstrated that achieved drug exposure was sufficient to inhibit >90% of murine Cat K during the study durationCitation49.
Combining previous research experience of introducing a 1,1′-cyclohexyl ring at P2 position and a piperazine moiety at P3 position, researchers from Novartis claimed the N-propylpiperazine substituted compound 25 (Balicatib), also known as AAE-581, in two patents ()Citation50,Citation51. Compound 25 as a basic peptidic nitrile inhibitor showed a potent human Cat K inhibition and significantly more selectivity in cell-based assays against other cathepsins (IC50 Cat K, 1.4 nM; Cat B, 4800 nM; Cat L, 503 nM; Cat S, 65000 nM)Citation52. Balicatib, in a clinical study, showed an increase in bone mineral density and reduction of biochemical markers of bone resorption in the lumbar spine, femur and hips over 18 months of treatmentCitation53. In phase I clinical trials, balicatib had a well dose-dependent suppression of Cat K and was tolerated with 90% suppression at the 25 mg dosage. In phase II clinical trials, a 50 mg dose of balicatib decreased bone resorption markers serum C-terminal cross-linking telopeptides of type I collagen (61%) and urinary N-terminal cross-linking telopeptides of type I collagen (55%) as normalised to creatineat 1 monthCitation54. However, compared to in vitro enzyme assays, selectivity of balicatib was dramatically decreased in cell-based enzyme assays, due to the lysosomotropic characters which results in basic compounds to accumulate in acidic compartmentsCitation52. Because the Cat B and L are highly expressed in the skin and skin-derived cells, balicatib has been reported to lead to incidences of skin rashes, pruritus, and rare morphea-like skin thickeningCitation55. With these reverse results, Novartis has claimed to stop the development of this compound.
In order to avoid such side effects caused by basic character, Marjana et al. designed a less basic N-(functionalized benzoyl)-homocycloleucyl-glycino-nitrile 26 as a Cat K inhibitor, which revealed high affinity for Cat K with Ki values and was highly selective for Cat K when compared with Cat L and S (Ki,app Cat K, 10 nM; Cat L, 11,000 nM; Cat S, >100 μM) ()Citation56. The kinetic studies showed that compound 26 exhibited reversible tight binding to Cat K, while the X-ray structural studies showed covalent and non-covalent binding between the nitrile group and the Cys25 site.
In the subsequent study, Wu et al. reported a new type of Cat K inhibitor 27 with azadipeptide nitrile and without the P2–P3 amide linker, which possessed the favourable balance between potency (Ki = 0.29 nm) and selectivity of Cat K over other cathepsins (Ki,app Cat K, 0.29 nM; Cat L, 93 nM; Cat S, 517 nM; Cat B, 2484 nM) ()Citation57. The study of covalent protein-ligand docking explained the improved selectivity of several representative compounds and this new approach of removing the hydrogen bond donated by the P2–P3 amide linkage of inhibitors to the backbone oxygen of Gly66 in the cathepsin enzymes resulted in highly Cat K-selective aza-nitrile inhibitors.
Black et al. disclosed a novel Cat K inhibitor 28 that a trifluoromethyl group replaced the carbonyl of an amide in P2 position and generated a metabolically stable, non-basic amine ()Citation58,Citation59. Compound 28 maintained the excellent hydrogen bond to Gly66 instead of an amide. Furthermore, it minimised the basicity of an NH donor to avoid formation of NH2+ moiety in the biological milieuCitation60. This compound was an admirable Cat K inhibitor with IC50 < 5 pM and good selectivity over other cathepsin enzymes (IC50 Cat B, 1111 nM; Cat L, 47 nM; Cat S, 451 nM). They have arrived at a conclusion that trifluoroethylamine is an excellent surrogate for the P2 amide bond in the inhibitors of Cat K.
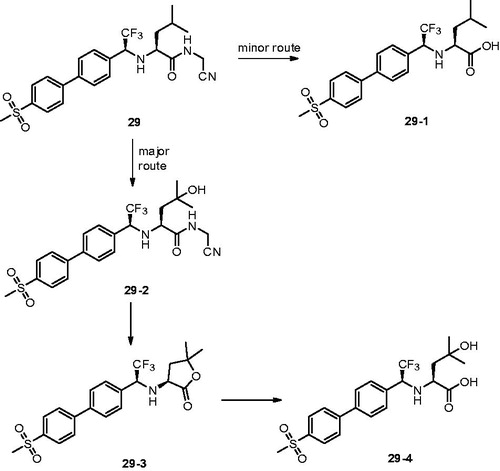
With the same design concept of keeping trifluoroethylamine amide isostere to enhance potency and selectivity, compound 29 (L-873724) was provided as a potent, selective, and orally bioavailable Cat K inhibitor, which possessed a non-basic P3 substituent ()Citation61. The methyl sulfone biphenyl analog 29 showed a high potency in the rabbit bone resorption assay and better selectivity than other cathepsins (IC50 Cat K, 0.2 nM; Cat B, 5239 nM; Cat L, 1480 nM; Cat S, 265 nM). From the X-ray crystal structure research, the phenyl group in compound 29 adjacent to the CF3 group was coplanar with the glycine shelf (Gly66–Gly65) creating a significant hydrophobic interaction. Preliminary pharmacokinetic study revealed a mean decrease of 68% in urinary uNTx/Cr was observed during the six-day on-treatment phase at 3 mg/kg with once-daily oral dosing in the ovariectomised (OVX) rhesus monkey modelCitation62. The short half-life (2 h) and clearance (Cl =7.5 ml/min/kg) in monkey prevented its further development, while compound 29 has two routes of metabolism.
Figure 8. Metabolic pathways for 29 based on in vitro and in vivo studies.
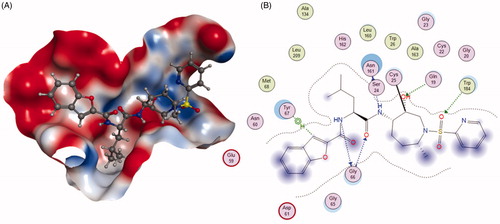
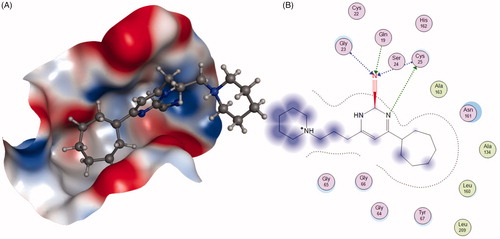
To address the metabolic liability, substituting P1 and modifying P2 moiety of inhibitor L-873724, to give 30 (odanacatib), produced a metabolically robust inhibitor with a long half-life in preclinical species ()Citation63. The odanacatib directly binds with the side chain of Cys25 of Cat K by forming a covalent bond. The nitrogen atom near the CF3 group and the residue Gly66 forms a hydrogen bond. Besides, the Asn161 forms a hydrogen bond with amide group of odanacatib. Furthermore, Gln19 and Gly66 also form hydrogen bonds with odanacatib, as shown in Citation64. The 4-fluoroleucine side chain at P2 position interacting within the S2 pocket played a crucial role in enhancing the potency and selectivity of odanacatib, which was evaluated in whole cell enzyme occupancy assays (IC50 Cat K, 0.2 nM; Cat B, 1034 nM; Cat L, 2995 nM; Cat S, 60 nM)Citation65. Compared with balicatib and relacatib, odanacatib still displayed a high potency for decreasing bone resorption markers and increasing BMD in rabbit and monkey ovariectomised osteoporosis modelsCitation66. The randomised placebo-controlled phase I studies showed odanacatib as oral Cat K inhibitor was well tolerated by postmenopausal female, who were treated for 3 weeks (at doses of 5, 25, 50, or 100 mg) or once daily for 21 days (at doses of 0.5, 2.5, or 10 mg) without any significant adverse effectsCitation67,Citation68. Interesting, data from phase II indicated women receiving combinations of odanacatib (10–50 mg) for 5 years had gains in spine and hip BMD and showed larger reductions in bone resorption than bone formation markersCitation69. Moreover, more than 16,000 patients received odanacatib 50 mg weekly, which increased BMD at the lumbar spine and total hip after 5 years by 11.2 and 9.5% in the phase III trial, respectively, and reduced the risk of hip fracture by 47%, non-vertebral fractures by 23%, and clinical vertebral fractures by 72%Citation70,Citation71. However, odanacatib has been discontinued due to a small increased risk of stroke in the postmenopausal patients, even possessing robust efficacy for treatment of osteoporosisCitation72,Citation73.
Figure 9. The interactions between 30 and Cat K from molecular modelling. (A) The pocket is shown in electrostatics representation. (B) The detailed interactions between 30 and Cat K. The molecular docking is calculated by AutoDock Vina. Green line: Sidechain hydrogen bond; Blue line: Backbone hydrogen bond; Black line: Covalent bond.
Further exploring the strategy that maintained the beneficial features of odanacatib in P1 (cyclopropane) and P2 (fluoroleucine), Isabel et al.Citation74 disclosed a series of odanacatib analogs 31–36 (), with various substituted groups in P3 in order to seek novel potent and selective inhibitors. The experimental data in vitro or in vivo revealed compound 31 with good potency and selectivity suffered from poor oral bioavailability and a complicated metabolic pathway, which led to significant developmental challenges (IC50 Cat K, 0.2 nM; Cat B, 288 nM; cathepsin F (Cat F), 718 nM; Cat L, 4266 nM; Cat S, 138 nM). Compound 32 with an alcohol moiety at the benzylic position was lack of selectivity of Cat S (IC50 Cat S, 21 nM). Although the dimethyl carbinol analog 33 was highly selective against Cat S with moderate bioavailability in rats of 24%, 33 suffered from a half-life in rats of less than 1 h. Compound 34 failed due to its poor bioavailability in rats (8%) and squirrel monkeys (5%). Interestingly, a pair of enantiomerically pure alcohols, 35 and 36, were potent and highly selective in in vitro assays and exhibited high oral bioavailability as well as long half-lives in rats, rabbits, and rhesus monkeys.
Crane et al.Citation75 described a series of novel cycloalkylcarboxamides compounds as Cat K inhibitors, in which P2 amide bond was replaced by the cyclohexane moiety, e.g. Citation37 (). The potent enantiomer 37 retained an acceptable selectivity profile against cathepsins L, B, and S (IC50 Cat K, 0.28 nM; Cat B, 10080 nM; Cat L, 218 nM; Cat S, 263 nM), and exhibited good potency in the functional bone resorption assay that evaluated the degradation of the type I collagen matrix of bovine bone by isolated rabbit osteoclastsCitation76. It appeared that both fluorine substituents in 37 acted cooperatively to produce a synergistic improvement of inhibitor binding to active site of S2 in the Cat K.
Subsequent to the preliminary research, further development found introduction of a methyl sulfone P3-subsitutent and incorporation of five-membered heterocycles as P2–P3 linkers could improve the potency and selectivity of inhibitor, such as 38, 39, which were minimally shifted in the bone resorption assay ()Citation77,Citation78. Compounds 38 and 39 were quite selective against a wider panel of human lysosomal cysteine proteases including human cathepsins (IC50 Cat K, 0.6 nM; Cat B, >9600 nM; Cat L, >2700 nM; Cat S, >2500 nM). Both 38 and 39 displayed acceptable PK profiles in multiple animal species, however, compound 39 exhibited superior bioavailability and half-life.
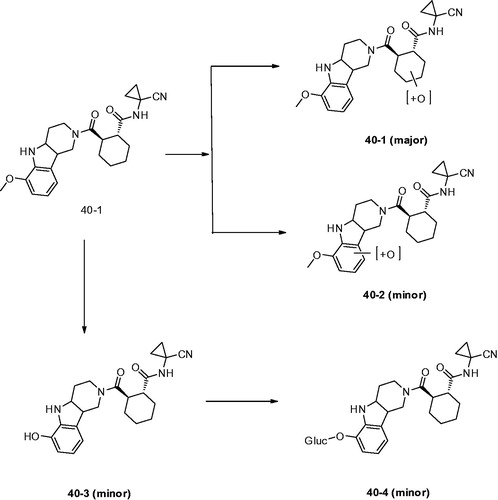
Dossetter et al.Citation79 optimised the structures by using small substituents and selected AZD4996 (40) as a highly potent and selective Cat K inhibitor (IC50 Cat K, <1 nM; Cat B, 2970 nM) (). The study of incubation of 40 with rat haptocytes showed a switch of major metabolite to oxidation of the cyclohexyl ring (40–1) and reduced oxidation of the carboline aromatic (40–2). In addition, a new metabolic route by oxidative demethylation and conjugation was found (40–3 and 40–4) compared with other compounds although they were minor metabolites (). They revealed key SAR and demonstrated that baseline physical properties and in vitro stability by synthesis of compounds with reduced molecular complexity.
Figure 10. Metabolites found after in vitro incubation of 40 with rat hepatocytes.
Despite the amidoacetonitrile warheads dominate the centre position in the development of Cat K inhibitors, a number of inhibitors based on nitrile heteroaryl chemotypes were still investigated and exhibited excellent potency and selectivityCitation80,Citation81. Novel Cat K inhibitors have been developed based on a 2-Cyano-pyrimidines or triazine. The triazine derivative 41 () as a Cat K inhibitor exhibited high potency against Cat K and selectivity over other cathepsins (IC50 Cat K, 1 nM; Cat B, 520 nM; Cat L, 1711 nM; Cat S, 158 nM)Citation82. Although 41 showed good stability in human microsomes (t½ >120 min) and hepatocytes (t½ = 118 min), it was rapidly cleared in rodent microsomes (rat t½ = 23 min; mouse t½ = 4 min) and hepatocytes (rat t½ = 6 min).
Rankovic et al.Citation83 disclosed a novel series of 2-cyano-pyrimidines as potent inhibitors of Cat K. Compound 42 () incorporating a piperidinyl group not only improved Cat K potency (IC50 Cat K, 4 nM; Cat B, 4200 nM; Cat L, 10,000 nM; Cat S, 23 nM), but also enhanced solubility (from <1 mg/L to 379 mg/L). Interestingly, 3-CF3-phenyl in 42 was replaced with cycloheptyl ring and obtained compound 43, which showed lower potency but higher selectivity profile against cathepsins (IC50 Cat K, 100 nM; Cat B, >10,000 nM; Cat L, 10,000 nM; Cat S, 10,000 nM). From the analysis of X-ray structure, the cycloheptyl ring bound in the S2 pocket, but not as deeply as the 3-CF3-phenyl. Moreover, the S2 pocket is slightly narrower in Cat S, which could explain the greater Cat S selectivity displayed by compound 43, as shown in .
Figure 11. The interactions between 43 and Cat K from molecular modelling. (A) The pocket is shown in electrostatics representation. (B) The detailed interactions between 43 and Cat K. Note: The molecular docking is calculated by AutoDock Vina. Green line: Sidechain hydrogen bond; Blue line: Backbone hydrogen bond.
Meanwhile, an additional series of Cat K inhibitors have been disclosed in which the central pyrimidine moiety was replaced with an imidazopyridine scaffoldCitation84. Compound 44 () as a promising Cat K inhibitor exhibited good inhibition activity and selectivity against other cathpsins (IC50 Cat K, <1 nM; Cat S, >1000 nM; Cat L, >1000 nM)Citation85. It prevented collagen degradation by 73.2% while produced a high concentration in serum. Meanwhile, compound 44 strongly reduced rat CTx with a high concentration even 25 h after administration in the target tissue and showed no signs of toxicity on prolonged administration. In the subsequent patents, extension of C6 and 3-trifluoromethylphenyl substituents has been claimed to provide some dual Cat K/S inhibitors, such as 45 and 46. It is interestingly observed that moving the basic functionality from imidazopyridine moiety to phenyl moiety can skew the potent selectivity toward Cat SCitation86.
Cat K inhibitors based on non-covalent interaction
Besides the ketone-based and nitrile-based inhibitors, researchers were also developing non-covalent amide derivatives as potent Cat K inhibitors. Those kinds of compounds do not possess an electrophilic warhead, which could form a covalent reversible bond with the active cysteine residue. However, the lipophilic interactions between aminoethylaniline moiety of the inhibitors and the prime sites in the Cat K furnish to some extent the binding affinity of these non-covalent reversible inhibitors. Meanwhile, the isobutyl or 1,1-cyclothexyl group preferred at P2 and ethyl group preferred at P1 are forming van der Waals with Leu157, Tyr67 etc.Citation7,Citation87.
In order to form a highly favourable ionic bonding interaction with Asp61, Setti et al. designed a 3,4-disubstituted azetidinones compound 47 with a basic substituent in the S3 pocket ()Citation88. Compound 47 exerted its inhibition through direct interaction of Cat K active site with the C-2 carbonyl of the β-lactam, similar to β-lactam reaction with serine proteasesCitation89. In addition, 47 exhibited excellent potency and selectivity against other cathepsins (IC50 Cat K, 4.8 nM; Cat B, 340 nM; Cat L, 2400 nM; Cat S, 17,000 nM), but rat pharmacokinetic results showed 47 possessed high clearance (92 ml/min/kg) and a short MRT (17 min). According to docking result, the Asn161 forms a hydrogen bond with amide group of 47. In addition, the nitrogen of azetidinone group forms a hydrogen bond with Gly20. The oxygen atom on azetidinone group forms a hydrogen bond with Gln19 ().
Figure 12. Molecular structures of Cat K inhibitors based on non-covalent interaction.
Figure 13. The interactions between 47 and Cat K from molecular modelling. (A) The pocket is shown in electrostatics representation. (B) The detailed interactions between 47 and Cat K. The molecular docking is calculated by AutoDock Vina. Green line: Sidechain hydrogen bond; Blue line: Backbone hydrogen bond.
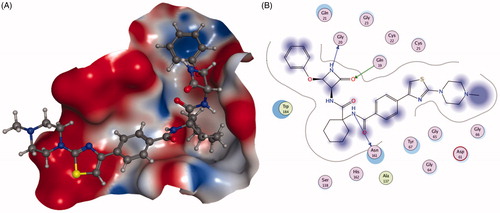
Altmann et al.Citation90 reported the discovery of arylaminoethyl amides as novel non-covalent inhibitors of Cat K and probed the structure–activity relationship through incorporation of extended P1 substituents and the replacement of the P3 moiety, exemplifying compound 48 (). Compound 48 emerged as the most attractive analog revealed high potency for Cat K inhibition and favourable selectivity profile (IC50 Cat K, <3 nM; Cat L,>10,000 nM; Cat S, >10,000 nM)Citation91.
Further optimisation of the structure based on the strategy discovered the morpholine derivative 49 (), which showed the good Cat K inhibition potency and excellent selectivity over Cat B and L except Cat S (IC50 Cat K, 6.8 nM; Cat B, >20,000 nM; Cat L, >20,000 nM; Cat S, 13 nM)Citation92.
Sankyo Co., Ltd claimed the 4-aminophenoxyacetic acid 50 as a novel Cat K inhibitor by optimizing the P1, P3, and P1′ units ()Citation93. The selectivity of inhibitor 50 was achieved as likely to be lack of a covalent interaction with the catalytic cysteine and the potency was maintained on both human and rat (IC50 Cat K, 4.8 nM; Cat B,>20,000 nM; Cat L, >20,000 nM; Cat S, 10,000 nM). With biochemical potency on the rat protein and reasonable PK following 50 mg/kg oral dosing (Cmax = 9.2 μg/mL; AUC =103.9 μgh/mL), 50 showed a trend toward increased BMD in OVX rat femur following b.i.d. dosing for 42 daysCitation14.
Preferred compound 51 () as one of a series of derivatives exemplified was claimed with replacement of trifluorethylamine in the P2–P3 moietyCitation94. The non-covalent inhibitor 51 showed the different activity ranges of Cat K inhibition.
With the fast development of computational method for discovery of various inhibitors, Antonio et al. identified the first small-molecule allosteric Cat K inhibitor 52 by high-throughput docking of compound libraries to surface sites on the peptidase ()Citation95. The crystal structure revealed 52 bound to a novel allosteric site on Cat K. Moreover, this compound completely inhibited collagen degradation and had good selectivity for Cat K over related enzymes (IC50 Cat K, 80 μM; Cat B, 2300 μM; Cat L, 1600 μM; Cat S, 3300 μM; Cat V, 7700 μM) .
Through screening, Hiroshi et al. obtained a potent orally active Cat K inhibitor 53, which suppressed osteoclastic bone resorption both in vivo and in vitroCitation96. Computer-assisted simulation of the Cat K/53 complex indicated that 53 blocked the active-site cleft where Cys25 and His162 of Cat K form the catalytic site (). Compound 53 with good selectivity is slightly acidic in aqueous solution and thus considered to have less off-target effects than basic inhibitors (IC50 Cat K, 34.5 nM; Cat B, 284 nM; Cat L, 582 nM; Cat S, 186 nM).
Cat K inhibitors based on natural products
As part of a search for novel inhibitors, traditional Chinese medicine has been used as an important source for treatment of various diseasesCitation97,Citation98. Rhizoma Drynariae (RD) known as “Gu-Sui-Bu” in folk medicine has been frequently utilised in the treatment of bone-related diseases in clinical formulasCitation99–101. Some natural products with novel structures from traditional Chinese medicine, microorganisms, or marine organisms exhibited good potency against Cat KCitation6,Citation102.
Haploscleridamine (54) () as a novel tryptamine-derived tetrahydro-β-carboline alkaloid was extracted and characterised by Patil et al.Citation103, which displayed the moderate activity against Cat K with an IC50 of 26 μM. Unfortunately, no information about selectivity and structureactivity relationship on this inhibitor of Cat K was supplied.
Figure 14. Molecular structures of Cat K inhibitors based on natural products.
Park et al. isolated a furanquinone compound 55 from Paulownia tomentosa stem ()Citation103. Compound 55 showed an IC50 value of 21 μM for Cat K. However, it was also found to be capable of inhibiting Cat L, which was closely related to Cat K. Moreover, 5-hydroxyl functional group of 55 could play an important role in the reduction potential of its Cat K inhibitory activity. Although this inhibitory potency of 55 might not be strong enough, it was believed that it still had the potential to be a candidate for designing a new class of Cat K inhibitors.
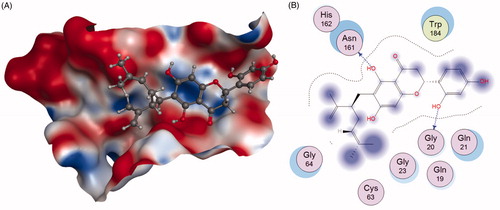
Qiu et al.Citation104 successfully identified Kushennol F(56) and Sophoraflavanone G(57) to be the active ingredients in Rhizoma drynariae that specifically acted on lysosomal enzyme Cat K. Compounds 56 and 57 were shown to suppress Cat K activities with IC50 of 8.80 and 27.24 µM, respectively (). Using molecular docking and dynamics method, the interactions between these two compounds and protease Cat K were confirmed. As shown in , compound 56 formed two hydrogen bonds with Gly20 and Asn161 of Cat K, respectively.
Figure 15. The interactions between 56 and Cat K from molecular modelling. (A) The pocket is shown in electrostatics representation. (B) The detailed interactions between 56 and Cat K. The molecular docking is calculated by AutoDock Vina. Blue line: Backbone hydrogen bond.
Screening natural product libraries, Brömme et al. identified para-dihydrotanshinone (DHT) 58 as a specific collagenase inhibitor of Cat K that did not interfere with the degradation of other biologically relevant substrates ()Citation102. Meanwhile, 58 revealed a similar outcome of selective inhibition of the collagenase activity of Cat K in osteoclasts when compared with odanacatib (IC50 = 6.2 µM)Citation105. The study of molecular docking and binding experiments suggested that 58 bound to a specific exosite in Cat K, which was crucial for the formation of collagenolytically active oligomers in the presence of GAGsCitation106.
Icariin (59), the major pharmacologically active flavonol diglycoside contained in the Epimedium family, had bone-strengthening properties and has been used to promote healing of bone fractures or prevent osteoporosis ()Citation107. Moreover, Hsieh et al.Citation108 reported 59 had the inhibition of osteoclast differentiation and bone resorption. Pang et al. revealed 59 with the maximum concentration tested 1 mM had 64% inhibition of Cat K activityCitation109. Unfortunately, there was no further report about the selectivity compared with other cathepsins.
Conclusions
Over the past decades, especially the confirmation of tight relationship between Cat K and bone-related diseases like osteoporosis, a large number of efforts have been devoted on developing the Cat K inhibitors. The nitrile and ketone warheads particularly played the prominent roles. Cat K possesses a series of active residues, among which catalytic cysteine Cys25 forms an irreversible or reversible covalent bond with different kinds of inhibitors through a hydrazide, vinylsulfone, epoxide, aldehyde, ketone, or nitrile. Irreversible covalent inhibitors at early stage used in preclinical study led to antigenic and immunologic complications characterised by the results of pcynodysostosis in humans and osteopetrosis in mouse modelCitation8,Citation110,Citation111. Subsequently, reversible covalent or non-covalent inhibitors started another chapter for the research of Cat K. Introducing basic groups or modulating basic functionality in the S3 pocket could compensate for decreased affinity of the non-covalent binder for Cat K. It was noted that the basic nitrogen provided the potential to interact with Asp61 in the S3 pocket of Cat K resulting in increased potency and selectivityCitation112. However, the inhibitors containing excessively basic functionality could reach concentrations in acidic lysosome and be sufficient to cause other off-target effects despite excellent biochemical selectivity. It is essential to control selectivity by making use of of non-covalent interactions for the initial molecular recognition. In addition, creating the suitable chemical moiety renders other interaction, like van der waals between P2 substituent and Leu157, Tyr67 in S2 pocket. Moreover, excellent binding affinity could be achieved by making use of the reversible binding nitrile and ketone warheads. In the view of overall potency, the synthesised compounds have obvious advantages against the Cat K in the selectivity and inhibition. However, some compounds in the preclinical or clinical trials were still facing with the challenges in unavoidable side effects and limitations. Some strategies of altering the delivery systems were demonstrated to improve the bioavailability and reduce side effects by forming the conjugate via encapsulation or suitable linkersCitation113–116. In a word, a large number of synthesised compounds and characterised natural products offered a relative clear structure–activity relationship for the future design and development of Cat K inhibitors.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Bossard MJ, Tomaszek TA, Thompson SK, et al. Proteolytic activity of human osteoclast cathepsin K expression, purification, activation, and substrate identification. J Biol Chem 1996;271:12517–24.
- Garnero P, Borel O, Byrjalsen I, et al. The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J Biol Chem 1998;273:32347–52.
- Consensus A. Consensus development conference: diagnosis, prophylaxis, and treatment of osteoporosis. Am J Med 1993;94:646–50.
- Brömme D, Okamoto K, Wang BB, Biroc S. Human cathepsin O2, a matrix protein-degrading cysteine protease expressed in osteoclasts functional expression of human cathepsin O2 in Spodoptera frugiperda and characterization of the enzyme. J Biol Chem 1996;271:2126–32.
- Zaidi M, Troen B, Moonga BS, Abe E. Cathepsin K, osteoclastic resorption, and osteoporosis therapy. J Bone Miner Res 2001;16:1747–9.
- Patil AD, Freyer AJ, Killmer L, et al. A new dimeric dihydrochalcone and a new prenylated flavone from the bud covers of Artocarpus altilis: potent inhibitors of Cathepsin K. J Nat Prod 2002;65:624–7.
- McGrath ME, Klaus JL, Barnes MG, Brömme D. Crystal structure of human cathepsin K complexed with a potent inhibitor. Nat Struct Mol Biol 1997;4:105–9.
- Stoch S, Wagner J. Cathepsin K inhibitors: a novel target for osteoporosis therapy. Clin Pharmacol Ther 2008;83:172–6.
- Mazid Helali A, Matin Iti F, Naina Mohamed I. Cathepsin K inhibitors: a novel target but promising approach in the treatment of osteoporosis. Curr Drug Targets 2013;14:1591–600.
- Duong LT, Leung AT, Langdahl B. Cathepsin K inhibition: a new mechanism for the treatment of osteoporosis. Calcif Tissue Int 2016;98:381–97.
- Hussein H, Dulin J, Smanik L, et al. Repeated oral administration of a cathepsin K inhibitor significantly suppresses bone resorption in exercising horses with evidence of increased bone formation and maintained bone turnover. J Vet Pharmacol Ther 2017;40:327–34.
- Tanaka M, Hashimoto Y, Hasegawa C, et al. Antiresorptive effect of a cathepsin K inhibitor ONO-5334 and its relationship to BMD increase in a phase II trial for postmenopausal osteoporosis. BMC Musculoskelet Disord 2017;18:267.
- Tu L, Rui K, Feng H, Wang Z. Safety of the cathepsin K inhibitor odanacatib in postmenopausal women with osteopenia or osteoporosis: a meta-analysis. Int J Clin Exp Med 2017;10:5977–84.
- Allen JG, Fotsch C, Babij P. Emerging targets in osteoporosis disease modification. J Med Chem 2010;53:4332–53.
- Wijkmans J, Gossen J. Inhibitors of cathepsin K: a patent review (2004–2010). Expert Opin Ther Pat 2011;21:1611–29.
- Yamashita DS, Dodds RA. Cathepsin K and the design of inhibitors of cathepsin K. Curr Pharm Des 2000;6:1–24.
- Yasuda Y, Kaleta J, Brömme D. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev 2005;57:973–93.
- Kim TS, Tasker AS. Non-covalent cathepsin K inhibitors for the treatment of osteoporosis. Curr Top Med Chem 2006;6:355–60.
- Yamashita DS, Smith WW, Zhao B, et al. Structure and design of potent and selective cathepsin K inhibitors. JACS 1997;119:11351–2.
- Shi G-P, Munger J, Meara J, et al. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J Biol Chem 1992;267:7258–62.
- DesJarlais RL, Yamashita DS, Oh HJ, et al. Use of X-ray co-crystal structures and molecular modeling to design potent and selective non-peptide inhibitors of cathepsin K. JACS 1998;120:9114–15.
- Marquis RW, Ru Y, LoCastro SM, et al. Azepanone-based inhibitors of human and rat cathepsin K. J Med Chem 2001;44:1380–95.
- Yamashita DS, Marquis RW, Xie R, et al. Structure activity relationships of 5-, 6-, and 7-methyl-substituted azepan-3-one cathepsin K inhibitors. J Med Chem 2006;49:1597–612.
- Kumar S, Dare L, Vasko-Moser J, et al. A highly potent inhibitor of cathepsin K (relacatib) reduces biomarkers of bone resorption both in vitro and in an acute model of elevated bone turnover in vivo in monkeys. Bone 2007;40:122–31.
- Marquis RW, Ru Y, Zeng J, et al. Cyclic ketone inhibitors of the cysteine protease cathepsin K. J Med Chem 2001;44:725–36.
- Boyd MJ, Crane SN, Robichaud J, et al. Investigation of ketone warheads as alternatives to the nitrile for preparation of potent and selective cathepsin K inhibitors. Bioorg Med Chem Lett 2009;19:675–9.
- Prasit P, Bayly CI, Robichaud JS, et al. Cathepsin cysteine protease inhibitors: Google Patents; 2006.
- Robichaud J, Oballa R, Prasit P, et al. A novel class of nonpeptidic biaryl inhibitors of human cathepsin K. J Med Chem 2003;46:3709–27.
- Grabowskal U, Chambers T, Shiroo M. Recent developments in cathepsin K inhibitor design. Curr Opin Drug Discovery Dev 2005;8:619–30.
- Fuller K, Lawrence KM, Ross JL, et al. Cathepsin K inhibitors prevent matrix-derived growth factor degradation by human osteoclasts. Bone 2008;42:200–11.
- Ochi Y, Yamada H, Mori H, et al. Effects of ONO-5334, a novel orally-active inhibitor of cathepsin K, on bone metabolism. Bone 2011;49:1351–6.
- Eastell R, Nagase S, Small M, et al. Effect of ONO-5334 on bone mineral density and biochemical markers of bone turnover in postmenopausal osteoporosis: 2-year results from the OCEAN study. J Bone Mine Res 2014;29:458–66.
- Quibell M, Benn A, Flinn N, et al. Bicyclic peptidomimetic tetrahydrofuro [3, 2-b] pyrrol-3-one and hexahydrofuro [3, 2-b] pyridine-3-one based scaffolds: synthesis and cysteinyl proteinase inhibition. Bioorg Med Chem 2004;12:5689–710.
- Quibell M, Watts JP, Furo [3, 2-b] pyrrol derivatives: Google Patents; 2010.
- Quibell M, Watts JP, Furo [3, 2-B] pyrrol-3-one derivatives and their use as cysteinyl proteinase inhibitors: Google Patents; 2014.
- Quibell M, Watts JP, Tetrahydrofuro (3, 2-B) pyrrol-3-one derivatives as inhibitors of cysteine proteinases: Google Patents; 2010.
- Quibell M, Watts JP, Tetrahydrofuro [3, 2-B] pyrrol-3-ones as cathepsin K inhibitors: Google Patents; 2010.
- Boros EE, Deaton DN, Hassell AM, et al. Exploration of the P 2–P 3 SAR of aldehyde cathepsin K inhibitors. Bioorg Med Chem Lett 2004;14:3425–9.
- Buysse A, Mendonca R, Palmer J, et al. Novel compounds and compositions as protease inhibitors: Google Patents; 2002.
- Altmann E, Betschart C, Gohda K, et al. Dipeptide nitriles: Google Patents; 2002.
- Greenspan PD, Clark KL, Tommasi RA, et al. Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. J Med Chem 2001;44:4524–34.
- Ward YD, Thomson DS, Frye LL, et al. Design and synthesis of dipeptide nitriles as reversible and potent cathepsin S inhibitors. J Med Chem 2002;45:5471–82.
- Lewis Jr CA, Wolfenden R. Thiohemiacetal formation by inhibitory aldehydes at the active site of papain. Biochemistry 1977;16:4890–5.
- Moon JB, Coleman RS, Hanzlik RP. Reversible covalent inhibition of papain by a peptide nitrile. Carbon-13 NMR evidence for a thioimidate ester adduct. JACS 1986;108:1350–1.
- Suzue S, Irikura T. Studies on hepatic agents. I. Synthesis of aminoacyl (and hydroxyacyl) aminoacetonitriles. Chem Pharm Bull 1968;16:1417–32.
- Robichaud J, Bayly C, Oballa R, et al. Rational design of potent and selective NH-linked aryl/heteroaryl cathepsin K inhibitors. Bioorg Med Chem Lett 2004;14:4291–5.
- Altmann E, Aichholz R, Betschart C, et al. Dipeptide nitrile inhibitors of cathepsin K. Bioorg Med Chem Lett 2006;16:2549–54.
- Palmer JT, Bryant C, Wang D-X, et al. Design and synthesis of tri-ring P3 benzamide-containing aminonitriles as potent, selective, orally effective inhibitors of cathepsin K. J Med Chem 2005;48:7520–34.
- Soung DY, Gentile MA, Duong LT, Drissi H. Effects of pharmacological inhibition of cathepsin K on fracture repair in mice. Bone 2013;55:248–55.
- Zimmermann J, Goessl C, Combinations of a cathepsin k inhibitor and a bisphosphonate in the treatment of bone metastasis, tumor growth and tumor-induced bone loss: Google Patents; 2004.
- Novartis A, Use of cathepsin K inhibitors in severe bone loss diseases. WO05049028; 2005.
- Falgueyret JP, Desmarais S, Oballa R, et al. Lysosomotropism of basic cathepsin K inhibitors contributes to increased cellular potencies against off-target cathepsins and reduced functional selectivity. J Med Chem 2005;48:7535–43.
- Adami S, Supronik J, Hala T, et al. Effect of one year treatment with the cathepsin-K inhibitor, balicatib, on bone mineral density (BMD) in postmenopausal women with osteopenia/osteoporosis. J Bone Miner Res 2006;21:S24.
- Papanastasiou P, Ortmann C, Olson M, et al. Effect of three month treatment with the cathepsin-k inhibitor, balicatib, on biochemical markers of bone turnover in postmenopausal women: evidence for uncoupling of bone resorption and bone formation. J Bone Miner Res 2006;21:S59.
- Bühling F, Röcken C, Brasch F, et al. Pivotal role of cathepsin K in lung fibrosis. Am J Pathol 2004;164:2203–16.
- Borišek J, Vizovišek M, Sosnowski P, et al. Development of N-(Functionalized benzoyl)-homocycloleucyl-glycinonitriles as potent cathepsin K inhibitors. J Med Chem 2015;58:6928–37.
- Yuan X-Y, Fu D-Y, Ren X-F, et al. Highly selective aza-nitrile inhibitors for cathepsin K, structural optimization and molecular modeling. Org Biomolec Chem 2013;11:5847–52.
- Gante J. Peptidomimetics-tailored enzyme inhibitors. Angew Chem Int Ed 1994;33:1699–720.
- Volonterio A, Bellosta S, Bravin F, et al. Synthesis, structure and conformation of partially-modified retro-and retro-inverso [NHCH (CF3)] Gly peptides. Chem Eur J 2003;9:4510–22.
- Black WC, Bayly CI, Davis DE, et al. Trifluoroethylamines as amide isosteres in inhibitors of cathepsin K. Bioorg Med Chem Lett 2005;15:4741–4.
- Li CS, Deschenes D, Desmarais S, et al. Identification of a potent and selective non-basic cathepsin K inhibitor. Bioorg Med Chem Lett 2006;16:1985–9.
- Guay J, Riendeau D, Mancini J. Cloning and expression of rhesus monkey cathepsin K. Bone 1999;25:205–9.
- Gauthier JY, Chauret N, Cromlish W, et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett.
- Law S, Andrault P-M, Aguda AH, et al. Identification of mouse cathepsin K structural elements that regulate the potency of odanacatib. Biochem J 2017;474:851–64.
- Falgueyret J-P, Black WC, Cromlish W, et al. An activity-based probe for the determination of cysteine cathepsin protease activities in whole cells. Analyt Biochem 2004;335:218–27.
- Pennypacker BL, Duong LT, Cusick TE, et al. Cathepsin K inhibitors prevent bone loss in estrogen‐deficient rabbits. J Bone Miner Res 2011;26:252–62.
- Eisman JA, Bone HG, Hosking DJ, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: three-year continued therapy and resolution of effect. J Bone Miner Res 2011;26:242–51.
- Stoch S, Zajic S, Stone J, et al. Effect of the cathepsin K inhibitor odanacatib on bone resorption biomarkers in healthy postmenopausal women: two double‐blind, randomized, placebo-controlled phase I studies. Clin Pharmacol Ther 2009;86:175–82.
- Langdahl B, Binkley N, Bone H, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase 2 study. J Bone Miner Res 2012;27:2251–8.
- Mcclung MR, Langdahl B, Papapoulos S, et al. Odanacatib anti-fracture efficacy and safety in postmenopausal women with osteoporosis: results from the phase III long-term odanacatib fracture trial. Arthritis Rheum 2014;66:S987.
- Harsløf T, Langdahl BL. New horizons in osteoporosis therapies. Curr Opin Pharmacol 2016;28:38–42.
- Cabal A, Williams DS, Jayakar RY, et al. Long-term treatment with odanacatib maintains normal trabecular biomechanical properties in ovariectomized adult monkeys as demonstrated by micro-CT-based finite element analysis. Bone Rep 2017;6:26–33.
- Hirsch HD, Sikon A, Thacker HL. Osteoporosis for the female patient. In: Falcone T, Hurd W, eds. Clinical reproductive medicine and surgery. Cham (Switzerland): Springer; 2017:195–208.
- Isabel E, Bateman KP, Chauret N, et al. The discovery of MK-0674, an orally bioavailable cathepsin K inhibitor. Bioorg Med Chem Lett 2010;20:887–92.
- Crane SN, Black WC, Palmer JT, et al. β-Substituted cyclohexanecarboxamide: a nonpeptidic framework for the design of potent inhibitors of cathepsin K. J Med Chem 2006;49:1066–79.
- Falgueyret J-P, Oballa RM, Okamoto O, et al. Novel, nonpeptidic cyanamides as potent and reversible inhibitors of human cathepsins K and L. J Med Chem 2001;44:94–104.
- Robichaud J, Bayly CI, Black WC, et al. β-Substituted cyclohexanecarboxamide cathepsin K inhibitors: modification of the 1,2-disubstituted aromatic core. Bioorg Med Chem Lett 2007;17:3146–51.
- Robichaud J, Black WC, Thérien M, et al. Identification of a nonbasic, nitrile-containing cathepsin K inhibitor (MK-1256) that is efficacious in a monkey model of osteoporosis. J Med Chem 2008;51:6410–20.
- Dossetter AG, Beeley H, Bowyer J, et al. (1 R, 2 R)-N-(1-Cyanocyclopropyl)-2-(6-methoxy-1, 3, 4, 5-tetrahydropyrido [4, 3-b] indole-2-carbonyl) cyclohexanecarboxamide (AZD4996): a potent and highly selective cathepsin K inhibitor for the treatment of osteoarthritis. J Med Chem 2012;55:6363–74.
- Altmann E, Aichholz R, Betschart C, et al. 2-Cyano-pyrimidines: a new chemotype for inhibitors of the cysteine protease cathepsin K. J Med Chem 2007;50:591–4.
- Teno N, Miyake T, Ehara T, et al. Novel scaffold for cathepsin K inhibitors. Bioorg Med Chem Lett 2007;17:6096–100.
- Organon N. 2-Cyano-1,3,5-triazine-4,6-diamine derivatives. WO05011703; 2005.
- Rankovic Z, Cai J, Kerr J, et al. Design and optimization of a series of novel 2-cyano-pyrimidines as cathepsin K inhibitors. Bioorg Med Chem Lett 2010;20:1524–7.
- Cai J, Baugh M, Black D, et al. 6-Phenyl-1H-imidazo [4, 5-c] pyridine-4-carbonitrile as cathepsin S inhibitors. Bioorg Med Chem Lett 2010;20:4350–4.
- Teno N, Masuya K, Ehara T, et al. Effect of cathepsin K inhibitors on bone resorption. J Med Chem 2008;51:5459–62.
- Organon N, 6-Phenyl-1H-imidazo [4, 5-c] pyridine-4-carbonitrile derivatives as cathepsin inhibitors. WO07080191; 2007.
- Shinozuka T, Shimada K, Matsui S, et al. 4-Aminophenoxyacetic acids as a novel class of reversible cathepsin K inhibitors. Bioorg Med Chem Lett 2006;16:1502–5.
- Setti EL, Davis D, Janc JW, et al. 4-Disubstituted azetidinones as selective inhibitors of the cysteine protease cathepsin K. Exploring P3 elements for potency and selectivity. Bioorg Med Chem Lett 2005;15:1529–34.
- Setti EL, Davis D, Chung T, McCarter J. 3, 4-Disubstituted azetidinones as selective inhibitors of the cysteine protease cathepsin K. Exploring P2 elements for selectivity. Bioorg Med Chem Lett 2003;13:2051–3.
- Altmann E, Renaud J, Green J, et al. Arylaminoethyl amides as novel non-covalent cathepsin K inhibitors. J Med Chem 2002;45:2352–4.
- Altmann E, Green J, Tintelnot-Blomley M. Arylaminoethyl amides as inhibitors of the cysteine protease cathepsin K-investigating P 1′ substituents. Bioorg Med Chem Lett 2003;13:1997–2001.
- Shinozuka T, Shimada K, Matsui S, et al. Arylamine based cathepsin K inhibitors: investigating P3 heterocyclic substituents. Bioorg Med Chem 2006;14:6807–19.
- Shinozuka T, Shimada K, Matsui S, et al. Potent and selective cathepsin K inhibitors. Bioorg Med Chem 2006;14:6789–806.
- Unoki G, Hayamizu T, Eguchi H, et al. Cysteine protease inhibitors: Google Patents; 2009.
- Novinec M, Korenč M, Caflisch A, et al. A novel allosteric mechanism in the cysteine peptidase cathepsin K discovered by computational methods. Nature Commun 2014;5:3287.
- Asagiri M, Hirai T, Kunigami T, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science 2008;319:624–7.
- Wang ZQ, Li JL, Sun YL, et al. Chinese herbal medicine for osteoporosis: a systematic review of randomized controlled trails. Evid-Based Complementary Altern Med 2013;2013:356260.
- Mukwaya E, Xu F, Wong MS, Zhang Y. Chinese herbal medicine for bone health. Pharmaceut Biol 2014;52:1223–8.
- Jeong JC, Lee BT, Yoon CH, et al. Effects of Drynariae rhizoma on the proliferation of human bone cells and the immunomodulatory activity. Pharmacol Res 2005;51:125–36.
- Chen LL, Lei LH, Ding PH, et al. Osteogenic effect of Drynariae rhizoma extracts and Naringin on MC3T3-E1 cells and an induced rat alveolar bone resorption model. Arch Oral Biol 2011;56:1655–62.
- Sun JS, Lin CY, Dong GC, et al. The effect of Gu-Sui-Bu (Drynaria fortunei J. Sm) on bone cell activities. Biomaterials 2002;23:3377–85.
- Guo Y, Li Y, Xue L, et al. Salvia miltiorrhiza: an ancient Chinese herbal medicine as a source for anti-osteoporotic drugs. J Ethnopharmacol 2014;155:1401–16.
- Patil AD, Freyer AJ, Carte B, et al. Haploscleridamine, a novel tryptamine-derived alkaloid from a sponge of the order Haplosclerida: an inhibitor of cathepsin K. J Natural Prod 2002;65:628–9.
- Qiu Z-C, Dong X-L, Dai Y, et al. Discovery of a new class of cathepsin K inhibitors in Rhizoma drynariae as potential candidates for the treatment of osteoporosis. Int J Mol Sci 2016;17:2116.
- Panwar P, Soe K, Guido RV, et al. A novel approach to inhibit bone resorption: exosite inhibitors against cathepsin K. Brit J Pharmacol 2016;173:396–410.
- Sharma V, Panwar P, O’Donoghue AJ, et al. Structural requirements for the collagenase and elastase activity of cathepsin K and its selective inhibition by an exosite inhibitor. Biochem J 2015;465:163–73.
- Chen K, Ge B, Liu X, et al. Icariin inhibits the osteoclast formation induced by RANKL and macrophage-colony stimulating factor in mouse bone marrow culture. Die Pharm 2007;62:388–91.
- Hsieh T-P, Sheu S-Y, Sun J-S, Chen M-H. Icariin inhibits osteoclast differentiation and bone resorption by suppression of MAPKs/NF-κB regulated HIF-1α and PGE2 synthesis. Phytomedicine 2011;18:176–85.
- Sun P, Liu Y, Deng X, et al. An inhibitor of cathepsin K, icariin suppresses cartilage and bone degradation in mice of collagen-induced arthritis. Phytomedicine 2013;20:975–9.
- Dean JH. Immunotoxicology and immunopharmacology. New York (NY): Raven Press; 1985.
- Lecaille F, Kaleta J, Brömme D. Human and parasitic papain-like cysteine proteases: their role in physiology and pathology and recent developments in inhibitor design. Chem Rev 2002;102:4459–88.
- McGrath ME, Klaus JL, Barnes MG, Brömme D. Crystal structure of human cathepsin K complexed with a potent inhibitor. Nat Struct Mol Biol 1997;4:105.
- Wang D, Li W, Pechar M, et al. Cathepsin K inhibitor-polymer conjugates: potential drugs for the treatment of osteoporosis and rheumatoid arthritis. Int. J. Pharm 2004;277:73–9.
- Wang D, Pechar M, Li W, et al. Inhibition of cathepsin K with lysosomotropic macromolecular inhibitors. Biochemistry 2002;41:8849–59.
- Lu J, Jiang F, Lu A, Zhang G. Linkers having a crucial role in antibody-drug conjugates. Int J Molec Sci 2016;17:561.
- Yu NY, Fathi A, Murphy CM, et al. Local co-delivery of rhBMP-2 and cathepsin K inhibitor L006235 in poly (d, l-lactide-co-glycolide) nanospheres. J Biomed Mater Res Part B Appl Biomater 2017;105:136–44.