
Abstract
The synthesis of a new series of sulfamides incorporating ortho-, meta, and para-benzenesulfamide moieties is reported, which were investigated for the inhibition of two human (h) isoforms of the zinc enzyme carbonic anhydrase (CA, EC 4.2.1.1), hCA I and II, and two Vibrio cholerae enzymes, belonging to the α- and β-CA classes (VchCAα, VchCAβ). The compounds were prepared by using the “tail approach”, aiming to overcome the scarcity of selective inhibition profiles associated to CA inhibitors belonging to the zinc binders. The built structure–activity relationship showed that the incorporation of benzhydryl piperazine tails on a phenyl sulfamide scaffold determines rather good efficacies against hCA I and VchCAα, with several compounds showing KIs < 100 nM. The activity was lower against hCA II and VchCAβ, probably due to the fact that the incorporated tails are quite bulky. The obtained evidences allow us to continue the investigations of different tails/zinc binding groups, with the purpose to increase the effectiveness/selectivity of such inhibitors against bacterial CAs from pathogens, affording thus potential new anti-infectives.
1. Introduction
Cholera is an infectious human disease of the small intestine and is caused by the gram-negative bacterium Vibrio choleraCitation1,Citation2. It is characterised by a massive loss of water and electrolytes which leads to severe dehydration and hypovolemic shock followed by death if the disease is not well treatedCitation2–4. In developing countries, cholera spreads among victims mainly through contaminated water sources, and countries without proper sanitation techniques have greater incidence of this diseaseCitation3,Citation5. In this regard, WHO reported 132,121 cases in 38 countries during 2016, which also included 2420 deathsCitation6. It has been demonstrated that a potential inducer of virulence gene expression is sodium bicarbonate, which is present at a high concentration in the upper small intestineCitation2. Consequently, bicarbonate is considered the first positive effector for ToxT, the major direct transcription activator of the virulence genesCitation2.
Many studies conducted by some of us have shown that specific carbonic anhydrase (CA, EC 4.2.1.1) inhibitors, may control the bicarbonate-mediated virulence induction, suggesting the conversion of CO2 into bicarbonate by CA plays a crucial role as a virulence factor for Vibrio choleraeCitation7,Citation8. Three different CAs have been found in Vibrio cholerae, belonging to the three enzyme classes found in bacteria, VchCAα, β, and γ. VchCAs have been suggested as potential targets for anti-infectives development with a novel mechanism of actionCitation9,Citation10.
Up to now, inhibition studies performed against VchCAs mainly considered sulfonamide-bearing derivatives, with sulfamates (R-OSO2NH2) and sulfamides (R-NHSO2NH2) being less investigated. These latter derivatives are the closest bioisosters and congeners to the primary sulfonamides (R-SO2NH2), which constitute the most important, clinically used class of CA inhibitors (CAIs)Citation11–14.
We recently reported a set of benzhydrylpiperazine benzenesulfamide showing effective and selective inhibition profiles against the cytosolic isoform hCA ICitation15. In the present study, we have oriented our efforts in developing sulfamide CAIs as potential antibacterial agents, continuing the development of the previously reported series of sulfamidesCitation15, and report the synthesis of new series of such derivatives. In addition to the bacterial enzyme VchCAα and β, these novel compounds were investigated for their property to inhibit the physiologically most important human cytosolic isoforms CA I and IICitation16,Citation17.
2. Materials and methods
2.1. General
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich (Milan, Italy), Alfa Aesar (Milan, Italy), and TCI (Milan, Italy). All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringes techniques to transfer solutions. Nuclear magnetic resonance spectra (1H NMR: 400 MHz; 13C NMR: 100 MHz; 19F NMR: 376 MHz) were recorded in DMSO-d6 using an Avance III 400 MHz spectrometer (Bruker, Milan, Italy). Chemical shifts are reported in parts per million (ppm) and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; brs, broad singlet; dd, double of doublets. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Analytical thin-layer chromatography (TLC) was carried out on silica gel F-254 plates (Merck, Milan, Italy). Melting points (m.p.) were carried out in open capillary tubes and are uncorrected. The solvents used in MS measures were acetone, acetonitrile (Chromasolv grade), purchased from Sigma-Aldrich (Milan, Italy) and mQ water 18 MX, obtained from Millipore’s Simplicity system (Milan, Italy). The mass spectra were obtained using a 1200 L triple quadrupole system (Varian, Palo Alto, CA) equipped by electrospray source (ESI) operating in both positive and negative ions. Stock solutions of analytes were prepared in acetone at 1.0 mg ml−1 and stored at 4 °C. Working solutions of each analyte were freshly prepared by diluting stock solutions in a mixture of mQ H2O/ACN 1:1 (v/v) up to a concentration of 1.0 µg ml−1. The mass spectra of each analyte were acquired by introducing, via syringe pump at 10 µl min−1, its working solution. Raw-data were collected and processed by Varian Workstation Vers. 6.8 software (Palo Alto, CA).
2.2. General procedure for the synthesis of compounds 2a–d
Compounds 1a,b (1.0 eq) and the appropriate N-Boc-protected carboxylic acid (1.1 eq) in DMF (10.0 ml) were treated with DIPEA (2.0 eq), and HATU (1.5 eq) at r.t. for 30 min. When the reaction was complete (TLC: monitoring), it was quenched with ice cold water and extracted with ethyl acetate (3 × 15 ml). The combined organic layers were washed with H2O (3 × 15 ml), dried over Na2SO4, filtered-off and concentrated under reduced pressure to afford the title compounds 2a–d as off-white solids.
2.2.1. Tert-butyl (2-4-benzhydrylpiperazin-1-yl)-2-oxoethyl)carbamate (2a)
Using 1a and N-Boc-glycine as starting materials, compound 2a was obtained with yield: 99%, off white solid. 1H NMR (DMSO-d6, 400 MHz): δ 1.36 (9H, s, 3 × CH3), 2.27 (4H, m, 2 × piperazine-CH2), 3.42 (4H, m, 2 × piperazine-CH2), 3.72 (2H, d, J = 5.7, COCH2), 4.32 (1H, s, CH), 6.71 (1H, m, NH), 7.19 (2H, t, J = 7.2, 2 × Ar-H), 7.30 (4H, t, J = 7.5, 4 × Ar-H), 7.43 (4H, d, J = 7.4, Ar-H).
2.2.2. Tert-butyl 4-4-benzhydrylpiperazine-1-carbonyl)piperidine-1-carboxylate (2b)
Using 1a and N-Boc-isonipecotic acid as starting materials, compound 2b was obtained with yield: 76%, off white solid. 1H NMR (DMSO-d6, 400 MHz): δ 1.38 (11H, s, 3 × CH3, piperidine-CH2), 1.55 (2H, m, piperidine-CH2), 2.25 (4H, br s, 2 × piperazine-CH2), 2.70 (3H, m, piperidine-CH2, COCH), 3.48 (4H, br d, 2 × piperazine-CH2), 3.87 (2H, m, piperidine-CH2), 4.40 (1H, s, CH), 7.19 (2H, t, J = 7.2, 2 × Ar-H), 7.30 (4H, t, J = 7.5, 4 × Ar-H), 7.43 (4H, d, J = 7.4, 4 × Ar-H).
2.2.3. Tert-butyl (2-(4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)carbamate (2c)
Using 1b and N-Boc-glycine as starting materials, compound 2c was obtained with yield: 99%, off white solid. 1H NMR (DMSO-d6, 400 MHz): δ 1.36 (9H, s, 3 × CH3), 2.27 (4H, m, 2 × piperazine-CH2), 3.42 (4H, m, 2 × piperazine-CH2), 3.72 (2H, d, J = 5.7, COCH2), 4.32 (1H, s, CH), 6.71 (1H, m, NH), 7.14 (4H, m, J = 7.2, 4 × Ar-H), 7.43 (4H, m, J = 7.4, 4 × Ar-H).
2.2.4. Tert-butyl 4-(4-(bis(4-fluorophenyl)methyl)piperazine-1-carbonyl)piperidine-1-carboxylate (2d)
Using 1b and N-Boc-isonipecotic acid as starting materials, compound 2d was obtained with yield: 71%, off white solid. 1H NMR (DMSO-d6, 400 MHz): δ 1.38 (11H, s, 3 × CH3, piperidine-CH2), 1.55 (2H, m, piperidine-CH2), 2.25 (4H, br s, 2 × piperazine-CH2), 2.70 (3H, m, piperidine-CH2, COCH), 3.48 (4H, br d, 2 × piperazine-CH2), 3.87 (2H, m, piperidine-CH2), 4.40 (1H, s, CH), 7.14 (4H, t, J = 8.7, 4 × Ar-H), 7.44 (4H, t, J = 8.1, 4 × Ar-H).
2.2.5. General procedure for the synthesis of compounds 4a–p
A stirred solution of compounds 2a–d (1.0 eq) in DCM (10.0 ml) was treated with TFA (3.0 eq) and stirred at r.t. for 2 h. The reaction mixture was concentrated to dry and co-distilled twice with DCM to afford the corresponding amines 3a–d as TFA salts (not isolated), which were readily dissolved in DCM (10.0 ml) and treated with DIPEA (5.0 eq) and the appropriate sulfonyl chloride (1.2 eq). The reaction solutions were stirred at r.t. for 1 h, then concentrated to dry and the residue obtained was purified by silica gel column chromatography using ethyl acetate in n-hexane (20–40% v/v) as eluent to afford the titled compounds 4a–p as off-white solids.
2.2.6. 1-Benzhydryl-4-((3-nitrophenyl)sulfonyl)piperazine (4a)
Using 1a and 3-nitrobenzenesulfonyl chloride as starting materials, compound 4a was obtained with yield 75%, off white solid; m.p. 170–172 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.36 (4H, br s, 2 × piperazine-CH2), 3.00 (4H, br s, 2 × piperazine-CH2), 4.31 (1H, s, CH), 7.14 (2H, t, J = 7.2, 2 × Ar-H), 7.23 (4H, t, J = 7.4, 4 × Ar-H), 7.33 (4H, d, J = 7.3, 4 × Ar-H), 7.96 (1H, t, J = 8.0, Ar-H), 8.15 (1H, d, J = 7.8, Ar-H), 8.35 (1H, s, Ar-H), 8.56 (1H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 46.0, 50.3, 74.1, 122.2, 126.9, 127.4, 127.8, 128.5, 131.5, 133.4, 136.5, 142.2, 148.2; MS (ESI positive) m/z = 437.9 [M + H]+.
2.2.7. 1-Benzhydryl-4-((4-nitrophenyl)sulfonyl)piperazine (4b)
Using 1a and 4-nitrobenzenesulfonyl chloride as starting materials, compound 4b was obtained with yield 46%, off white solid; m.p. 216–218 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.36 (4H, br s, 2 × piperazine-CH2), 2.99 (4H, br s, 2 × piperazine-CH2), 4.31 (1H, s, CH), 7.14 (2H, t, J = 7.2, 2 × Ar-H), 7.23 (4H, t, J = 7.4, 4 × Ar-H), 7.33 (4H, d, J = 7.3, 4 × Ar-H), 7.98 (2H, d, J = 8.8, 2 × Ar-H), 8.45 (2H, d, J = 8.8, 2 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 46.0, 50.2, 74.0, 124.7, 126.9, 127.4, 128.5, 129.1, 140.5, 142.1, 150.1; MS (ESI positive) m/z = 437.9 [M + H]+.
2.2.8. N-(2-(4-Benzhydrylpiperazin-1-yl)-2-oxoethyl)-2-nitrobenzenesulfonamide (4c)
Using 3a and 2-nitrobenzenesulfonyl chloride as starting materials, compound 4c was obtained with yield 61%, off white solid; m.p. 188–192 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.20 (4H, br d, 2 × piperazine-CH2), 3.37 (4H, br s, 2 × piperazine-CH2), 3.89 (2H, d, J = 4.8, CH2), 4.28 (1H, s, CH), 7.17 (2H, t, J = 7.3, 2 × Ar-H), 7.28 (4H, t, J = 7.4, 4 × Ar-H), 7.40 (4H, d, J = 7.3, 4 × Ar-H), 7.81 (2H, m, 2 Ar-H), 7.93 (2H, m, Ar-H, NH), 8.01 (1H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 41.5, 43.9, 44.0, 50.9, 51.3, 74.6, 124.4, 126.9, 127.5, 128.5, 129.8, 132.6, 133.1, 133.8, 142.3, 147.4, 165.4; MS (ESI positive) m/z = 494.9 [M + H]+.
2.2.9. N-(2-(4-Benzhydrylpiperazin-1-yl)-2-oxoethyl)-3-nitrobenzenesulfonamide (4d)
Using 3a and 3-nitrobenzenesulfonyl chloride as starting materials, compound 4d was obtained with yield 56%, off white solid; m.p. 208–212 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.13 (2H, s, piperazine-CH2), 2.20 (2H, s, piperazine-CH2), 3.30 (4H, br s, 2 × piperazine-CH2), 3.81 (2H, d, J = 5.5, CH2), 4.24 (1H, s, CH), 7.17 (2H, t, J = 7.3, 2 × Ar-H), 7.28 (4H, t, J = 7.4, 4 × Ar-H), 7.39 (4H, d, J = 7.2, 4 × Ar-H), 7.84 (1H, t, J = 8.0, Ar-H), 8.17 (2H, m, Ar-H, NH), 8.43 (1H, m, Ar-H), 8.54 (1H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 41.4, 43.6, 44.1, 50.9, 51.5, 74.7, 121.5, 126.8, 126.9, 127.5, 128.5, 130.9, 132.8, 142.3, 147.6, 165.3; MS (ESI positive) m/z = 494.9 [M + H]+.
2.2.10. (4-Benzhydrylpiperazin-1-yl)(1-((2-nitrophenyl)sulfonyl)pipe-ridin-4-yl)methanone (4e)
Using 3b and 2-nitrobenzenesulfonyl chloride as starting materials, compound 4e was obtained with yield 49%, off white solid; m.p. 158–160 °C; 1H NMR (DMSO-d6, 400 MHz): δ 1.48 (2H, m, piperidine-CH2), 1.66 (2H, m, piperidine-CH2), 2.22 (4H, br s, 2 × piperazine-CH2), 2.72 (3H, m, piperidine-CH2, COCH), 3.44 (4H, br d, 2 × piperazine-CH2), 3.66 (2H, d, piperidine-CH2), 4.29 (1H, s, CH), 7.17 (2H, t, J = 7.3, 2 × Ar-H), 7.28 (4H, t, J = 7.4, 4 × Ar-H), 7.40 (4H, d, J = 7.5, 4 × Ar-H), 7.85 (2H, m, 2 × Ar-H), 7.97 (2H, m, 2 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 27.7, 35.7, 41.3, 44.7, 45.0, 51.2, 52.0, 074.6, 124.0, 126.9, 127.5, 128.5, 129.2, 130.3, 132.1, 134.7, 142.4, 147.8, 171.6; MS (ESI positive) m/z = 548.9 [M + H]+.
2.2.11. (4-Benzhydrylpiperazin-1-yl)(1-((3-nitrophenyl)sulfonyl)pipe-ridin-4-yl)methanone (4f)
Using 3b and 3-nitrobenzenesulfonyl chloride as starting materials, compound 4f was obtained with yield 48%, off white solid; m.p. 222–226 °C; 1H NMR (DMSO-d6, 400 MHz): δ 1.52 (2H, m, piperidine-CH2), 1.64 (2H, m, piperidine-CH2), 2.20 (4H, br s, 2 × piperazine-CH2), 2.37 (2H, m, piperidine-CH2), 2.53 (1H, m, COCH), 3.41 (4H, 4 s, 2 × piperazine-CH2), 3.63 (2H, d, piperidine-CH2), 4.27 (1H, s, CH), 7.17 (2H, t, J = 7.3, 2 × Ar-H), 7.27 (4H, t, J = 7.4, 4 × Ar-H), 7.39 (4H, d, J = 7.5, 4 × Ar-H), 7.93 (1H, t, J = 8.0, Ar-H), 8.16 (1H, d, J = 7.8, Ar-H), 8.34 (1H, br s, Ar-H), 8.52 (1H, d, J = 8.1, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 27.5, 35.6, 41.1, 44.6, 45.2, 51.2, 52.0, 74.6, 121.8, 126.8, 126.9, 127.6, 128.5, 131.5, 133.4, 137.1, 142.3, 147.9, 171.5; MS (ESI positive) m/z = 548.9 [M + H]+.
2.2.12. (4-Benzhydrylpiperazin-1-yl)(1-((4-nitrophenyl)sulfonyl)pipe-ridin-4-yl)methanone (4g)
Using 3b and 4-nitrobenzenesulfonyl chloride as starting materials, compound 4g was obtained with yield 59%, off white solid; m.p. 182–186 °C; 1H NMR (DMSO-d6, 400 MHz): δ 1.49 (2H, m, piperidine-CH2), 1.64 (2H, m, piperidine-CH2), 2.20 (4H, br s, 2 × piperazine-CH2), 2.37 (2H, m, piperidine-CH2), 2.57 (1H, m, COCH), 3.41 (4H, br s, 2 × piperazine-CH2), 3.62 (2H, m, piperidine-CH2), 4.27 (1H, s, CH), 7.16 (2H, t, J = 7.3, 2 × Ar-H), 7.27 (4H, t, J = 7.4, 4 × Ar-H), 7.39 (4H, d, J = 7.5, 4 × Ar-H), 7.99 (2H, d, J = 6.9, 2 × Ar-H), 8.42 (2H, d, J = 6.9, 2 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 26.5, 35.6, 41.1, 44.7, 45.1, 51.2, 51.9, 74.6, 124.6, 126.9, 127.5, 128.5, 128.9, 141.2, 142.3, 149.9, 171.5; MS (ESI positive) m/z = 548.9 [M + H]+.
2.2.13. 1-(Bis(4-Fluorophenyl)methyl)-4-((2-nitrophenyl)sulfonyl)pip-erazine (4h)
Using 1b and 2-nitrobenzenesulfonyl chloride as starting materials, compound 4h was obtained with yield: 77%, off white solid; m.p. 120–124 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.34 (4H, br s, 2 × piperazine-CH2), 3.18 (4H, br s, 2 × piperazine-CH2), 4.43 (1H, s, CH), 7.09 (4H, t, J = 8.7, 4 × Ar-H), 7.39 (4H, t, J = 8.2, 4 Ar-H), 7.91 (4H, m, 4 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 45.9, 50.3, 72.0, 115.2, 115.4, 124.1, 128.4, 129.3, 129.4, 130.4, 132.2, 134.9, 138.1, 147.9, 159.9, 162.3; MS (ESI positive) m/z = 474.04 [M + H]+.
2.2.14. 1-(Bis(4-Fluorophenyl)methyl)-4-((3-nitrophenyl)sulfonyl)pip-erazine (4i)
Using 1b and 3-nitrobenzenesulfonyl chloride as starting materials, compound 4i was obtained with yield: 87%, off white solid; m.p. 168–172 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.34 (4H, br s, 2 × piperazine-CH2), 2.99 (4H, br s, 2 × piperazine-CH2), 4.40 (1H, s, CH), 7.07 (4H, t, J = 8.8, 4 × Ar-H), 7.35 (4H, q, J = 5.7, 4 × Ar-H), 7.96 (1H, t, J = 8.0, Ar-H), 8.15 (1H, d, J = 7.8, Ar-H), 8.35 (1H, s, Ar-H), 8.56 (1H, d, J = 8.1, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 45.9, 50.0, 71.9, 115.2, 115.4, 124.7, 129.1, 129.2, 129.4, 138.1, 143.5, 155.1, 159.8, 162.2; MS (ESI positive) m/z = 473.9 [M + H]+.
2.2.15. 1-(Bis(4-Fluorophenyl)methyl)-4-((4-nitrophenyl)sulfonyl)pip-erazine (4j)
Using 1b and 4-nitrobenzenesulfonyl chloride as starting materials, compound 4j was obtained with yield: 73%, yellow solid; m.p. 222–226 °C; 1H NMR (DMSO-d6, 400 MHZ): δ 2.34 (4H, br s, 2 × piperazine-CH2), 2.99 (4H, br s, 2 × piperazine-CH2), 4.40 (1H, s, CH), 7.07 (4H, t, J = 8.8, 4 Ar-H), 7.34 (4H, m, J = 5.7, 4 × Ar-H), 7.98 (2H, d, J = 8.7, 2 × Ar-H), 8.45 (2H, d, J = 8.7, 2 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 46.0, 50.0, 71.9, 115.2, 115.4, 124.7, 129.1, 129.2, 129.3, 138.1, 140.5, 150.1, 159.8, 162.2; MS (ESI positive) m/z = 473.9 [M + H]+.
2.2.16. N-(2-(4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)-2-nitrobenzenesulfonamide (4k)
Using 3c and 2-nitrobenzenesulfonyl chloride as starting materials, compound 4k was obtained with yield: 69%, off white solid; m.p. 136–140 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.20 (4H, br d, 2 × pipeazine-CH2), 3.37 (4H, br s, 2 × piperazine-CH2), 3.91 (2H, d, J = 5.38, CH2), 4.39 (1H, s, CH), 7.13 (4H, t, J = 8.7, 4 × Ar-H), 7.42 (4H, q, J = 5.8, 4 × Ar-H), 7.83 (2H, m, 2 × Ar-H), 8.02 (3H, m, Ar-H, NH), 8.01 (1H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 41.5, 43.9, 43.9, 50.9, 51.3, 72.5, 115.2, 115.5, 124.4, 129.4, 129.5, 129.9, 132.6, 133.1, 133.9, 138.3, 159.5, 162.7, 165.4; MS (ESI positive) m/z = 531.12 [M + H]+.
2.2.17. N-(2-(4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)-3-nitrobenzenesulfonamide (4l)
Using 3c and 3-nitrobenzenesulfonyl chloride as starting materials, compound 4l was obtained with yield: 58%, off white solid; m.p. 202–206 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.15 (4H, br s, 2 × piperazine-CH2), 3.30 (4H, br s, 2 × piperazine-CH2), 3.82 (2H, d, J = 5.16, CH2), 4.34 (1H, s, CH), 7.11 (4H, t, J = 8.7, 4 × Ar-H), 7.40 (4H, t, J = 6.1, 4 × Ar-H), 7.84 (1H, t, J = 8.01, Ar-H), 8.15 (1H, t, J = 5.09, NH), 8.19 (1H, d, J = 7.78, Ar-H), 8.44 (1H, d, J = 8.16, Ar-H), 8.53 (1H, s, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 23.6, 26.6, 37.3, 41.1, 44.8, 45.9, 48.1, 51.0, 51.8, 72.6, 115.3, 115.6, 124.5, 129.4, 129.5, 129.9, 132.6, 133.2, 133.8, 138.3, 159.5, 162.8, 165.5; MS (ESI positive) m/z = 531.04 [M + H]+.
2.2.18. N-(2-(4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)-4-nitrobenzenesulfonamide (4m)
Using 3c and 4-nitrobenzenesulfonyl chloride as starting materials, compound 4m was obtained with yield: 47%, off white solid; m.p. 176–180 °C; 1H NMR (DMSO-d6, 400 MHz): δ 2.17 (4H, br d, 2 × piperazine-CH2), 3.33 (4H, br s, 2 × piperazine-CH2), 3.81 (2H, d, J = 5.5, CH2), 4.37 (1H, s, CH), 7.11 (4H, t, J = 8.6, 4 × Ar-H), 7.41 (4H, t, J = 6.1, 4 × Ar-H), 8.03 (2H, d, J = 8.6, 2 × Ar-H), 8.15 (1H, t, J = 5.4, NH), 8.36 (2H, d, J = 8.6, 2 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 41.4, 43.7, 44.1, 50.7, 51.2, 72.5, 115.5, 124.2, 129.3, 132.4, 138.2, 146.3, 149.4, 159.8, 162.3, 165.3; MS (ESI positive) m/z = 531.04 [M + H]+.
2.2.19. (4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)(1-((2-nitrophenyl)sulfonyl)piperidin-4-yl)methanone (4n)
Using 3d and 2-nitrobenzenesulfonyl chloride as starting materials, compound 4n was obtained with yield: 59%, off white solid; m.p. 170–174 °C; 1H NMR (DMSO-d6, 400 MHz): δ 1.48 (2H, m, piperidine-CH2), 1.66 (2H, m, piperidine-CH2), 2.20 (4H, br s, 2 × piperazine-CH2), 2.71 (3H, m, piperidine-CH2, COCH), 3.44 (4H, br d, 2 × piperazine-CH2), 3.67 (2H, m, piperidine-CH2), 4.38 (1H, s, CH), 7.11 (4H, t, J = 8.7, 4 × Ar-H), 7.41 (4H, m, 4 × Ar-H), 7.91 (4H, m, 4 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 27.7, 35.7, 40.1, 44.8, 44.9, 51.0, 51.8, 72.5, 115.2, 115.4, 124.0, 129.2, 129.3, 129.4, 130.3, 132.1, 134.7, 138.3, 147.8, 159.8, 162.2, 171.6; MS (ESI positive) m/z = 585.11 [M + H]+.
2.2.20. (4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)(1-((3-nitrophenyl)sulfonyl)piperidin-4-yl)methanone (4o)
Using 3d and 3-nitrobenzenesulfonyl chloride as starting materials, compound 4o was obtained with yield: 48%, off white solid; m.p. 180–184 °C; 1H NMR (DMSO-d6, 400 MHz): δ 1.52 (2H, m, piperdine-CH2), 1.65 (2H, d, piperidine-CH2), 2.20 (4H, s, 2 × piperazine-CH2), 2.37 (2H, m, piperidne-CH2), 2.54 (1H, m, COCH), 3.41 (4H, br s, 2 × piperazine-CH2), 3.64 (2H, m, piperidine-CH2), 4.29 (1H, s, CH), 7.12 (4H, t, J = 8.7, 4 × Ar-H), 7.43 (4H, t, J = 5.7, 4 × Ar-H), 7.94 (1H, t, J = 8.7, Ar-H), 8.16 (1H, d, J = 8.8, Ar-H), 8.34 (1H, s, Ar-H), 8.53 (1H, d, J = 8.7, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 27.5, 35.7, 41.1, 44.7, 45.2, 51.2, 51.9, 74.6, 115.3, 115.5, 121.9, 127.8, 129.4, 129.3, 129.4, 129.5, 131.5, 133.3, 137.5, 138.4, 138.4, 148.1, 159.5, 162.7, 170.6; MS (ESI positive) m/z = 585.09 [M + H]+.
2.2.21. (4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)(1-((4-nitrophenyl)sulfonyl)piperidin-4-yl)methanone (4p)
Using 3d and 4-nitrobenzenesulfonyl chloride as starting materials, compound 4p was obtained with yield: 46%, off white solid; m.p. 232–236 °C; 1H NMR (DMSO-d6, 400 MHz): δ 1.54 (2H, m, piperidine-CH2), 1.66 (2H, m, CH2), 2.21 (4H, br s, 2 × piperazine-CH2), 2.37 (2H, m, piperidine-CH2), 2.80 (1H, m, COCH), 3.41 (4H, br s, 2 × piperazine-CH2), 3.65 (2H, m, piperdine-CH2), 4.30 (1H, s, CH), 7.12 (4H, t, J = 8.8, 4 × Ar-H), 7.44 (4H, t, J = 5.7, 4 × Ar-H), 7.98 (2H, d, J = 9.2, 2 × Ar-H) 8.42 (2H, d, J = 9.2, 2 × Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 26.6, 35.6, 41.2, 44.7, 45.2, 51.3, 51.9, 74.7, 115.2, 115.4, 124.7, 128.7, 129.3, 129.4, 138.3, 141.4, 149.9, 159.8, 162.3, 170.3; MS (ESI positive) m/z = 585.10 [M + H]+.
2.2.22. General procedure for the synthesis of compounds 5a–p
The appropriate nitrobenzenesulfonamides 4a–p (1.0 eq) in a solution of H2O (0.4 ml) and EtOH (0.3 ml) was treated with glacial AcOH (0.05 ml) and Fe (0) (12.0 eq). The reaction mixture was stirred at 75 °C for 1 h (TLC monitoring), then cooled to r.t. and diluted with EtOAc (10.0 ml). The mixture was filtered through Celite 521®, washed with a saturated NaHCO3 aqueous solution (3 × 15 ml), brine (3 × 10 ml) and dried over Na2SO4. The organic solvent was evaporated in vacuo to give an oil residue, which was triturated from Et2O, to afford the titled compounds 5a–p as white solids.
2.2.23. 3-((4-Benzhydrylpiperazin-1-yl)sulfonyl)aniline (5a)
Compound 5a was obtained in 70% yield; m.p. 120–122 °C; TLC: Rf = 0.17 (ethyl acetate/n-hexane 20% v/v); 1H NMR (DMSO-d6, 400 MHZ) (DMSO-d6, 400 MHZ) (DMSO-d6, 400 MHz): δ 2.40 (4H, m, 2 × piperazine-CH2), 2.92 (4H, m, 2 × piperazine-CH2), 4.34 (1H, s, CH), 5.70 (2H, s, exchange with D2O, NH2), 6.81 (1H, m, Ar-H), 6.91 (2H, m, Ar-H), 7.20 (2H, t, J = 7.2, Ar-H), 7.29 (5H, m, Ar-H), 7.40 (4H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.4, 75.3, 114.5, 117.3, 118.4, 127.9, 128.4, 129.4, 130.9, 138.0, 142.7, 148.2; MS (ESI positive) m/z = 408.2 [M + H]+.
2.2.24. 4-((4-Benzhydrylpiperazin-1-yl)sulfonyl)aniline (5b)
Compound 5b was obtained in 80% yield; m.p. 196–198 °C; TLC: Rf = 0.25 (ethyl acetate/n-hexane 20% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.38 (4H, m, 2 × piperazine-CH2), 2.85 (4H, m, 2 × piperazine-CH2), 4.33 (1H, s, CH), 6.16 (2H, s, exchange with D2O, NH2), 6.70 (2H, m, Ar-H), 7.20 (2H, m, Ar-H), 7.29 (4H, t, J = 7.4, Ar-H), 7.40 (6H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.4, 75.3, 113.6, 126.8, 127.8, 128.4, 129.3, 130.6, 143.4, 154.2; MS (ESI positive) m/z = 408.2 [M + H]+.
2.2.25. 2-Amino-N-(2-(4-benzhydrylpiperazin-1-yl)-2-oxoethyl)benzenesulfonamide (5c)
Compound 5c was obtained in 85% yield; m.p. 140–142 °C; TLC: Rf = 0.39 (ethyl acetate/n-hexane 50% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.25 (4H, m, 2 × piperazine-CH2), 3.42 (4H, m, 2 × piperazine-CH2), 3.66 (2H, d, J = 5.4, COCH2NH), 4.33 (1H, s, CH), 5.99 (2H, s, exchange with D2O, NH2), 6.59 (1H, t, J = 7.4, Ar-H), 6.80 (1H, d, J = 8.0, Ar-H), 7.24 (3H, m, Ar-H), 7.33 (4H, m, Ar-H), 7.47 (6H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 42.4, 45.1, 52.2, 75.5, 115.7, 117.8, 120.3, 127.8, 128.4, 129.4, 130.0, 134.4, 143.3, 147.3, 166.43; MS (ESI positive) m/z = 465.2 [M + H]+.
2.2.26. 3-Amino-N-(2-(4-benzhydrylpiperazin-1-yl)-2-oxoethyl)benzenesulfonamide (5d)
Compound 5d was obtained in 78% yield; m.p. 135–137 °C; TLC: Rf = 0.33 (ethyl acetate/n-hexane 50% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.26 (4H, m, 2 × piperazine-CH2), 3.33 (4H, overlap with water peak, 2 × piperazine-CH2), 3.67 (2H, s, COCH2NH), 4.34 (1H, s, CH), 5.58 (2H, s, exchange with D2O, NH2), 6.76 (1H, d, J = 6.8, Ar-H), 6.90 (1H, d, J = 7.0, Ar-H), 6.99 (1H, m, Ar-H), 7.21 (3H, m, Ar-H), 7.33 (4H, m, Ar-H), 7.45 (5H, m, 4 × Ar-H, exchange with D2O, CH2NHSO2); 13C NMR (DMSO-d6, 100 MHz): δ 42.2, 44.9, 51.9, 75.7, 116.4, 120.7, 122.5, 126.2, 127.6, 128.2, 129.4, 141.1, 141.6, 148.3, 164.3; MS (ESI positive) m/z = 465.2 [M + H]+.
2.2.27. (1-((2-Aminophenyl)sulfonyl)piperidin-4-yl)(4-benzhydrylpiperazin-1-yl)methanone (5e)
Compound 5e was obtained in 47% yield; m.p. 202–234 °C; TLC: Rf = 0.54 (ethyl acetate/n-hexane 60% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.52 (2H, m, piperidine-CH2), 1.67 (2H, m, piperidine-CH2), 2.26 (4H, m, 2 × piperazine-CH2), 2.54 (3H, overlap with DMSO peak, COCH, piperidine-CH2), 3.47 (4H, m, 2 × piperazine-CH2), 3.63 (2H, m, piperidine-CH2), 4.32 (1H, s, CH), 6.07 (2H, s, exchange with D2O, NH2), 6.65 (1H, m, Ar-H), 6.86 (1H, d, J = 7.6, Ar-H), 7.22 (2H, m, Ar-H), 7.32 (5H, m, Ar-H), 7.38 (1H, t, J = 7.6, Ar-H) 7.42 (4H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 42.0, 45.9, 53.0, 75.6, 116.6, 127.8, 128.5, 129.4, 130.9, 132.5, 134.6, 134.7, 135.5, 143.3, 167.8; MS (ESI positive) m/z = 519.7 [M + H]+.
2.2.28. (1-((3-Aminophenyl)sulfonyl)piperidin-4-yl)(4-benzhydrylpiperazin-1-yl)methanone (5f)
Compound 5f was obtained in 30% yield; m.p. 124–126 °C; TLC: Rf = 0.13 (ethyl acetate/n-hexane 60% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.54 (2H, m, piperidine-CH2), 1.68 (2H, m, piperidine-CH2), 2.29 (6H, m, 2 × piperazine-CH2, piperidine-CH2), 2.54 (1H, overlap with DMSO peak, COCH) 3.33 (4H, m, overlap with water peak, 2 × piperazine-CH2), 3.56 (2H, m, piperidine-CH2), 4.32 (1H, s, CH), 5.64 (2H, s, exchange with D2O, NH2) 6.81 (2H, m, Ar-H), 6.91 (1H, m, Ar-H), 7.23 (3H, m, Ar-H), 7.30 (4H, t, J = 7.6, Ar-H), 7.44 (4H, d, J = 7.2, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8 (overlap with DMSO peak), 40.4 (overlap with DMSO peak), 46.2, 65.8, 75.6, 112.6, 114.9, 118.7, 127.8, 128.5, 129.5, 130.5, 136.5, 143.4, 150.4, 172.7; MS (ESI positive) m/z = 519.2 [M + H]+.
2.2.29. (1-((4-Aminophenyl)sulfonyl)piperidin-4-yl)(4-benzhydrylpiperazin-1-yl)methanone (5g)
Compound 5g was obtained in 70% yield; m.p. 174–176 °C; TLC: Rf = 0.40 (ethyl acetate/n-hexane 70% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.54 (2H, m, piperidine-CH2), 1.68 (2H, m, piperidine-CH2), 2.29 (6H, m, 2 × piperazine-CH2, piperidine-CH2), 2.54 (1H, overlap with DMSO peak, COCH), 3.51 (6H, m, 2 × piperazine-CH2, piperidine-CH2), 4.32 (1H, s, CH), 6.06 (2H, s, exchange with D2O, NH2) 6.65 (2H, m, Ar-H), 6.91 (1H, m, Ar-H), 7.23 (3H, m, Ar-H), 7.30 (4H, t, J = 7.6, Ar-H), 7.44 (4H, d, J = 7.2, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 40.4 (overlap with DMSO peak), 46.2, 62.2, 75.6, 113.6, 127.8, 128.5, 129.5, 130.5, 132.5, 143.4, 153.9, 172.7; MS (ESI positive) m/z = 519.2 [M + H]+.
2.2.30. 2-((4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)sulfonyl)aniline (5h)
Compound 5h was obtained in 94% yield; m.p. 176–179 °C (dec); TLC: Rf = 0.46 (ethyl acetate/n-hexane 30% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.41 (4H, m, 2 × piperazine-CH2), 3.03 (4H, m, 2 × piperazine-CH2), 4.44 (1H, s, CH), 6.07 (2H, s, exchange with D2O, NH2), 6.69 (1H, t, J = 8.0, Ar-H), 6.91 (1H, d, J = 8.0, Ar-H), 7.12 (4H, m, Ar-H), 7.39 (6H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.1, 73.2, 115.4, 116.2 (d, Citation2JC–F 21), 116.4, 118.2, 130.2 (d, 3JC–F 8), 130.8, 135.2, 139.3, 148.4, 161.9 (d, Citation1JC–F 242); 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 444.2 [M + H]+.
2.2.31. 3-((4-(Bis(4-Fluorophenyl)methyl)piperazin-1-yl)sulfonyl)aniline (5i)
Compound 5i was obtained in 53% yield; m.p. 183–186 °C (dec); TLC: Rf = 0.21 (ethyl acetate/n-hexane 30% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.39 (4H, m, 2 × piperazine-CH2), 2.91 (4H, m, 2 × piperazine-CH2), 4.42 (1H, s, CH), 5.69 (2H, s, exchange with D2O, NH2), 6.81 (1H, d, J = 7.8, Ar-H), 6.90 (2H, m, Ar-H), 7.12 (4H, m, Ar-H), 7.30 (1H, t, J = 7.8, Ar-H), 7.42 (4H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.2, 73.2, 112.7, 115.1, 116.2 (d, Citation2JC–F 21), 118.8, 130.2 (d, 3JC–F 8), 130.5, 135.7, 139.2, 148.4, 161.9 (d, Citation1JC–F 242); 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 444.2 [M + H]+.
2.2.32. 4-((4-(Bis(4-Fluorophenyl)methyl)piperazin-1-yl)sulfonyl)aniline (5j)
Compound 5j was obtained in 64% yield; m.p. 155–157 °C (dec); TLC: Rf = 0.18 (ethyl acetate/n-hexane 30% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.37 (4H, m, 2 × piperazine-CH2), 2.85 (4H, m, 2 × piperazine-CH2), 4.46 (1H, s, CH), 6.16 (2H, s, exchange with D2O, NH2), 6.71 (2H, d, J = 8.8, Ar-H), 7.12 (4H, m, Ar-H), 7.38 (2H, d, J = 8.8, Ar-H), 7.42 (4H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.1, 73.2, 113.6, 116.2 (d, Citation2JC–F 21), 125.6, 130.2 (d, 3JC–F 8), 130.5, 139.2, 154.1, 161.9 (d, Citation1JC–F 242); 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 444.2 [M + H]+.
2.2.33. 2-Amino-N-(2-(4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)benzenesulfonamide (5k)
Compound 5k was obtained in 79% yield; m.p. 137–139 °C; TLC: Rf = 0.16 (ethyl acetate/n-hexane 60% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.24 (4H, m, 2 × piperazine-CH2), 3.33 (4H, m, overlap with water peak, 2 × piperazine-CH2), 3.66 (2H, s, COCH2NH) 4.42 (1H, s, CH), 5.99 (2H, s, exchange with D2O, NH2), 6.59 (1H, t, J = 7.2, Ar-H), 6.79 (2H, d, J = 8.0, Ar-H), 7.17 (4H, m, Ar-H), 7.28 (1H, m, Ar-H), 7.36 (1H, t, J = 7.2, exchange with D2O, SO2NHCH2), 7.46 (4H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 42.4, 45.1, 52.2, 75.5, 116.1, 116.3 (d, Citation2JC–F 21), 116.6, 118.2, 130.2, (d, 3JC–F 9), 130.8, 135.6, 139.2, 148.1, 162.1 (d, Citation1JC–F 242), 171.3; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 501.2 [M + H]+.
2.2.34. 3-Amino-N-(2-(4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)benzenesulfonamide (5l)
Compound 5l was obtained in 32% yield; m.p. 162–164 °C; TLC: Rf = 0.40 (ethyl acetate/n-hexane 70% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.24 (4H, m, 2 × piperazine-CH2), 3.41 (4H, overlap with water peak, 2 × piperazine-CH2), 3.66 (2H, s, COCH2NH) 4.42 (1H, s, CH), 6.0 (2H, s, exchange with D2O, NH2), 6.77 (1H, d, J = 7.4, Ar-H), 6.90 (1H, d, J = 7.4, Ar-H), 6.99 (1H, s, Ar-H), 7.16 (5H, m, Ar-H), 7.46 (5H, m, 4 × Ar-H, exchange with D2O, SO2NHCH2); 13C NMR (DMSO-d6, 100 MHz): δ 42.4, 44.9, 51.9, 73.5, 112.0, 114.2, 116.9 (d, Citation2JC–F 21), 118.1, 130.3 (d, 3JC–F 8), 130.4, 139.2, 141.4, 150.1, 161.9 (d, Citation1JC–F 242), 166.3; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 501.2 [M + H]+.
2.2.35. 4-Amino-N-(2-(4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)benzenesulfonamide (5m)
Compound 5m was obtained in 44% yield; m.p. 182–184 °C; TLC: Rf = 0.28 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.21 (4H, m, 2 × piperazine-CH2), 3.37 (4H, m, overlap with water peak, 2 × piperazine-CH2), 3.59 (2H, s, COCH2NH) 4.40 (1H, s, CH), 6.0 (2H, s, exchange with D2O, NH2), 6.61 (2H, d, J = 8.8, Ar-H), 7.16 (4H, m, Ar-H), 7.28 (1H, m, exchange with D2O, SO2NHCH2), 7.45 (6H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 42.4, 44.9, 51.7, 73.5, 113.4, 116.3 (2JC–F 21), 125.6, 129.6, 130.3 (d, 3JC–F 8), 139.2, 153.5, 162.0 (d, Citation1JC–F 242), 166.6; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 501.2 [M + H]+.
2.2.36. (1-((2-Aminophenyl)sulfonyl)piperidin-4-yl)(4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)methanone (5n)
Compound 5n was obtained in 70% yield; m.p. 140–142 °C; TLC: Rf = 0.35 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.54 (2H, m, piperidine-CH2), 1.66 (2H, m, piperidine-CH2), 2.47 (4H, m, 2 × piperazine-CH2), 2.54 (3H, m, overlap with DMSO peak, piperidine-CH2, COCH), 3.46 (4H, m, 2 × piperazine-CH2), 3.64 (2H, m, piperidine-CH2), 3.42 (1H, s, CH), 6.06 (2H s, exchange with D2O, NH2), 6.66 (1H, t, J = 8.0, Ar-H), 6.87 (1H, d, J = 8.4, Ar-H), 7.16 (4H, t, J = 8.8, Ar-H), 7.38 (1H, m, Ar-H), 7.55 (4H, m, Ar-H), 7.59 (1H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 42.0, 45.9, 53.0, 75.6, 116.1, 116.3 (d, Citation2JC–F 21), 116.6, 118.2, 130.2, (d, 3JC–F 9), 130.8, 135.6, 139.2, 148.1, 162.1 (d, Citation1JC–F 242), 171.3; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 555.2 [M + H]+.
2.2.37. (1-((3-Aminophenyl)sulfonyl)piperidin-4-yl)(4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)methanone (5o)
Compound 5o was obtained in 52% yield; m.p. 142–144 °C; TLC: Rf = 0.31 (ethyl acetate/n-hexane 70% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.54 (2H, m, piperidine-CH2), 1.68 (2H, m, piperidine-CH2), 2.23 (4H, m, 2 × piperazine-CH2), 2.33 (2H, m, piperidine-CH2), 2.60 (1H, m, COCH), 3.52 (4H, m, 2 × piperazine-CH2), 3.60 (2H, m, piperidine-CH2), 4.42 (1H, s, CH), 5.64 (2H, s, exchange with D2O, NH2), 6.82 (2H, m, Ar-H), 6.92 (1H, s, Ar-H), 7.16 (4H, m, Ar-H), 7.25 (1H, m, Ar-H); 7.46 (4H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 41.9, 46.2, 52.4, 73.5, 112.6, 114.9, 116.2 (d, Citation2JC–F 21), 118.7, 130.3 (d, 3JC–F 8), 130.4, 136.5,139.2, 150.4, 162.0 (d, Citation1JC–F 242), 172.6; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 555.2 [M + H]+.
2.2.38. (1-((4-Aminophenyl)sulfonyl)piperidin-4-yl)(4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)methanone (5p)
Compound 5p was obtained in 74% yield; m.p. 162–164 °C (dec); TLC: Rf = 0.43 (ethyl acetate/n-hexane 70% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.55 (2H, m, piperidine-CH2), 1.66 (2H, m, piperidine-CH2), 2.22 (6H, m, 2 × piperazine-CH2, piperidine-CH2), 2.54 (1H, m, overlap with DMSO peak, COCH), 3.51 (6H, m, 2 × piperazine-CH2, piperidine-CH2), 4.41 (1H, s, CH), 5.64 (2H, s, exchange with D2O, NH2), 6.66 (2H, d, J = 8.8, Ar-H), 7.16 (4H, m, Ar-H), 7.35 (2H, d, J = 8.8, Ar-H), 7.46 (4H, m, Ar-H); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 41.9, 46.2, 52.4, 73.5, 113.5, 116.2 (d, Citation2JC–F 21), 120.6, 130.2, 130.3 (d, 3JC–F 8), 139.2, 153.9, 162.4 (d, Citation1JC–F 242), 172.7; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 555.2 [M + H]+.
2.3. General procedure for the synthesis of compounds 6a–p
The appropriate aminobenzensulfonamides 5a–p (1.0 eq) dissolved in dry DMA (5.0 ml) at 0 °C were treated with Et3N (1.3 eq) and freshly prepared sulfamoyl chloride until consumption of starting material was confirmed (TLC monitoring). Then the solution was quenched with slush and extracted with EtOAc (3 × 20 ml). The combined organic layers were washed with NaHCO3 aqueous solution (3 × 10 ml), HCl aqueous solution 1.0 M (1 × 10 ml), brine (3 × 10 ml), dried over Na2SO4, filtered-off and concentrated under vacuo. The obtained residue was purified by trituration from Et2O to afford the titled sulfamides 6a–p as white solids.
2.3.1. 1-Benzhydryl-4-((3-sulfamoylaminophenyl)sulfonyl)piperazine (6a)
Compound 6a was obtained in 14% yield; m.p. 125–127 °C; TLC: Rf = 0.55 (ethyl acetate/n-hexane 60% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.40 (4H, m, 2 × piperazine-CH2), 2.94 (4H, m, 2 × piperazine-CH2), 4.34 (1H, s, CH), 7.20 (2H, t, J = 7.2, Ar-H), 7.29 (5H, m, Ar-H), 7.40 (5H, m, Ar-H), 7.53 (3H, m, Ar-H, exchange with D2O, NHSO2NH2), 7.61 (1H, t, J = 7.6, Ar-H), 10.04 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.4, 75.3, 117.3, 121.6, 122.7, 127.9, 128.4, 129.5, 129.6, 136.2, 141.3, 143.3; MS (ESI positive) m/z = 487.0 [M + H]+.
2.3.2. 1-Benzhydryl-4-((4-sulfamoylaminophenyl)sulfonyl)piperazine (6b)
Compound 6b was obtained in 42% yield; m.p. 207–210 °C; TLC: Rf = 0.30 (ethyl acetate/n-hexane 50% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.40 (4H, m, 2 × piperazine-CH2), 2.91 (4H, m, 2 × piperazine-CH2), 4.33 (1H, s, CH), 7.19 (2H, t, J = 7.2, Ar-H), 7.28 (4H, t, J = 7.2, Ar-H), 7.38 (8H, m, 6 × Ar-H, exchange with D2O, NHSO2NH2), 7.65 (2H, d, J = 8.4, Ar-H), 10.35 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.3, 75.3, 117.5, 127.3, 127.9, 128.4, 129.5, 130.0, 143.3, 144.9; MS (ESI positive) m/z = 487.0 [M + H]+.
2.3.3. N-(2-(4-Benzhydrylpiperazin-1-yl)-2-oxoethyl)-2-(sulfamoylamino)benzenesulfonamide (6c)
Compound 6c was obtained in 10% yield; m.p. 150–152 °C; TLC: Rf = 0.64 (ethyl acetate/n-hexane 60% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.26 (4H, m, 2 × piperazine-CH2), 3.40 (4H, m, 2 × piperazine-CH2), 3.80 (2H, s, COCH2NH), 4.35 (1H, s, CH), 7.21 (3H, m, Ar-H), 7.33 (4H, m, Ar-H), 7.45 (4H, m, Ar-H), 7.50 (2H, s, exchange with D2O, NHSO2NH2), 7.64 (2H, m, Ar-H), 7.81 (1H, d, J = 8.0, Ar-H), 8.22 (1H, s, exchange with D2O, SO2NHCH2), 8.84 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 42.2, 45.3, 52.0, 75.6, 117.8, 123.1, 127.2, 127.9, 128.5, 129.5, 130.2, 134.6, 137.4, 143.3, 166.2; MS (ESI positive) m/z = 487.0 [M + H]+.
2.3.4. N-(2-(4-Benzhydrylpiperazin-1-yl)-2-oxoethyl)-3-(sulfamoylamino)benzenesulfonamide (6d)
Compound 6d was obtained in 29% yield; m.p. 154–156 °C; TLC: Rf = 0.18 (methanol/dichloromethane 10% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.26 (4H, m, 2 × piperazine-CH2), 3.13 (4H, m, 2 × piperazine-CH2), 3.72 (2H, s, COCH2NH), 4.34 (1H, s, CH), 7.24 (3H, m, Ar-H, exchange with D2O, SO2NHCH2), 7.33 (6H, m, Ar-H), 7.45 (7H, m, 5 × Ar-H, exchange with D2O, NHSO2NH2), 7.62 (1H, m, Ar-H), 8.99 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 42.2, 44.9, 51.9, 75.7, 116.4, 120.7, 122.5, 127.9, 128.6, 129.5, 130.5, 141.1, 141.6, 143.3, 171.3; MS (ESI positive) m/z = 544.0 [M + H]+.
2.3.5. (4-Benzhydrylpiperazin-1-yl)(1-((2-sulfamoylaminophenyl)sulfonyl)piperidin-4-yl)methanone (6e)
Compound 6e was obtained in 65% yield; m.p. 162–164 °C; TLC: Rf = 0.41 (methanol/dichloromethane 10% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.58 (2H, m, piperidine-CH2), 1.70 (2H, m, piperidine-CH2), 2.26 (4H, m, 2 × piperazine-CH2), 2.54 (2H, overlap with DMSO peak, piperidine-CH2), 2.60 (1H, overlap with DMSO peak, COCH), 3.47 (4H, m, 2 × piperazine-CH2), 3.65 (2H, m, piperidine-CH2), 4.33 (1H, s, CH), 7.23 (3H, m, Ar-H), 7.32 (5H, m, Ar-H), 7.43 (5H, m, Ar-H), 7.70 (3H, m, Ar-H, exchange with D2O, NHSO2NH2), 8.95 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 28.5, 36.6, 41.9, 46.0, 53.0, 75.6, 118.8, 121.7, 127.9, 128.6, 129.5, 130.9, 135.5, 138.3, 141.3, 143.3, 172.5; MS (ESI positive) m/z = 598.0 [M + H]+.
2.3.6. (4-Benzhydrylpiperazin-1-yl)(1-((3-sulfamoylaminophenyl)sulfonyl)piperidin-4-yl)methanone (6f)
Compound 6f was obtained in 65% yield; m.p. 230–232 °C; TLC: Rf = 0.13 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.58 (2H, m, piperidine-CH2), 1.68 (2H, m, piperidine-CH2), 2.25 (4H, m, 2 × piperazine-CH2), 2.33 (2H, m, piperidine-CH2), 2.54 (1H, overlap with DMSO peak, COCH), 3.4 (4H, m, 2 × piperazine-CH2), 3.59 (2H, m, piperidine-CH2), 4.32 (1H, s, CH), 7.21 (2H, m, Ar-H), 7.32 (7H, m, 5 × Ar-H, exchange with D2O, NHSO2NH2), 7.45 (5H, m, Ar-H), 7.54 (2H, m, Ar-H), 9.97 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 40.4 (overlap with DMSO peak), 46.2, 52.4, 75.6, 117.1, 121.3, 122.5, 126.2, 127.9, 128.5, 129.5, 130.8, 136.9, 141.3, 172.7; MS (ESI positive) m/z = 598.0 [M + H]+.
2.3.7. (4-Benzhydrylpiperazin-1-yl)(1-((4-sulfamoylaminophenyl)sulfonyl)piperidin-4-yl)methanone (6g)
Compound 6 g was obtained in 44% yield; m.p. 160–162 °C; TLC: Rf = 0.26 (ethyl acetate/n-hexane 70% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.55 (2H, m, piperidine-CH2), 1.67 (2H, m, piperidine-CH2), 2.25 (6H, m, 2 × piperazine-CH2, piperidine-CH2), 2.54 (1H, m, overlap with DMSO peak, COCH), 3.47 (4H, m, 2 × piperazine-CH2), 3.58 (2H, m, piperidine-CH2), 4.31 (1H, s, CH), 7.21 (2H, t, J = 8.6, Ar-H), 7.32 (7H, m, Ar-H), 7.44 (4H, m, 2 × Ar-H, exchange with D2O, NHSO2NH2), 7.64 (2H, d, J = 8.2, Ar-H), 10.28 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.9, 40.4 (overlap with DMSO peak), 46.2, 62.2, 75.6, 113.6, 127.8, 128.4, 129.5, 130.2, 132.6, 143.1, 153.9, 172.70; MS (ESI positive) m/z = 598.0 [M + H]+.
2.3.8. 1-(Bis(4-Fluorophenyl)methyl)-4-((2-sulfamoylaminophenyl)sulfonyl)piperazine (6h)
Compound 6h was obtained in 34% yield; m.p. 170–172 °C (dec); TLC: Rf = 0.56 (methanol/dichloromethane 10% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.41 (4H, m, 2 × piperazine-CH2), 3.03 (4H, m, 2 × piperazine-CH2), 4.44 (1H, s, CH), 7.13 (4H, m, Ar-H), 7.29 (1H, t, J = 7.4, Ar-H), 7.43 (5H, m, Ar-H) , 7.75 (4H, m, 2 × Ar-H, exchange with D2O, NHSO2NH2), 8.95 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 46.8, 51.1, 73.1, 115.9, 116.2 (d, Citation2JC–F 21), 118.3, 123.0, 130.2 (d, 3JC–F 7), 130.8, 135.1, 138.6, 139.1, 161.9 (d, Citation1JC–F 241); 19F NMR (DMSO-d6, 376 MHz): δ –115.4 (2F, s); MS (ESI positive) m/z = 523.0 [M + H]+.
2.3.9. 1-(Bis(4-Fluorophenyl)methyl)-4-((3-sulfamoylaminophenyl)sulfonyl)piperazine (6i)
Compound 6i was obtained in 43% yield; m.p. 132–135 °C; TLC: Rf = 0.30 (ethyl acetate/n-hexane 50% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.39 (4H, m, 2 × piperazine-CH2), 2.96 (4H, m, 2 × piperazine-CH2), 4.43 (1H, s, CH), 7.12 (4H, t, J = 8.8, Ar-H), 7.37 (7H, m, 5 × Ar-H, exchange with D2O, NHSO2NH2), 7.52 (2H, m, Ar-H), 7.59 (1H, m, Ar-H), 10.01 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.2, 73.2, 116.2 (d, Citation2JC–F 21), 117.2, 121.5, 122.7, 130.2 (d, 3JC–F 8), 130.8, 136.1, 139.2, 141.3, 161.9 (d, Citation1JC–F 242); 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 523.0 [M + H]+.
2.3.10. 1-(Bis(4-Fluorophenyl)methyl)-4-((4-sulfamoylaminophenyl)sulfonyl)piperazine (6j)
Compound 6j was obtained in 42% yield; m.p. 176–179 °C (dec); TLC: Rf = 0.28 (methanol/dichloromethane 10% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.38 (4H, m, 2 × piperazine-CH2), 2.90 (4H, m, 2 × piperazine-CH2), 4.42 (1H, s, CH), 7.12 (4H, m, Ar-H), 7.41 (8H, m, 6 × Ar-H, exchange with D2O, NHSO2NH2), 7.65 (2H, d, J = 8.4, Ar-H), 10.33 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 47.0, 51.1, 73.2, 116.2 (d, Citation2JC–F 21), 117.5, 127.2, 129.9, 130.2 (d, 3JC–F 8), 139.2, 144.8, 161.9 (d, Citation1JC–F 242); 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 523.0 [M + H]+.
2.3.11. N-(2-(4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)-2-(sulfamoylamino)benzenesulfonamide (6k)
Compound 6k was obtained in 80% yield; m.p. 183–186 °C (dec); TLC: Rf = 0.23 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.20 (4H, m, 2 × piperazine-CH2), 3.45 (4H, m, 2 × piperazine-CH2), 3.75 (2H, s, COCH2NH) 4.43 (1H, s, CH), 7.17 (5H, m, Ar-H), 7.46 (6H, m, 4 × Ar-H, exchange with D2O, NHSO2NH2), 7.63 (2H, m, Ar-H), 7.80 (1H, d, J = 8.0, Ar-H), 8.23 (1H, m, exchange with D2O, SO2NHCH2), 8.84 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 42.4, 44.9, 51.9, 73.5, 116.3 (d, Citation2JC–F 21), 116.7, 118.2, 123.1, 130.3 (d, 3JC–F 10), 130.5, 134.9, 138.2, 139.3, 162.0 (d, Citation1JC–F 242), 171.4; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 580.0 [M + H]+.
2.3.12. N-(2-(4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)-3-(sulfamoylamino)benzenesulfonamide (6l)
Compound 6l was obtained in 29% yield; m.p. 155–157 °C (dec); TLC: Rf = 0.28 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.27 (4H, m, 2 × piperazine-CH2), 3.41 (4H, overlap with water peak, 2 × piperazine-CH2), 3.76 (2H, s, COCH2NH) 4.43 (1H, s, CH), 7.16 (5H, m, Ar-H), 7.26 (2H, s, exchange with D2O, NHSO2NH2), 7.36 (1H, m, exchange with D2O, SO2NHCH2), 7.42 (6H, m, Ar-H), 7.61 (1H, s, Ar-H), 9.93 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 42.4, 44.9, 51.9, 73.5, 116.9 (d, Citation2JC–F 21), 118.4, 122.3, 123.2, 130.4 (d, 3JC–F 8), 130.9, 137.2, 139.1, 141.2, 161.9 (d, Citation1JC–F 242), 167.2; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 580.0 [M + H]+.
2.3.13. N-(2-(4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)-2-oxoethyl)-4-(sulfamoylamino)benzene sulfonamide (6m)
Compound 6m was obtained in 35% yield; m.p. 157–159 °C; TLC: Rf = 0.12 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 2.22 (4H, m, 2 × piperazine-CH2), 3.37 (4H, m, overlap with water peak, 2 × piperazine-CH2), 3.68 (2H, s, COCH2NH), 4.42 (1H, s, CH), 7.16 (4H, m, Ar-H), 7.22 (2H, d, J = 8.8, Ar-H), 7.40 (2H, s, exchange with D2O, NHSO2NH2), 7.47 (5H, m, 4 × Ar-H, exchange with D2O, SO2NHCH2), 7.76 (2H, d, J = 8.8, Ar-H), 10.18 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 42.4, 44.9, 51.7, 73.5, 116.3 (2JC–F 21), 116.6, 129.6, 130.1, 130.3 (d, 3JC–F 8), 139.3, 142.9, 162.0 (d, Citation1JC–F 242), 166.6; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 580.0 [M + H]+.
2.3.14. (4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)(1-((2-sulfamo-ylaminophenyl)sulfonyl)piperidin-4-yl)methanone (6n)
Compound 6n was obtained in 44% yield; m.p. 140–142 °C; TLC: Rf = 0.50 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.56 (2H, m, piperidine-CH2), 1.70 (2H, m, piperidine-CH2), 2.24 (4H, m, 2 × piperazine-CH2), 2.58 (2H, m, piperidine-CH2), 2.68 (1H, m, COCH), 3.48 (4H, m, 2 × piperazine-CH2), 3.66 (2H, m, piperidine-CH2), 4.42 (1H, s, CH), 7.22 (5H, m, Ar-H), 7.43 (4H, m, Ar-H), 7.60 (2H, s, exchange with D2O, NHSO2NH2), 7.65 (2H, m, Ar-H), 7.76 (1H, m, Ar-H), 8.95 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 41.9, 46.2, 52.4, 73.5, 115.1, 115.4 (d, Citation2JC–F 21), 118.0, 123.0, 129.4 (d, 3JC–F 8), 130.8, 135.4, 138.4, 139.6, 161.1 (d, Citation1JC–F 242), 168.4; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 634.0 [M + H]+.
2.3.15. (4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)(1-((3-sulfamoylaminophenyl)sulfonyl)piperidin-4-yl)methanone (6o)
Compound 6o was obtained in 35% yield; m.p. 160–162 °C (dec); TLC: Rf = 0.12 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.54 (2H, m, piperidine-CH2), 1.68 (2H, m, piperidine-CH2), 2.23 (4H, m, 2 × piperazine-CH2), 2.33 (2H, m, piperidine-CH2), 2.54 (1H, m, overlap with DMSO peak, COCH), 3.47 (4H, m, 2 × piperazine-CH2), 3.60 (2H, m, piperidine-CH2), 4.42 (1H, s, CH), 7.16 (5H, m, Ar-H), 7.32 (2H, s, exchange with D2O, NHSO2NH2), 7.54 (7H, m, Ar-H), 9.81 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 41.9, 46.2, 52.4, 73.5, 116.2 (d, Citation2JC–F 21), 118.7, 122.0, 122.9, 130.3 (d, 3JC–F 8), 130.9, 135.3, 137.4, 139.6, 162.0 (d, Citation1JC–F 242), 172.6; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 634.0 [M + H]+.
2.3.16. (4-(bis(4-Fluorophenyl)methyl)piperazin-1-yl)(1-((4-sulfamo-ylaminophenyl)sulfonyl)piperidin-4-yl)methanone (6p)
Compound 6p was obtained in 46% yield; m.p. 150–152 °C (dec); TLC: Rf = 0.16 (methanol/dichloromethane 5% v/v); 1H NMR (DMSO-d6, 400 MHz): δ 1.55 (2H, m, piperidine-CH2), 1.67 (2H, m, piperidine-CH2), 2.26 (6H, m, 2 × piperazine-CH2, piperidine-CH2), 2.54 (1H, m, overlap with DMSO peak, COCH), 3.46 (4H, m, 2 × piperazine-CH2), 3.59 (2H, m, piperidine-CH2), 4.41 (1H, s, CH), 7.15 (4H, t, J = 8.6, Ar-H), 7.32 (2H, d, J = 8.2, Ar-H), 7.46 (6H, m, 4 × Ar-H, exchange with D2O, NHSO2NH2), 7.64 (2H, d, J = 8.2, Ar-H) 10.28 (1H, s, exchange with D2O, NHSO2NH2); 13C NMR (DMSO-d6, 100 MHz): δ 28.6, 36.8, 41.9, 46.2, 52.4, 73.5, 116.2 (d, Citation2JC–F 21), 116.6, 129.7, 130.2, 130.3 (d, 3JC–F 8), 139.2, 143.1, 162.4 (d, Citation1JC–F 242), 172.70; 19F NMR (DMSO-d6, 376 MHz): δ –115.6 (2F, s); MS (ESI positive) m/z = 634.0 [M + H]+.
2.4. Carbonic anhydrase inhibition assay
An Applied Photophysics (Leatherhead, UK) stopped-flow instrument has been used for assaying the CA-catalysed CO2 hydration activityCitation18. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlier, and represent the mean from at least three different determinationsCitation19,Citation20. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation21.
3. Results and discussion
3.1. Chemistry
The sulfamide moiety is able to establish a more extended network of hydrogen bonds within the enzymatic pocket compared to classical sulfonamides, due to the presence of the additional nitrogen atom, but unfortunately this feature does not confer selectivity against different CA isoformsCitation16,Citation17. Therefore, the purpose of this study is not only to explore the activity of the sulfamides against the two CA isoforms of Vibrio cholerae, but also to propose specific structural changes to obviate to the problem of selectivity.
The novel sulfamides reported here feature the sulfamides groups (in ortho-, meta-, or para positions on the benzene ring), connected into a highly flexible alkyl-aryl scaffolds. In particular, we designed two series of compounds which differ by the spacer connecting the 1-benzhydrylpiperazin tail with the benzene-sulfamide zinc binding functionality (Scheme 1)Citation16.
Scheme 1. General synthetic scheme of compounds 6a–p.
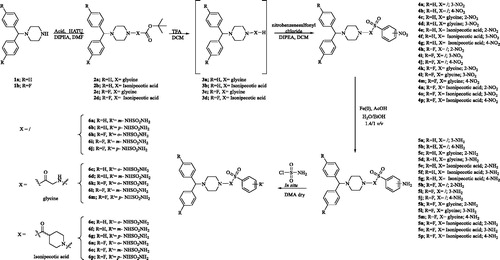
The planned synthesis of the compounds 6a–p provides a first step during which the coupling of the benzhydryl piperazines 1a,b with the appropriate acids takes place to give the intermediates 2a–d, followed by treatment with trifluoroacetic acid (TFA) to afford the alkylamines 3a–d. In the second step, the free amines were coupled with commercially available nitrobenzenesulfonyl chlorides and the obtained nitro derivatives 4a–p were reduced with iron, Fe(0), in acidic media, to afford the amines 5a–p. Finally, the desired compounds 6a–p () were obtained by treatment of amines 5a–p with freshly prepared sulfamoyl chlorideCitation16. All compounds were purified by trituration from Et2O to afford products with high purity in yields between 10 and 80%; the analytical and spectroscopic data of the purified compounds are in agreement with the purposed structures (see section 2 for details).
Figure 1. Structures of the compounds 6a–p.
3.2. Carbonic anhydrase inhibition
All synthesised compounds 6a–p, were tested in vitro for their inhibitory activity against the cytosolic isoenzymes hCA I and II and bacterial enzymes VchCAα and VchCAβ by means of a stopped-flow carbon dioxide hydration assayCitation18, and their activities were compared to the standard CAI acetazolamide (AAZ). The following structure–activity–relationship (SARs) can be drawn from the data of :
Table 1. Inhibition data of hCA I, hCA II, VchCA α, VchCA β with compounds reported here and the standard sulfonamide inhibitor acetazolamide (AAZ) by a stopped flow CO2 hydrase assayCitation18.
hCA I was effectively inhibited by most compounds investigated here, with inhibition constants spanning between 43.1 and 839.9 nM. It is interesting to note the case of compounds 6m and 6p that demonstrate the best activity (KIs = 47.8 and 49.1 nM), both having the sulfamide group in para position of the aromatic ring but differing for the glycine and isonipecotic acid spacer, respectively. The removal of the fluorine atoms in the aromatic rings of compound 6g (corresponding to the non-halogenated analogue of 6d), decreased the activity (KI = 601.3 nM), suggesting that substitution with halogens as –F, in this case, is advantageous for the inhibitory activity. The same trend concerned compounds 6j and its corresponding non-halogenated 6b, which showed the following inhibition data KIs of 64.6 and 555.1 nM, respectively. On the other hand, the presence of fluorine atoms in aromatic rings of compound 6i (KI = 75.0 nM), did not have a significant impact on its activity, both compound having a similar activity to that of 6a (KI = 66.2 nM). The inhibition potencies were dependent not only on the presence of halogens but also on the sulfamide regioisomer considered. In fact, the potency ranking for compounds 6j, 6i, and 6h (KIs = 64.6, 75.0, 842.6 nM), 6m, 6l, and 6k (KIs = 47.8, 71.1, 337.8 nM) and for compounds 6p, 6o, and 6n (KIs = 49.1, 72.0, 682.6 nM) was para>meta>ortho.
The ubiquitous hCA II was inhibited by sulfamides 6a–p, with KIs spanning between range 181.7 and 1430.7 nM. Likewise hCA I, the derivatives with sulfamide group in para position of the aromatic ring showed better activity than the corresponding meta and ortho analogues. In fact, compound 6g shows a lower KI = 226.7 nM than its meta-substituted analogue 6f (KI = 576.4 nM) or ortho-substituted derivative 6e (KI = 1360.1 nM). The introduction of fluorine atoms on the phenyl rings of compounds 6g, 6f, and 6e to give derivatives 6o, 6p, and 6n, caused a slight change in the inhibition trend, with a better KI of the ortho-substituted compared to the meta-substituted derivative. It is interesting to underline the case of the compound 6p (KI = 181.7 nM), which is the best inhibitor against hCA II. The substitution of the isonipecotic acid spacer of 6p with the glycine spacer of 6m did not show any impact on the inhibitory activity. The removal of the fluorine atoms in 6g did not substantially altered the activity compared to the perhydro derivative. Equally to hCA I, for hCA II the most considerable differences of inhibitory activity concerned the position of the ZBG in the phenyl ring, as demonstrated for example by compounds 6d (meta-substituted) and 6c (ortho-substituted) with KIs of 396.4 and 850.2 nM, respectively.
The general tendencies described above were also applicable for the VchCAα, with this latter being moderately inhibited by benzenesulfamide derivatives prepared in this study (KIs values in the range of 91.4 and 3748.6 nM). As reported for the human isoforms discussed above, also for VchCAα, the worst inhibitors are reconfirmed to be the compounds that present the ZBG in the ortho position of the phenyl ring (6c, 6e, 6h, 6k, and 6n with KIs = 930.8, 879.3, 2797.0, 1747.0, and 3748.6 nM, respectively). The most active compounds were the halogen derivative 6l and the non-halogen analogue 6d, which exhibited KI values of 91.4 and 95.7 nM, respectively. It is interesting to note that the introduction of the rigid linker of isonipecotic acid determined a lower efficacy against this isoform, as shown by the compounds 6f (KI = 654.2 nM) and 6o (KI = 497.2 nM), compared with the above-mentioned derivatives 6d and 6l. This improvement of VchCAα inhibition potency might be due to the flexible glycine spacer, which probably increases the interactions of the inhibitor with active site amino acids residues.
VchCAβ was the least inhibited among the enzymes herein considered and showed KIs spanning between 1578.2 and 4676.1 nM. Interestingly, the meta derivatives 6o and 6i (KIs = 1578.2 and 1742.2 nM) were the most potent inhibitors against the VchCAβ, compared to the corresponding para analogues 6p and 6j (KIs = 4533.7 and 4349.2 nM). On the contrary, the meta derivatives 6a and 6f (KIs = 4676.1 and 3792.0 nM) were less effective with respect to para derivatives 6b and 6g (KIs = 1880.7 and 2538.1 nM). It is rather difficult to explain this result but we could speculate that the introduction of fluorine atoms leads to a decrease in the inhibitory potency of sulfamides against VchCAβ. In fact the fluorination of compounds 6g, 6b, and 6d (KIs = 2538.1, 1880.7, and 3782.8 nM), to afford 6p, 6j, and 6l, respectively, decreased the inhibition efficacy (KIs = 4533.7, 4349.2, and >10,000 nM, respectively), with the potency of compound 6l being very low. Finally, it is important to emphasise the selectivity of some compounds against VchCAα with respect to the β-bacterial isoform. For instance, compounds 6l and 6d showed a nanomolar efficacy against VchCAα (KIs of 91.4 and 95.7 nM) but 6l was totally ineffective against the β-isoform, whereas compound 6d showed a KI of 3782.8 nM against VchCAβ.
4. Conclusions
In this work, we reported the investigation of benzenesulfamide derivatives (–NH-SO2NH2) as an alternative zinc binding class of inhibitors to the primary aromatic sulfonamides in the design of new potential bacterial CAIsCitation17. The “tail approach”, the method that consists in varying the terminal portions of the molecule to selectively enhance the inhibition of the target isoform(s), was applied to the phenyl sulfamide scaffold. All compounds were synthesised by a convenient and efficient method and were screened for in vitro inhibition against cytosolic isoenzymes hCA I, II, VchCAα and VchCAβ by using a stopped-flow CO2 hydrase assay method. The obtained results suggest that the benzhydryl piperazine moiety appended to benzene-sulfamide functionalities are not favourable for the interaction with Vibrio cholerae CA isoforms. Conversely, the same moiety is slightly more favourable for the inhibition of hCA I, as demonstrated by compounds 6f, 6m, 6p, and 6d that showed interesting activities against this isoform, with a nanomolar inhibition constant (KIs = 43.1, 47.8, 49.1, and 58.1 nM, respectively), being thus more potent than AAZ (as standard inhibitor).
Disclosure statement
The authors do not report conflict of interest.
Related Research Data
References
- Silva AJ, Benitez JA. Vibrio cholerae biofilms and cholera pathogenesis. PLoS Negl Trop Dis 2016;10:e0004330.
- Abuaita BH, Withey JH. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect Immun 2009;77:4111–20.
- Alafeefy AM, Ceruso M, Al-Tamimi AM, et al. Quinazoline–sulfonamides with potent inhibitory activity against the α-carbonic anhydrase from Vibrio cholerae. Bioorg Med Chem 2014;22:5133–40.
- Unterweger D, Miyata ST, Bachmann V, et al. The Vibrio cholerae type VI secretion system employs diverse effector modules for intraspecific competition. Nat Commun 2014;5:3549.
- Zuckerman JN, Rombo L, Fisch A. The true burden and risk of cholera: implications for prevention and control. Lancet Infect Dis 2007;7:521–30.
- Cholera Annual Report 2017. Weekly Epidemiological Record 8 September 2017; 92, 36:521–36. Available from: http://www.who.int/wer/2017/wer9236/en/.
- Thomson JJ, Withey JH. Bicarbonate increases binding affinity of Vibrio cholerae ToxT to virulence gene promoters. J Bacteriol 2014;196:3872–80.
- Del Prete S, Isik S, Vullo D, et al. DNA cloning, characterization, and inhibition studies of an α-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. J Med Chem 2012;55:10742–8.
- Del Prete S, De Luca V, Scozzafava A, et al. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium, Vibrio cholerae. J Enzyme Inhib Med Chem 2014;29:23–7.
- Del Prete S, Vullo D, De Luca V, et al. Sulfonamide inhibition studies of the β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem 2016;24:1115–20.
- Nocentini A, Moi D, Balboni G, et al. Discovery of thiazolin-4-one-based aromatic sulfamates as a new class of carbonic anhydrase isoforms I, II, IV, and IX inhibitors. Bioorg Chem 2018;77:293–9.
- (a) Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem 2014;6:1149–65. (b) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91. (c) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16. (d) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. (d) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:E25.
- (a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- (a) D'Ambrosio K, Smaine FZ, Carta F, et al. Development of potent carbonic anhydrase inhibitors incorporating both sulfonamide and sulfamide groups. J Med Chem 2012;55:6776–83. (b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (c) Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (d) Abbate F, Winum JY, Potter BV, et al. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett 2004;14:231–4.
- Berrino E, Bua S, Mori M, et al. Novel sulfamide-containing compounds as selective carbonic anhydrase I inhibitors. Molecules 2017;22:pii:1049.
- (a) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. (b) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48. (c) Ward C, Langdon SP, Mullen P, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev 2013;39:171–9. (d) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. (e) Casey JR, Morgan PE, Vullo D, et al. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004;47:2337–47.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (b) Capasso C, Supuran CT. An overview of the alpha-, beta-and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. (c) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704. (d) Vermelho AB, da Silva CV, Ricci JE, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46. (e) de Menezes DR, Calvet CM, Rodrigues GC, et al. Hydroxamic acid derivatives: a promising scaffold for rational compound optimization in Chagas disease. J Enzyme Inhib Med Chem 2016;31:964–73. (f) Nocentini A, Cadoni R, Dumy P, et al. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J Enzyme Inhib Med Chem 2018;33:286–9.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7. (b) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54. (c) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. (d) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9. (e) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47. (f) Dogne JM, Hanson J, Supuran C, Pratico D. Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des 2006;12:971–5.
- (a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. (b) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3. (c) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (d) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11. (e) Di Fiore A, De Simone G, Alterio V, et al. The anticonvulsant sulfamide JNJ-26990990 and its S,S-dioxide analog strongly inhibit carbonic anhydrases: solution and X-ray crystallographic studies. Org Biomol Chem 2016;14:4853–8.
- (a) Krall N, Pretto F, Decurtins W, et al. A small‐molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. (b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. (c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure–activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. (d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. (e) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. (f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8. (g) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.