
Abstract
We report the synthesis and biological evaluation of a new series of 3- or 4-(substituted)phenylisoxazolones as HNE inhibitors. Due to tautomerism of the isoxazolone nucleus, two isomers were obtained as final compounds (2-NCO and 5-OCO) and the 2-NCO derivatives were the most potent with IC50 values in the nanomolar range (20–70 nM). Kinetic experiments indicated that 2-NCO 7d and 5-OCO 8d are both competitive HNE inhibitors. Molecular modelling on 7d and 8d suggests for the latter a more crowded region about the site of the nucleophilic attack, which could explain its lowered activity. In addition molecular dynamics (MD) simulations showed that the isomer 8d appears more prone to form H-bond interactions which, however, keep the reactive sites quite distant for the attack by Ser195. By contrast the amide 7d appears more mobile within the active pocket, since it makes single H-bond interactions affording a favourable orientation for the nucleophilic attack.
Introduction
Proteases are enzymes implicated in cellular reactions involving the cleavage of protein substratesCitation1. Serine proteases are characterised by the presence of a serine residue at the active siteCitation2. They are divided into four classes: chymotrypsin, subtilisin, carboxypeptidase Y, and caseinolytic proteaseCitation3. Human neutrophil elastase (HNE), proteinase 3 (PR3), cathepsin G, and the recently discovered NSP4Citation4 are serine proteases belonging to the chymotrypsin family and represent neutrophil serine proteases (NSP)Citation5,Citation6. NSP are synthesised and expressed in neutrophil azurophilic granules. Neutrophils play a pivotal role in host defence, inflammation and tissue remodelling, and HNE is a key mediator of neutrophil-driven inflammationCitation7.
HNE is a small, basic, and soluble glycoprotein of about 30 kDaCitation8 that performs many functions in our body. For example, HNE is involved in the maintenance of tissue homeostasis; it degrades a variety of structural proteins of the extracellular matrix, such as elastin, fibronectin, collagen, proteoglycan, and laminin; and it repairs damaged tissueCitation9. Moreover, HNE plays an important and dual role in inflammation by degrading pro-inflammatory cytokines to reduce the intensity of the inflammation but also increasing the secretion of pro-inflammatory factorsCitation10. In the case of infection, HNE plays as an intracellular function in destroying phagocytosed pathogens, as well as an extracellular function through the formation of neutrophil extracellular traps, which can trap and kill microorganismsCitation11,Citation12. The powerful HNE activity is tightly controlled by the presence of extracellular neutralizing endogenous serine protease inhibitors, such as α-1 antitrypsin, secretory leucocyte protease inhibitor (SLPI), and elafinCitation13. α1-Antitrypsin is a water-soluble glycoprotein classified as systemic HNE inhibitor that is synthesised in the liver and is particularly abundant in the lungsCitation14. In contrast, SLPI and elafin are classified as alarm inhibitors because they are produced and released directly into the airway epithelium in response to the release of cytokines, regulating the immune response and inflammatory processesCitation15–17. Under physiological conditions, the balance between protease and anti-protease supports the maintenance of tissue homeostasis. However, if this balance fails, excessive HNE activity can cause tissue the damage associated with some serious chronic diseasesCitation4,Citation6. Among the pathologies associated with increased HNE activity are adult respiratory distress syndrome (ARDS)Citation18, chronic obstructive pulmonary disease (COPD)Citation19,Citation20, cystic fibrosis (CF)Citation21,Citation22, and other disorders with an inflammatory component, such as rheumatoid arthritisCitation23, atherosclerosisCitation24, psoriasis, and dermatitisCitation25. Recently, it was demonstrated that HNE is also implicated in the progression of non-small cell lung cancerCitation26.
Although a large number of molecules have been reported as HNE inhibitorsCitation27,Citation28, only two drugs are currently available for clinical use: Prolastin (purified α1-AT), a peptide drug synthesised by recombinant DNA techniquesCitation29 and used for the treatment of α1-antitripsin deficiency (AATD)Citation30, and Sivelestat (Elaspol®100), a non-peptide low molecular weight compound, belonging to the second generation of HNE inhibitorsCitation31. Sivelestat has an IC50 = 44 nM and is currently marketed only in Japan and South KoreaCitation32,Citation33 (). AZD9668 (Alvelestat, AstraZeneca)Citation34 and BAY 85-8501 (Bayer HealthCare)Citation35 are two potent HNE inhibitors, belonging to the third and fifth generations of HNE inhibitorsCitation31, respectively, that have recently reached Phase II of clinical trials for COPD, CF, and BE ().
Figure 1. Potent HNE inhibitors.
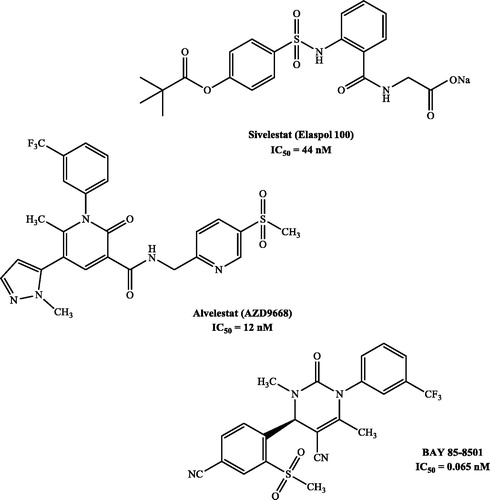
In recent research focused on the development of new HNE inhibitors, we investigated various bicyclic scaffolds, such as indazoleCitation36,Citation37, indoleCitation38, and cinnolinoneCitation39, compounds. The most interesting inhibitors were effective in the nanomolar range, with a potency comparable to Sivelestat. Subsequently, we focused our research on the design and synthesis of monocyclic derivatives with an isoxazol-5(2H)-one core, and the first series of 3/4-alkyl-(di)substituted isoxazolones was recently publishedCitation40. We report here the synthesis of a new series of isoxazolone derivatives bearing a (substituted)phenyl at positions 3 and 4 and biological evaluation of their HNE inhibitory activity.
Material and methods
All melting points were determined on a Büchi apparatus (New Castle, DE) and are uncorrected. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. Merck F-254 commercial plates (Merck, Durham, NC) were used for analytical TLC to follow the course of reactions. Silica gel 60 (Merck 70–230 mesh, Merck, Durham, NC) was used for column chromatography. 1H NMR, 13C NMR, HSQC, HMBC, and NOESY bidimensional spectra were recorded on an Avance 400 instrument (Bruker Biospin Version 002 with SGU, Bruker Inc., Billerica, MA). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using the solvent as an internal standard. Coupling constants (J values) are given in Hz and were calculated using TopSpin 1.3 software (Nicolet Instrument Corp., Madison, WI) and are rounded to the nearest 0.1 vHz. Mass spectra (m/z) were recorded on an ESI-TOF mass spectrometer (Brucker Micro TOF, Bruker Inc., Billerica, MA), and reported mass values are within the error limits of ± 5 ppm mass units. Microanalyses indicated by the symbols of the elements or functions were performed with a Perkin–Elmer 260 elemental analyser (PerkinElmer, Inc., Waltham, MA) for C, H, and N, and the results were within ± 0.4% of the theoretical values, unless otherwise stated. Reagents and starting material were commercially available.
Chemistry
3-Methyl-2-(3-methylbenzyl)-4-phenylisoxazol-5(2H)-one (2)
A mixture of the appropriate intermediate (1aCitation41) (0.57 mmol), K2CO3 (1.14 mmol), and 1-(chloromethyl)-3-methylbenzene (0.86 mmol) in 2 ml of anhydrous acetonitrile was stirred at reflux for 2 h. After cooling, the mixture was concentrated in vacuo, diluted with ice-cold water (10 ml), and extracted with ethyl acetate (3 × 15 ml). The organic phase was dried over sodium sulphate and the solvent was evaporated in vacuo to afford the final compound 2, which was purified by column chromatography using cyclohexane/ethyl acetate 2:1 as eluent. Yield =57%; oil. 1H NMR (CDCl3-d1) δ 2.35 (s, 6H, 2 × CH3), 4.79 (s, 2H, CH2), 7.07–7.14 (m, 3H, Ar), 7.22–7.28 (m, 2H, Ar), 7.38 (t, 2H, Ar, J = 7.8 Hz), 7.45 (d, 2H, Ar, J = 7.2 Hz). 13C NMR (CDCl3-d1) δ 12.44 (CH3), 21.63 (CH3), 54.90 (CH2), 103.58 (C), 125.16 (CH), 127.17 (CH), 128.21 (CH), 128.58 (CH), 128.82 (CH), 128.90 (CH), 129.40 (CH), 129.77 (C), 133.45 (C), 138.75 (C), 158.65 (C), 169.53 (C). ESI-MS calcd. for C18H17NO2, 279.33; found: m/z 280.13 [M + H]+. Anal. C18H17NO2 (C, H, N).
General procedure for compounds (3a–c)
To a suspension of the appropriate 4-substituted benzensulfonyl chloride (0.16 mmol) in 3 ml of anhydrous pyridine, 0.79 mmol of intermediate 1aCitation41 was added. The mixture was stirred at room temperature for 4 h. The solvent was concentrated in vacuo to afford the final compounds 3a–c which were purified by column chromatography using cyclohexane/ethyl acetate in different ratio (2:1 for 3a, 4:1 for 3b) or toluene/ethyl acetate 9:1 for 3c as eluents.
2-((4-Hydroxyphenyl)sulfonyl)-3-methyl-4-phenylisoxazol-5(2H)-one (3a)
Yield = 30%; mp = 50–51 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.57 (s, 3H, CH3), 6.93 (d, 2H, Ar, J = 8.8 Hz), 7.30–7.40 (m, 5H, Ar), 7.78 (d, 2H, Ar, J = 8.8 Hz). 13C NMR (CDCl3-d1) δ 14.59 (CH3), 113.94 (C), 116.39 (CH), 121.92 (C), 126.91 (C), 128.77 (CH), 128.87 (CH), 129.19 (CH), 131.88 (CH), 158.94 (C), 162.88 (C), 168.80 (C). ESI-MS calcd. for C16H13NO5S, 331.34; found: m/z 332.05 [M + H]+. Anal. C16H13NO5S (C, H, N).
4-((3-Methyl-5-oxo-4-phenylisoxazol-2(5H)-yl)sulfonyl)phenyl pivalate (3b)
Yield = 15%; mp = 115–116 °C (EtOH). 1H NMR (CDCl3-d1) δ 1.35 (s, 9H, C(CH3)3), 2.58 (s, 3H, CH3), 7.26 (d, 2H, Ar, J = 8.0 Hz), 7.32 (d, 2H, Ar, J = 8.6 Hz), 7.35–7.40 (m, 3H, Ar), 7.95 (d, 2H, Ar, J = 8.6 Hz). 13C NMR (CDCl3-d1) δ 14.50 (CH3), 26.97 (CH3), 29.37 (C), 39.36 (C), 114.27 (C), 122.81 (CH), 127.03 (C), 128.45 (CH), 128.81 (CH), 129.11 (CH), 130.39 (CH), 130.97 (CH), 156.83 (C), 157.77 (C), 167.45 (C), 175.75 (C). ESI-MS calcd. for C21H21NO6S, 415.46; found: m/z 416.11 [M + H]+. Anal. C21H21NO6S (C, H, N).
N-(4-((3-methyl-5-oxo-4-phenylisoxazol-2(5H)-yl)sulfonyl)phenyl)pivalamide (3c)
Yield = 72%; mp = 153–155 °C (EtOH). 1H NMR (CDCl3-d1) δ 1.30 (s, 9H, C(CH3)3), 2.58 (s, 3H, CH3), 7.26 (d, 1H, Ar, J = 6.8 Hz), 7.36–7.42 (m, 4H, Ar), 7.70 (exch br s, 1H, NH), 7.79 (d, 2H, Ar, J = 8.8 Hz), 7.84 (d, 2H, Ar, J = 8.8 Hz). 13C NMR (CDCl3-d1) δ 14.61 (CH3), 27.42 (CH3), 44.65 (C), 113.95 (C), 119.39 (CH), 125.36 (C), 125.85 (CH), 128.43 (CH), 128.81 (CH), 128.97 (CH), 129.88 (CH), 130.68 (CH), 130.91 (CH), 134.00 (C), 144.98 (C), 158.06 (C), 165.00 (C), 177.20 (C). ESI-MS calcd. for C21H22N2O5S, 414.47; found: m/z 415.13 [M + H]+. Anal. C21H22N2O5S (C, H, N).
General procedure for compounds (4a–h, 4n–t)
To a suspension of the appropriate substrates 1a–e (1aCitation41, 1bCitation42, 1c,dCitation43, and 1eCitation44) (0.86 mmol) in 10 ml of anhydrous THF, 1.72 mmol of sodium hydride (60% dispersion in mineral oil), and 1.03 mmol of the appropriate acyl/aroyl chloride were added. The mixture was stirred at room temperature overnight. The solvent was concentrated in vacuo to obtain the final compounds 4a–h and 4n–t which were purified by column chromatography using hexane/ethyl acetate (5:1 for 4a,c,d; 5:2 for 4e,g), cyclohexane/ethyl acetate (1:1 for 4f; 3:1 for 4t; 4:1 for 4h; 5:1 for 4o–s; 6:1 for 4n), or toluene/ethyl acetate 9.5:0.5 (for 4b) as eluents.
3-Methyl-2-(3-methylbenzoyl)-4-phenylisoxazol-5(2H)-one (4a)
Yield = 52%; mp = 85–88 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.43 (s, 3H, m-CH3-Ph), 2.79 (s, 3H, CH3), 7.35–7.40 (m, 3H, Ar), 7.43–7.51 (m, 4H, Ar), 7.70–7.75 (m, 2H, Ar). 13C NMR (CDCl3-d1) δ 15.09 (CH3), 21.58 (CH3), 108.39 (C), 127.08 (CH), 127.62 (C), 128.28 (CH), 128.50 (CH), 128.81 (CH), 129.08 (CH), 130.28 (CH), 131.14 (C), 134.07 (CH), 138.32 (C), 154.62 (C), 163.79 (C), 165.90 (C). IR (υ) = 1690 cm−1 (CO amide), 1750 cm−1 (CO ester). ESI-MS calcd. for C18H15NO3, 293.32; found: m/z 294.11 [M + H]+. Anal. C18H15NO3 (C, H, N).
2-(Cyclopropanecarbonyl)-3-methyl-4-phenylisoxazol-5(2H)-one (4b)
Yield = 63%; mp = 83–86 °C (EtOH). 1H NMR (DMSO-d6) δ 1.02–1.07 (m, 2H, CH2 cC3H5), 1.09–1.15 (m, 2H, CH2 cC3H5), 2.36–2.41 (m, 1H, CH cC3H5), 2.58 (s, 3H, CH3), 7.35–7.41 (m, 1H, Ar), 7.43–7.48 (m, 4H, Ar). 13C NMR (DMSO-d6) δ 10.82 (CH2), 13.25 (CH3), 15.14 (CH), 106.28 (C), 128.25 (C), 128.58 (CH), 129.10 (CH), 129.35 (CH), 154.69 (C), 166.05 (C), 169.02 (C). IR (υ) = 1695 cm−1 (CO amide), 1755 cm−1 (CO ester). ESI-MS calcd. for C14H13NO3, 243.26; found: m/z 244.09 [M + H]+. Anal. C14H13NO3 (C, H, N).
3-Methyl-2-(4-methylbenzoyl)-4-phenylisoxazol-5(2H)-one (4c)
Yield = 35%; mp = 134–136 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.51 (s, 3H, p-CH3-Ph), 2.87 (s, 3H, CH3), 7.38 (d, 2H, Ar, J = 8.0 Hz), 7.43–7.48 (m, 1H, Ar), 7.51–7.58 (m, 4H, Ar), 7.92 (d, 2H, Ar, J = 8.4 Hz). 13C NMR (CDCl3-d1) δ 15.37 (CH3), 22.02 (CH3), 108.19 (C), 127.85 (C), 128.46 (CH), 128.59 (CH), 128.95 (CH), 129.19 (CH), 128.19 (CH), 130.28 (CH), 144.55 (C), 154.60 (C), 158.00 (C), 163.62 (C), 166.13 (C). IR (υ) = 1672 cm−1 (CO amide), 1755 cm−1 (CO ester). ESI-MS calcd. for C18H15NO3, 293.32; found: m/z 294.11 [M + H]+. Anal. C18H15NO3 (C, H, N).
3-Methyl-2-(2-methylbenzoyl)-4-phenylisoxazol-5(2H)-one (4d)
Yield = 42%; mp = 99–100 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.47 (s, 3H, o-CH3-Ph), 2.85 (s, 3H, CH3), 7.32 (d, 2H, Ar, J = 6.8 Hz), 7.40–7.49 (m, 7H, Ar). 13C NMR (CDCl3-d1) δ 15.37 (CH3), 20.22 (CH3), 108.95 (C), 126.27 (CH), 128.10 (C), 128.74 (CH), 129.14 (CH), 129.43 (CH), 129.60 (CH), 131.53 (CH), 132.10 (CH), 132.52 (C), 137.22 (C), 153.99 (C), 164.91 (C), 166.31 (C). IR (υ) = 1688 cm−1 (CO amide), 1767 cm−1 (CO ester). ESI-MS calcd. for C18H15NO3, 293.32; found: m/z 294.11 [M + H]+. Anal. C18H15NO3 (C, H, N).
3-Methyl-4-phenyl-2-(3-(trifluoromethyl)benzoyl)isoxazol-5(2H)-one (4e)
Yield = 42%; mp = 96–97 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.85 (s, 3H, CH3), 7.40–7.45 (m, 1H, Ar), 7.48–7.54 (m, 4H, Ar), 7.69 (t, 1H, Ar, J = 8.0 Hz), 7.90 (d, 1H, Ar, J = 7.8 Hz), 8.15 (d, 1H, Ar, J = 8.0 Hz), 8.20 (s, 1H, Ar). 13C NMR (CDCl3-d1) δ 15.61 (CH3), 109.53 (C), 125.39 (C), 127.34 (CH), 127.38 (C), 127.84 (CH), 129.33 (CH), 129.49 (CH), 129.63 (CH), 129.71 (CH), 130.28 (C), 131.69 (C), 132.75 (CH), 133.47 (CH), 154.88 (C), 162.74 (C), 166.07 (C). 19F NMR (CDCl3-d1) δ − 62.82. IR (υ) = 1689 cm−1 (CO amide), 1738 cm−1 (CO ester). ESI-MS calcd. for C18H12F3NO3, 347.29; found: m/z 348.08 [M + H]+. Anal. C18H12F3NO3 (C, H, N).
3-Methyl-2-(3-(methylsulfonyl)benzoyl)-4-phenylisoxazol-5(2H)-one (4f)
Yield = 15%; mp = 185–186 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.83 (s, 3H, CH3), 3.13 (s, 3H, CH3SO2), 7.40–7.45 (m, 1H, Ar), 7.48–7.55 (m, 4H, Ar), 7.75 (t, 1H, Ar, J = 8.0 Hz), 8.19 (d, 2H, Ar, J = 7.6 Hz), 8.46 (s, 1H, Ar). 13C NMR (CDCl3-d1) δ 14.98 (CH3), 44.52 (CH3), 109.12 (C), 127.11 (C), 128.84 (CH), 128.93 (CH), 129.02 (CH), 129.75 (CH), 131.47 (CH), 132.84 (C), 134.47 (CH), 141.35 (C), 154.22 (C), 161.55 (C), 165.36 (C). ESI-MS calcd. for C18H15NO5S, 357.38; found: m/z 358.07 [M + H]+. Anal. C18H15NO5S (C, H, N).
3-(3-Methyl-5-oxo-4-phenyl-2,5-dihydroisoxazole-2-carbonyl)benzonitrile (4g)
Yield = 7%; mp = 118–119 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.83 (s, 3H, CH3), 7.45–7.55 (m, 5H, Ar), 7.67 (t, 1H, Ar, J = 7.2 Hz), 7.89 (d, 1H, Ar, J = 6.4 Hz), 8.16 (d, 1H, Ar, J = 7.6 Hz), 8.20 (s, 1H, Ar). 13C NMR (CDCl3-d1) δ 14.94 (CH3), 107.22 (C), 112.76 (C), 118.63 (C), 127.98 (CH), 128.67 (CH), 128.91 (CH), 129.54 (CH), 130.71 (CH), 131.86 (CH), 134.54 (C), 134.95 (C), 135.62 (CH), 139.52 (C), 157.64 (C), 165.87 (C). ESI-MS calcd. for C18H12N2O3, 304.30; found: m/z 305.09 [M + H]+. Anal. C18H12N2O3 (C, H, N).
4-(3-Methyl-5-oxo-4-phenyl-2,5-dihydroisoxazole-2-carbonyl)benzonitrile (4h)
Yield = 46%; mp = 178–180 °C dec. (EtOH). 1H NMR (CDCl3-d1) δ 2.82 (s, 3H, CH3), 7.37–7.42 (m, 1H, Ar), 7.45–7.50 (m, 4H, Ar), 7.80 (d, 2H, Ar, J = 8.4 Hz), 8.00 (d, 2H, Ar, J = 8.4 Hz). 13C NMR (CDCl3-d1) δ 14.94 (CH3), 109.39 (C), 116.49 (C), 117.69 (C), 127.10 (C), 128.85 (CH), 128.93 (CH), 129.03 (CH), 130.41 (CH), 132.32 (CH), 135.19 (C), 154.20 (C), 161.73 (C), 165.45 (C). ESI-MS calcd. for C18H12N2O3, 304.30; found: m/z 305.09 [M + H]+. Anal. C18H12N2O3 (C, H, N).
2-(Cyclopropanecarbonyl)-3,4-diphenylisoxazol-5(2H)-one (4n)
Yield = 21%; mp = 100–103 °C (EtOH). 1H NMR (CDCl3-d1) δ 1.06–1.11 (m, 2H, CH2 cC3H5), 1.16–1.21 (m, 2H, CH2 cC3H5), 1.80–1.86 (m, 1H, CH cC3H5), 7.17–7.22 (m, 2H, Ar), 7.31–7.40 (m, 5H, Ar), 7.39 (d, 1H, Ar, J = 7.2 Hz), 7.46 (d, 2H, Ar, J = 7.6 Hz). 13C NMR (CDCl3-d1) δ 10.39 (CH2), 12.51 (CH), 103.59 (C), 128.03 (CH), 128.31 (CH), 128.37 (CH), 128.72 (CH), 129.16 (CH), 129.84 (CH), 134.92 (C), 142.60 (C), 170.20 (C), 180.73 (C). ESI-MS calcd. for C19H15NO3, 305.33; found: m/z 306.11 [M + H]+. Anal. C19H15NO3 (C, H, N).
4-Methyl-2-(3-methylbenzoyl)-3-phenylisoxazol-5(2H)-one (4o)
Yield = 27%; oil. 1H NMR (CDCl3-d1) δ 1.96 (s, 3H, CH3), 2.40 (s, 3H, m-CH3-Ph), 7.32–7.40 (m, 2H, Ar), 7.44–7.50 (m, 5H, Ar), 7.70 (d, 2H, Ar, J = 6.8 Hz). 13C NMR (CDCl3-d1) δ 7.57 (CH3), 21.63 (CH3), 105.74 (C), 127.37 (CH), 128.01 (CH), 128.33 (CH), 128.67 (CH), 128.82 (C), 130.51 (CH), 130.60 (CH), 131.07 (C), 134.31 (CH), 138.46 (C), 156.85 (C), 165.24 (C), 169.02 (C). ESI-MS calcd. for C18H15NO3, 293.32; found: m/z 294.11 [M + H]+. Anal. C18H15NO3 (C, H, N).
2-(Cyclopropanecarbonyl)-4-methyl-3-phenylisoxazol-5(2H)-one (4p)
Yield = 31%; oil. 1H NMR (CDCl3-d1) δ 1.04–1.09 (m, 2H, CH2 cC3H5), 1.10–1.15 (m, 2H, CH2 cC3H5), 1.86 (s, 3H, CH3), 2.38–2.44 (m, 1H, CH cC3H5), 7.36–7.41 (m, 2H, Ar), 7.43–7.48 (m, 3H, Ar). 13C NMR (CDCl3-d1) δ 7.30 (CH3), 10.55 (CH2), 12.71 (CH), 104.82 (C), 127.78 (C), 128.21 (CH), 128.35 (CH), 128.51 (CH), 130.44 (CH), 154.89 (C), 168.03 (C), 168.99 (C). ESI-MS calcd. for C14H13NO3, 243.26; found: m/z 244.09 [M + H]+. Anal. C14H13NO3 (C, H, N).
2-(3-Methylbenzoyl)-3-phenylisoxazol-5(2H)-one (4q)
Yield = 72%; oil. 1H NMR (CDCl3-d1) δ 2.46 (s, 3H, m-CH3-Ph), 6.54 (s, 1H, CH), 7.43–7.50 (m, 5H, Ar), 7.82–7.87 (m, 2H, Ar), 8.11–8.16 (m, 2H, Ar). 13C NMR (CDCl3-d1) δ 21.24 (CH3), 85.88 (CH), 126.76 (CH), 126.98 (C), 127.94 (CH), 129.04 (CH), 129.18 (C), 130.38 (CH), 131.23 (CH), 135.69 (CH), 139.06 (C), 160.47 (C), 164.26 (C), 165.70 (C). IR (υ) = 1600 cm−1 (CO amide), 1757 cm−1 (CO ester). ESI-MS calcd. for C17H13NO3, 279.29; found: m/z 280.09 [M + H]+. Anal. C17H13NO3 (C, H, N).
2-(Cyclopropanecarbonyl)-3-phenylisoxazol-5(2H)-one (4r)
Yield = 57%; oil. 1H NMR (CDCl3-d1) δ 1.10–1.16 (m, 2H, CH2 cC3H5), 1.23–1.28 (m, 2H, CH2 cC3H5), 1.84–1.90 (m, 1H, CH cC3H5), 6.34 (s, 1H, CH), 7.42–7.47 (m, 3H, Ar), 7.74–7.79 (m, 2H, Ar). 13C NMR (CDCl3-d1) δ 10.82 (CH2), 12.71 (CH), 85.75 (CH), 126.55 (CH), 128.87 (CH), 129.27 (C), 130.26 (CH), 164.15 (C), 165.51 (C), 168.48 (C). ESI-MS calcd. for C13H11NO3, 229.23; found: m/z 230.08 [M + H]+. Anal. C13H11NO3 (C, H, N).
2-(3-Methylbenzoyl)-3-(4-nitrophenyl)isoxazol-5(2H)-one (4s)
Yield = 40%; mp = 177–180 °C dec. (EtOH). 1H NMR (DMSO-d6) δ 2.42 (s, 3H, m-CH3-Ph), 7.13 (s, 1H, CH), 7.54 (t, 1H, Ar, J = 7.6 Hz), 7.64 (d, 1H, Ar, J = 7.6 Hz), 7.95–8.00 (m, 2H, Ar), 8.20 (d, 2H, Ar, J = 8.8 Hz), 8.36 (d, 2H, Ar, J = 8.8 Hz). 13C NMR (DMSO-d6) δ 21.27 (CH3), 124.60 (CH), 125.76 (C), 126.91 (CH), 127.99 (CH), 128.28 (CH), 128.91 (CH), 130.18 (CH), 131.20 (C), 133.90 (CH), 136.50 (C), 138.35 (C), 148.54 (C), 160.45 (C), 167.84 (C). ESI-MS calcd. for C17H12N2O5, 324.29; found: m/z 325.08 [M + H]+. Anal. C17H12N2O5 (C, H, N).
2-(Cyclopropanecarbonyl)-3-(4-nitrophenyl)isoxazol-5(2H)-one (4t)
Yield = 23%; mp = 160–163 °C (EtOH). 1H NMR (CDCl3-d1) δ 1.16–1.21 (m, 2H, CH2 cC3H5), 1.27–1.32 (m, 2H, CH2 cC3H5), 1.87–1.94 (m, 1H, CH cC3H5), 6.43 (s, 1H, CH), 7.96 (d, 2H, Ar, J = 8.8 Hz), 8.31 (d, 2H, Ar, J = 8.8 Hz). 13C NMR (CDCl3-d1) δ 10.55 (CH2), 12.71 (CH), 86.27 (CH), 121.42 (C), 124.67 (CH), 127.37 (CH), 135.22 (C), 148.74 (C), 162.53 (C), 168.75 (C). ESI-MS calcd. for C13H10N2O5, 274.23; found: m/z 275.06 [M + H]+. Anal. C13H10N2O5 (C, H, N).
General procedure for compounds (4i, 4l)
To a suspension of 4-(pivaloyoxy)benzoic acidCitation45 or 4-(pivalamido)benzoic acidCitation46 (0.32 mmol) in 1 ml of anhydrous toluene, 0.64 mmol of SOCl2, and a catalytic amount of DMF (0.05 mmol) were added. The mixture was stirred at reflux for 2 h. The solvent was concentrated in vacuo and the crude compound was used without purification and added to a previously prepared solution composed of 0.29 mmol of intermediate 1aCitation41 and 0.64 mmol of sodium hydride in 5 ml of anhydrous THF. The mixture was stirred at room temperature overnight. After evaporation of the solvent, the product was purified by column chromatography using toluene/ethyl acetate 9:1 for 4i and hexane/acetone 4:1 for 4l as eluents.
N-(4-(3-methyl-5-oxo-4-phenyl-2,5-dihydroisoxazole-2-carbonyl)phenyl)pivalamide (4i)
Yield = 10%; mp = 132–134 °C (EtOH). 1H NMR (CDCl3-d1) δ 1.37 (s, 9H, C(CH3)3), 2.82 (s, 3H, CH3), 7.41–7.46 (m, 1H, Ar), 7.49–7.55 (m, 4H, Ar), 7.73 (d, 2H, Ar, J = 8.4 Hz), 7.99 (d, 2H, Ar, J = 8.8 Hz). 13C NMR (CDCl3-d1) δ 15.11 (CH3), 27.56 (CH3), 39.95 (C), 118.86 (CH), 126.01 (C), 127.54 (C), 127.66 (CH), 128.47 (CH), 128.82 (CH), 129.05 (CH), 131.65 (CH), 142.83 (C), 154.74 (C), 162.65 (C), 166.04 (C), 176.92 (C). ESI-MS calcd. for C22H22N2O4, 378.42; found: m/z 379.16 [M + H]+. Anal. C22H22N2O4 (C, H, N).
4-(3-Methyl-5-oxo-4-phenyl-2,5-dihydroisoxazole-2-carbonyl)phenyl pivalate (4l)
Yield = 41%; mp = 130–132 °C (EtOH). 1H NMR (CDCl3-d1) δ 1.37 (s, 9H, C(CH3)3), 2.80 (s, 3H, CH3), 7.22 (d, 2H, Ar, J = 8.8 Hz), 7.35–7.40 (m, 1H, Ar), 7.45–7.50 (m, 4H, Ar), 7.99 (d, 2H, Ar, J = 8.8 Hz). 13C NMR (CDCl3-d1) δ 15.07 (CH3), 27.07 (CH3), 39.26 (C), 108.25 (C), 121.70 (CH), 127.56 (C), 128.16 (CH), 128.53 (CH), 128.83 (CH), 129.04 (CH), 131.70 (CH), 154.55 (C), 155.05 (C), 162.54 (C), 165.80 (C), 176.34 (C). IR (υ) = 1678 cm−1 (CO amide), 1753 cm−1 (CO ester), 1768 cm−1 (CO ester). ESI-MS calcd. for C22H21NO5, 379.41; found: m/z 380.15 [M + H]+. Anal. C22H21NO5 (C, H, N).
General procedure for compounds (4m, 4u)
To a solution of intermediate 1bCitation42 or 1fCitation47 (0.32 mmol) in 2 ml of t-BuOH, 0.35 mmol of K2CO3 and 0.64 mmol of m-toluoyl chloride were added. The mixture was stirred at reflux for 3 h. The solvent was concentrated in vacuo, diluted with ice-cold water (10 ml), and extracted with DCM (3 × 15 ml). The organic phase was dried over sodium sulphate and the solvent was evaporated in vacuo to afford the final compounds 4m,u, which were purified by column chromatography using cyclohexane/ethyl acetate in different ratio: 5:1 for 4m and 1:1 for 4u as eluent.
2-(3-Methylbenzoyl)-3,4-diphenylisoxazol-5(2H)-one (4m)
Yield = 14%; mp = 160–163 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.42 (s, 3H, m-CH3-Ph), 7.26–7.32 (m, 5H, Ar), 7.37–7.48 (m, 7H, Ar), 7.72–7.77 (m, 2H, Ar). 13C NMR (CDCl3-d1) δ 21.40 (CH3), 101.40 (C), 127.48 (CH), 128.31 (CH), 128.46 (CH), 128.53 (CH), 128.83 (CH), 130.74 (CH), 132.49 (C), 132.63 (C), 134.21 (C), 134.52 (CH), 138.50 (C), 142.65 (C), 157.66 (C), 165.80 (C). ESI-MS calcd. for C23H17NO3, 355.39; found: m/z 356.12 [M + H]+. Anal. C23H17NO3 (C, H, N).
N-(4-(2-(3-methylbenzoyl)-5-oxo-2,5-dihydroisoxazol-3-yl)phenyl)acetamide (4u)
Yield = 21%; oil. 1H NMR (CDCl3-d1) δ 2.20 (s, 3H, CH3CO), 2.45 (s, 3H, m-CH3-Ph), 6.50 (s, 1H, CH), 7.42 (t, 1H, Ar, J = 7.8 Hz), 7.50 (d, 1H, Ar, J = 7.6 Hz), 7.56 (exch br s, 1H, NH), 7.62 (d, 2H, Ar, J = 8.0 Hz), 7.76 (d, 2H, Ar, J = 8.4 Hz), 7.98–8.13 (m, 2H, Ar). 13C NMR (CDCl3-d1) δ 21.30 (CH3), 24.81 (CH3), 85.70 (C), 119.72 (CH), 127.45 (CH), 127.93 (CH), 128.88 (CH), 129.65 (C), 131.17 (CH), 134.11 (C), 135.75 (CH), 138.99 (C), 139.73 (C), 156.05 (C), 157.65 (C), 167.14 (C), 168.90 (C). ESI-MS calcd. for C19H16N2O4, 336.34; found: m/z 337.11 [M + H]+. Anal. C19H16N2O4 (C, H, N).
General procedure for compounds (5e, 5f)
To a cooled (0 °C) suspension of 5dCitation48 (0.68 mmol) in anhydrous CH2Cl2 (2 ml), Et3N (1.36 mmol) and 2.04 mmol of appropriate acyl chloride were added. The mixture was stirred at 0 °C for 2 h and then at room temperature for an additional 2 h. The solvent was evaporated, cold water was added and the mixture was neutralised with 0.5 N NaOH. The reaction mixture was extracted with CH2Cl2 (3 × 15 ml), the solvent was dried over sodium sulphate, evaporated in vacuo, and compounds 5e and 5f were purified by column chromatography using dichloromethane/methanol (9:1 for 5e; 99:1 for 5f) as eluents.
Ethyl 2-(4-acetamidophenyl)-3-oxobutanoate (5e)
Yield = 67%; mp = 112–113 °C (EtOH). 1H NMR (CDCl3-d1) showed a 3:1 mixture of aldo-enol tautomers: δ 1.24 (t, 3H, CH2CH3, J = 7.0 Hz), 1.61 (t, 1H, CH2CH3, J = 7.0 Hz), 1.81 (s, 1H, COCH3), 2.12 (s, 3H, COCH3), 2.15 (s, 4H, NHCOCH3), 4.16 (q, 2.7H, CH2CH3, J = 7.2 Hz), 4.65 (s,1H, CH), 7.06 (d, 0.7H, Ar, J = 8.0 Hz), 7.23 (d, 2H, Ar, J = 8.4 Hz), 7.48 (d, 2.7H, Ar, J = 8.0 Hz), 7.92 (exch br s, 1.3H, NH), 13.07 (exch br s, 0.3H, OH). ESI-MS calcd. for C14H17NO4, 263.29; found: m/z 264.12 [M + H]+. Anal. C14H17NO4 (C, H, N).
Ethyl 2-(4-(ciclopropanecarboxamido)phenyl)-3-oxobutanoate (5f)
Yield = 78%; oil. 1H NMR (CDCl3-d1) showed the only aldo tautomer: δ 0.72–0.77 (m, 2H, CH2 cC3H5), 0.80–0.85 (m, 2H, CH2 cC3H5), 1.20 (t, 3H, CH2CH3, J = 7.2 Hz), 1.39–1.46 (m, 1H, CH cC3H5), 2.35 (s, 3H, COCH3), 4.17 (q, 2H, CH2CH3, J = 7.2 Hz), 4.63 (s, 1H, CH), 7.19 (d, 2H, Ar, J = 8.4 Hz), 7.46 (d, 2H, Ar, J = 7.6 Hz), 7.52 (exch br s, 1H, NH). ESI-MS calcd. for C16H19NO4, 289.33; found: m/z 290.13 [M + H]+. Anal. C16H19NO4 (C, H, N).
General procedure for compounds (6a–e)
A 3.00 mmol of appropriate intermediate 5a–cCitation48–50 and 5e,f was dissolved in 1.3 ml of water and heated at 80 °C. To this solution, 3.3 mmol of hydroxylamine hydrochloride in 6.5 ml of methanol was added. The mixture was stirred at reflux for 5 h. After evaporation of the solvent, the residue was mixed with ice-cold water (20 ml). Compounds 6a,c were recovered by extraction with ethyl acetate (3 × 15 ml), while compounds 6b,d,e were recovered by vacuum filtration. The final compounds 6b,d were purified by crystallisation with ethanol, while the compounds 6a,c,e were purified by column chromatography using dichoromethane/methanol 9:1 as eluent.
3-Methyl-4-(p-tolyl)isoxazol-5(2H)-one (6a)
Yield = 35%; oil. 1H NMR (CDCl3-d1) δ 2.17 (s, 3H, CH3Ph), 2.28 (s, 3H, CH3), 7.09 (d, 2H, Ar, J = 6.0 Hz), 7.28 (d, 2H, Ar, J = 6.0 Hz), 9.03 (exch br s, 1H, NH). ESI-MS calcd. for C11H11NO2, 189.21; found: m/z 190.08 [M + H]+. Anal. C11H11NO2 (C, H, N).
4-(3-Methyl-5-oxo-2,5-dihydroisoxazol-4-yl)benzonitrile (6b)
Yield = 72%; mp = 114–116 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.43 (s, 3H, CH3), 7.70 (s, 4H, Ar). IR (υ) = 2270 cm−1 (CN). ESI-MS calcd. for C11H8N2O2, 200.19; found: m/z 201.06 [M + H]+. Anal. C11H8N2O2 (C, H, N).
3-Methyl-4-(4-nitrophenyl)isoxazol-5(2H)-one (6c)
Yield = 27%; mp = 208–209 °C (EtOH). 1H NMR (DMSO-d6) δ 2.19 (s, 3H, CH3), 7.83 (d, 2H, Ar, J = 9.2 Hz), 7.94 (d, 2H, Ar, J = 8.4 Hz). ESI-MS calcd. for C10H8N2O4, 220.18; found: m/z 221.05 [M + H]+. Anal. C10H8N2O4 (C, H, N).
N-(4-(3-methyl-5-oxo-2,5-dihydroisoxazol-4-yl)phenyl)acetamide (6d)
Yield = 75%; mp = 210–212 °C (EtOH). 1H NMR (DMSO-d6) δ 2.01 (s, 3H, NHCOCH3), 2.27 (s, 3H, CH3), 7.43 (d, 2H, Ar, J = 8.0 Hz); 7.56 (d, 2H, Ar, J = 8.0 Hz), 9.92 (exch br s, 1H, NHCOCH3), 12.54 (exch br s, 1H, NH). ESI-MS calcd. for C12H12N2O3, 232.24; found: m/z 233.09 [M + H]+. Anal. C12H12N2O3 (C, H, N).
N-(4-(3-methyl-5-oxo-2,5-dihydroisoxazol-4-yl)phenyl)cyclopropanecarboxamide (6e)
Yield = 58%; mp = 208–210 °C (EtOH). 1H NMR (DMSO-d6) δ 0.70–0.75 (m, 4H, 2 × CH2 cC3H5), 1.70–1.75 (m,1H, CH), 2.12 (s, 3H, CH3), 7.40 (d, 2H, Ar, J = 8.4 Hz), 7.47 (d, 2H, Ar, J = 8.4 Hz), 9.97 (exch br s, 1H, NHCO cC3H5), 12.50 (exch br s, 1H, NH). ESI-MS calcd. for C14H14N2O3, 258.27; found: m/z 259.10 [M + H]+. Anal. C14H14N2O3 (C, H, N).
General procedure for compounds (7a–e, 8a,b,d,e)
Compounds 7a–e and 8a,b,d,e were obtained following the same procedure performed for compounds 4a–h, 4n–t but starting from precursors 6a–e. The solvent was concentrated in vacuo to obtain the final compounds which were purified by column chromatography using petroleum ether/ethyl acetate 10:1 for 7a/8a, hexane/ethyl acetate (5:1 for 7b/8b; 5:2 for 7c), dichloromethane/methanol (98:2 for 7d/8d; 99:1 for 7e/8e) as eluents.
3-Methyl-2-(3-methylbenzoyl)-4-(p-tolyl)isoxazol-5(2H)-one (7a)
Yield = 60%; oil. 1H NMR (CDCl3-d1) δ 2.39 (s, 3H, p-CH3Ph), 2.43 (s, 3H, m-CH3Ph), 2.79 (s, 3H, CH3), 7.27 (d, 2H, Ar, J = 8.4 Hz), 7.37–7.42 (m, 4H, Ar), 7.70 (d, 2H, Ar, J = 6.8 Hz). 13 C NMR (CDCl3-d1) δ 15.05 (CH3), 21.33 (CH3), 108.19 (C), 124.63 (C), 127.03 (CH), 128.30 (CH), 128.89 (CH), 129.52 (CH), 130.24 (CH), 131.27 (C), 134.00 (CH), 138.28 (C), 138.48 (C), 154.09 (C), 163.82 (C), 166.07 (C). ESI-MS calcd. for C19H17NO3, 307.34; found: m/z 308.12 [M + H]+. Anal. C19H17NO3 (C, H, N).
4-(3-Methyl-2-(3-methylbenzoyl)-5-oxo-2,5-dihydroisoxazol-4-yl)benzonitrile (7b)
Yield = 26%; mp = 108–110 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.44 (s, 3H, CH3Ph), 2.84 (s, 3H, CH3), 7.38–7.48 (m, 2H, Ar), 7.65 (d, 2H, Ar, J = 8.4 Hz), 7.71 (d, 2H, Ar, J = 8.0 Hz), 7.76 (d, 2H, Ar, J = 8.4 Hz). 13C NMR (CDCl3-d1) δ 15.19 (CH3), 21.37 (CH3), 29.70 (C), 106.24 (C), 112.03 (C), 118.41 (C), 127.13 (CH), 128.43 (CH), 129.43 (CH), 130.34 (CH), 130.72 (C), 132.52 (CH), 132.74 (C), 134.45 (CH), 138.49 (C), 155.56 (C), 163.79 (C), 165.15 (C). ESI-MS calcd. for C19H14N2O3, 318.33; found: m/z 319.10 [M + H]+. Anal. C19H14N2O3 (C, H, N).
3-Methyl-2-(3-methylbenzoyl)-4-(4-nitrophenyl)isoxazol-5(2H)-one (7c)
Yield = 6%; mp = 152–154 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.45 (s, 3H, CH3Ph), 2.86 (s, 3H, CH3), 7.39–7.46 (m, 2H, Ar), 7.72 (d, 4H, Ar, J = 8.8 Hz), 8.32 (d, 2H, Ar, J = 8.8 Hz). 13C NMR (CDCl3-d1) δ 13.21 (CH3), 21.32 (CH3), 29.70 (C), 124.02 (CH), 127.16 (CH), 128.45 (CH), 129.56 (CH), 130.36 (CH), 130.65 (C), 134.51 (CH), 134.69 (C), 138.52 (C), 147.37 (C), 155.78 (C), 163.80 (C). ESI-MS calcd. for C18H14N2O5, 338.31; found: m/z 339.09 [M + H]+. Anal. C18H14N2O5 (C, H, N).
N-(4-(3-methyl-2-(3-methylbenzoyl)-5-oxo-2,5-dihydroisoxazol-4-yl)phenyl)acetamide (7d)
Yield = 37%; mp = 159–160 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.14 (s, 3H, NHCOCH3), 2.41 (s, 3H, CH3Ph), 2.75 (s, 3H, CH3), 7.34–7.40 (m, 4H, Ar), 7.58 (d, 2H, Ar, J = 8.4 Hz), 7.67 (s, 2H, Ar), 7.97 (exch br s, 1H, NH). 13C NMR (CDCl3-d1) δ 15.06 (CH3), 21.35 (CH3), 24.53 (CH3), 120.11 (CH), 123.11 (C), 126.99 (CH), 128.33 (CH), 128.94 (CH), 129.61 (CH), 130.23 (CH), 131.11 (C), 131.27 (CH), 134.12 (CH), 138.29 (C), 154.40 (C), 163.79 (C), 166.26 (C), 168.87 (C). ESI-MS calcd. for C20H18N2O4, 350.37; found: m/z 351.13 [M + H]+. Anal. C20H18N2O4 (C, H, N).
N-(4-(3-methyl-2-(3-methylbenzoyl)-5-oxo-2,5-dihydroisoxazol-4-yl)phenyl)ciclopropanecarboxamide (7e)
Yield = 6%; mp = 120–123 °C (EtOH). 1H NMR (CDCl3-d1) δ 0.84–0.89 (m, 2H, CH2 cC3H5), 1.07–1.12 (m, 2H, CH2 cC3H5), 1.24 (s, 1H, CH), 2.43 (s, 3H, CH3Ph), 2.79 (s, 3H, CH3), 7.36–7.41 (m, 2H, Ar), 7.46 (d, 2H, Ar, J = 8.4 Hz), 7.61 (d, 2H, Ar, J = 8.4 Hz), 7.70 (d, 2H, Ar, J = 7.2 Hz). 13C NMR (CDCl3-d1) δ 8.24 (CH2), 15.13 (CH3), 15.88 (CH), 21.41 (CH3), 107.20 (C), 119.79 (CH), 127.03 (CH), 128.33 (CH), 129.68 (CH), 130.25 (CH), 130.30 (C), 134.09 (CH), 134.10 (C), 137.70 (C), 138.33 (C), 139.50 (C), 157.61 (C), 163.83 (C), 180.70 (C). ESI-MS calcd. for C22H20N2O4, 376.41; found: m/z 377.15 [M + H]+. Anal. C22H20N2O4 (C, H, N).
3-Methyl-4-(p-tolyl)isoxazol-5-yl 3-methylbenzoate (8a)
Yield = 12%; oil. 1H NMR (CDCl3-d1) δ 2.33 (s, 3H, p-CH3Ph), 2.38 (s, 3H, CH3), 2.41 (s, 3H, m-CH3Ph), 7.18 (d, 2H, Ar, J = 8.0 Hz), 7.24 (d, 2H, Ar, J = 8.4 Hz), 7.38 (t, 1H, Ar, J = 8.0 Hz), 7.47 (d, 1H, Ar, J = 7.2 Hz), 7.93 (s, 2H, Ar). 13 C NMR (CDCl3-d1) δ 12.21 (CH3), 21.23 (CH3), 100.50 (C), 127.97 (CH), 128.10 (CH), 128.75 (CH), 129.63 (CH), 130.10 (C), 131.26 (CH), 133.30 (C), 135.57 (CH), 138.84 (C), 154.20 (C), 158.90 (C), 165.20 (C). ESI-MS calcd. for C19H17NO3, 307.34; found: m/z 308.12 [M + H]+. Anal. C19H17NO3 (C, H, N).
4-(4-Cyanophenyl)-3-methylisoxazol-5-yl 3-methylbenzoate (8b)
Yield = 20%; mp = 98–100 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.41 (s, 3H, CH3), 2.43 (s, 3H, CH3Ph), 7.43 (t, 1H, Ar, J = 8.0 Hz), 7.47 (d, 2H, Ar, J = 8.4 Hz), 7.51 (d, 1H, Ar, J = 7.6 Hz), 7.67 (d, 2H, Ar, J = 8.4 Hz), 7.92 (s, 2H, Ar). 13 C NMR (CDCl3-d1) δ 12.31 (CH3), 21.25 (CH3), 29.70 (C), 111.68 (C), 118.37 (C), 126.33 (C), 128.02 (CH), 128.61 (CH), 128.95 (CH), 131.30 (CH), 132.75 (CH), 133.27 (C), 136.04 (CH), 139.11 (C), 160.61 (C), 161.90 (C). ESI-MS calcd. for C19H14N2O3, 318.33; found: m/z 319.10 [M + H]+. Anal. C19H14N2O3 (C, H, N).
4-(4-Acetamidophenyl)-3-methylisoxazol-5-yl3-methylbenzoate (8d)
Yield = 10%; mp = 57–60 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.15 (s, 3H, NHCOCH3), 2.36 (s, 3H, CH3), 2.41 (s, 3H, CH3Ph), 7.29 (d, 3H, Ar, J = 8.4 Hz), 7.38 (t, 1H, Ar, J = 8.0 Hz), 7.46 (exch br s, 1H, NH), 7.51 (d, 2H, Ar, J = 8.4 Hz), 7.91 (s, 2H, Ar). 13C NMR (CDCl3-d1) δ 12.23 (CH3), 21.24 (CH3), 24.59 (CH3), 29.69 (C), 120.11 (CH), 123.90 (C), 126.71 (C), 127.97 (CH), 128.83 (CH), 129.67 (CH), 131.26 (CH), 135.70 (CH), 137.61 (C), 138.91 (C), 161.21 (C), 162.31 (C), 168.39 (C). ESI-MS calcd. for C20H18N2O4, 350.37; found: m/z 351.13 [M + H]+. Anal. C20H18N2O4 (C, H, N).
4-(4-(Ciclopropanecarboxamido)phenyl)-3-methylisoxazol-5-yl3-me-thylbenzoate (8e)
Yield = 8%; mp = 80–82 °C (EtOH). 1H NMR (CDCl3-d1) δ 0.80–0.90 (m, 4H, 2 × CH2 cC3H5), 1.35–1.40 (m, 1H, CH), 2.37 (s, 3H, CH3), 2.41 (s, 3H, CH3Ph), 7.28 (d, 2H, Ar, J = 8.0 Hz), 7.38 (t, 1H, Ar, J = 7.6 Hz), 7.40 (exch br s,1H, NH), 7.47 (d, 1H, Ar, J = 7.6 Hz), 7.52 (d, 2H, Ar, J = 7.2 Hz), 7.92 (s, 2H, Ar). 13C NMR (CDCl3-d1) δ 8.15 (CH2), 12.24 (CH3), 14.90 (CH), 21.25 (CH3), 100.52 (C), 119.91 (CH), 127.97 (CH), 128.79 (CH), 128.85 (CH), 131.25 (CH), 132.05 (C), 135.68 (CH), 138.70 (C), 138.89 (C), 154.20 (C), 158.90 (C), 165.20 (C), 180.70 (C). ESI-MS calcd. for C22H20N2O4, 376.41; found: m/z 377.15 [M + H]+. Anal. C22H20N2O4 (C, H, N).
General procedure for compounds (10a–c)
To suspension of the substrate 9Citation51 (0.37 mmol) in tert-Butanol (3 ml), K2CO3 (0.41 mmol), and 0.74 mmol of the appropriate acyl chloride were added. The mixture was stirred at reflux for 3 h. After evaporation of the solvent, the residue was mixed with ice-cold water (20 ml) and extracted with ethyl acetate (3 × 15 ml). The organic phase was dried over sodium sulphate, and the solvent was evaporated in vacuo to afford the final compounds 10a–c, which were purified by column chromatography using cyclohexane/ethyl acetate (5:1) as eluent.
1-Propionylbenzo[c]isoxazol-3(1H)-one (10a)
Yield = 7%; oil. 1H NMR (CDCl3-d1) δ 1.28 (t, 3H, CH2CH3, J = 7.4 Hz), 2.83 (q, 2H, CH2CH3, J = 7.2 Hz), 7.38 (t 1H, Ar, J = 7.6 Hz), 7.78 (t 1H, Ar, J = 7.6 Hz), 7.89 (d, 1H, Ar, J = 8.0 Hz), 8.11 (d, 1H, Ar, J = 8.4 Hz). 13C NMR (CDCl3-d1) δ 9.72 (CH3), 20.70 (CH2), 120.35 (CH), 122.50 (C), 124.09 (CH), 130.31 (CH), 133.90 (CH), 142.44 (C), 166.02 (C), 172.05 (C). ESI-MS calcd. for C10H9NO3, 191.18; found: m/z 192.06 [M + H]+. Anal. C10H9NO3 (C, H, N).
1-Pentanoylbenzo[c]isoxazol-3(1H)-one (10b)
Yield = 12%; oil. 1H NMR (CDCl3-d1) δ 0.87 (t, 3H, CH3CH2CH2CH2CO, J = 6.8 Hz), 1.53–1.58 (m, 2H, CH3CH2CH2CH2CO), 1.98–2.03 (m, 2H, CH3CH2CH2CH2CO), 3.63 (t, 2H, CH3CH2CH2CH2CO, J = 6.8 Hz), 7.22 (d, 1H, Ar, J = 8.4 Hz), 7.31 (t, 1H, Ar, J = 7.4 Hz), 7.68 (t, 1H, Ar, J = 7.6 Hz), 7.86 (d, 1H, Ar, J = 7.6 Hz). 13C NMR (CDCl3-d1) δ 13.10 (CH3), 22.15 (CH2), 27.65 (CH2), 28.21 (CH2), 120.31 (CH), 122.49 (C), 124.00 (CH), 130.33 (CH), 133.90 (CH), 142.41 (C), 165.31 (C), 172.22 (C). ESI-MS calcd. for C12H13NO3, 219.24; found: m/z 220.09 [M + H]+. Anal. C12H13NO3 (C, H, N).
1-(3-Methylbenzoyl)benzo[c]isoxazol-3(1H)-one (10c)
Yield = 53%; mp = 116–119 °C (EtOH). 1H NMR (CDCl3-d1) δ 2.44 (s, 3H, CH3), 7.39–7.45 (m, 3H, Ar), 7.75–7.80 (m, 2H, Ar), 7.83 (t 1H, Ar, J = 8.4 Hz), 7.92 (d, 1H, Ar, J = 8.0 Hz), 8.22 (d, 1H, Ar, J = 8.4 Hz). 13C NMR (CDCl3-d1) δ 21.40 (CH3), 115.83 (CH), 117.51 (C), 125.60 (CH), 126.03 (CH), 126.87 (CH), 128.34 (CH), 130.12 (CH), 133.40 (C), 133.81 (CH), 136.54 (CH), 137.80 (C), 151.60 (C), 158.51 (C), 172.03 (C). ESI-MS calcd. for C15H11NO3, 253.25; found: m/z 254.08 [M + H]+. Anal. C15H11NO3 (C, H, N).
HNE inhibition assay
Compounds were dissolved in 100% DMSO at 5 mM stock concentrations. The final concentration of DMSO in the reactions was 1%, and this level of DMSO had no effect on enzyme activity. The HNE inhibition assay was performed in black flat-bottom 96-well microtiter plates. Briefly, a buffer solution containing 200 mM Tris–HCl, pH 7.5, 0.01% bovine serum albumin, and 0.05% Tween-20 and 20 mU/mL of HNE (Calbiochem) was added to wells containing different concentrations of each compound. The reaction was initiated by addition of 25 µM elastase substrate (N-methylsuccinyl-Ala-Ala-Pro-Val-7-amino-4-methylcoumarin, Calbiochem) in a final reaction volume of 100 µl/well. Kinetic measurements were obtained every 30 s for 10 min at 25 °C using a Fluoroskan Ascent FL fluorescence microplate reader (Thermo Electron, MA) with excitation and emission wavelengths set at 355 and 460 nm, respectively. For all compounds tested, the concentration of inhibitor that caused 50% inhibition of the enzymatic reaction (IC50) was calculated by plotting % inhibition versus logarithm of inhibitor concentration (at least six points). The data are presented as the mean values of at least three independent experiments with relative standard deviations of <15%.
Analysis of compound stability
Spontaneous hydrolysis of selected derivatives was evaluated at 25 °C in 0.05 M phosphate buffer, pH 7.3. Kinetics of hydrolysis were monitored by measuring changes in the absorbance spectra over time using a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). Absorbance (At) at the characteristic absorption maxima of each compound was measured at the indicated times until no further absorbance decreases occurred (A∞)Citation52. Using these measurements, we created semilogarithmic plots of log(At–A∞) versus time, and k′ values were determined from the slopes of these plots. Half-conversion times were calculated using t1/2 = 0.693/k′, as described previouslyCitation36,Citation37.
Molecular modelling procedures
The programs used for the energy minimisation, MD, and docking were the simulation protocols Minimisation, Standard Dynamics Cascade, Analyse Trajectory, and CDocker implemented in Accelrys Discovery Studio 2.1Citation53. The Force Field used for all simulations was CHARMmCitation54.
The following parameters were used for MD simulations, both in vacuum and in implicit solvent (the latter was simulated by using distance dependent dielectric constant set to 4r): time step = 1 fs, equilibration time = 100 ps, production time = 1000 ps (5000 ps for the inhibitor-HNE assembly), T = 300 and 600 K. Ten snapshot conformations with evenly spaced intervals were extracted from each MD trajectory and subsequently minimised (using the steepest descent and conjugate gradient algorithms) in order to obtain the starting geometries for the subsequent quantum chemical calculations (QC) and MD simulations with the receptor.
gaussian09 (rev. C01)Citation55 was used for quantum chemical calculations (QC) on 7d and 8d by using the B3LYPCitation56,Citation57 and B97DCitation58 functionals. The basis set was 6-31 + G(d,p)Citation59, and the Berny algorithm was usedCitation60. Reliability of the stationary points was assessed by evaluation of the vibrational frequencies. For each inhibitor, different conformational isomers, chosen from amongst the low lying energy conformers (as found in MD simulations and subsequent geometry optimisation) were considered.
Molecular plots were produced by the program Discovery Studio Visualiser (v 4.5)Citation61.
Results and discussion
Chemistry
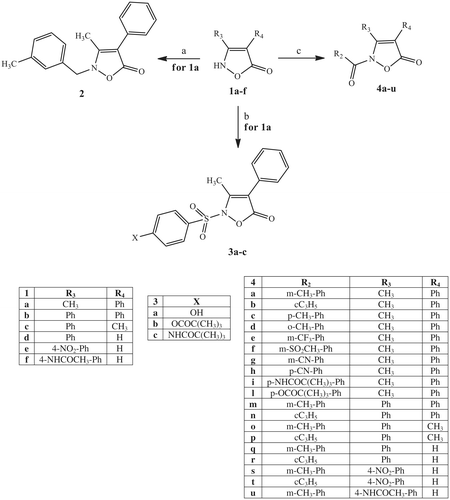
It is well-known that the isoxazolone nucleus exhibits three tautomersCitation43,Citation62,Citation63, as illustrated below. The NH form seems to be the most representative, especially in polar solventsCitation62,Citation64–66. However, it is possible to find examples of alkylation and acylation products in the literature, as shown by the NH and the OH formsCitation67. Taking into account this information, we performed the synthesis of our final compounds, as shown in Schemes 1–3, and the structures were confirmed on the basis of analytical and spectral data.
Scheme 1 depicts the synthetic pathway followed to obtain the final 2-N-substituted isoxazolones of type 2, 3, and 4, which have different groups at positions 3 and 4. The previously described key intermediate isoxazol-5-(2H)-ones of type 1 were treated under various conditions to obtain the final compounds 2, 3, and 4. The alkylation of 1aCitation41 with 3-methylbenzyl chloride and K2CO3 in anhydrous acetonitrile at reflux resulted in compound 2. On the other hand, treatment of compounds 1a–f (1aCitation41, 1bCitation42, 1c,dCitation43, 1eCitation44, and 1fCitation47) with the appropriate acyl/aroyl chloride and NaH in anhydrous THF at room temperature (compounds 4a–l,n–t) or with m-toluyl chloride, and K2CO3 in t-ButOH at 80 °C (compounds 4m,u) resulted in the corresponding 2-NCO derivatives of type 4. Likewise, we synthesised the sulfonamide derivatives 3a–c by treatment of intermediate 1aCitation41 with the appropriate commercially available phenyl sulfonyl chloride in pyridine at room temperature. All of these reactions led to a single derivative originating from the NH form of the isoxazolone nucleus, in agreement with previous data reported in literature.
Scheme 1. Reagents and conditions: (a) 3-methylbenzyl chloride, K2CO3, anhydrous CH3CN, 80 °C, 2 h; (b) 4-X-Ph-SO2Cl, anhydrous pyridine, r.t., 4 h; (c) for 4a–l, n–t: R2-COCl, NaH, anhydrous THF, r.t., 24 h; for 4m,u: m-toluoyl chloride, K2CO3, t-BuOH, 80 °C, 3 h.
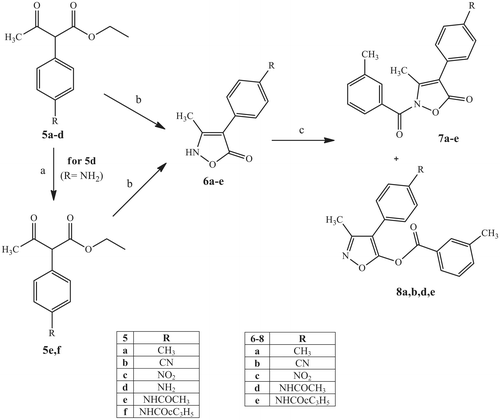
Scheme 2 shows the synthetic procedures used to obtain the final compounds of type 7 and 8, which have a 4-substituted phenyl at position 4. β-Ketoesters 5a–d (5aCitation49, 5b,dCitation48, and 5cCitation50) were synthesised as described previouslyCitation48–50, while compounds 5e,f were obtained by acylation of 5dCitation48 with the appropriate acyl chloride and Et3N in dichloromethane. These compounds served as the starting material for synthesis of the key intermediates of type 6 with an isoxazolone nucleus. Cyclisation of 5a–c,e,f with hydroxylamine hydrochloride in a mixture MeOH/H2O 1:1 at reflux resulted in the intermediates 6a–e which, in turn, were treated with m-toluoyl chloride under the same conditions reported in Scheme 1. Unexpectedly this last step resulted the pair of isomers of type 7 and 8 (NCO/OCO ratio 3:1), with the only exception being the nitro derivative 7c, which was obtained only in the N-CO form.
Scheme 2. Reagents and conditions: (a) R-COCl, Et3N, anhydrous CH2Cl2, 0 °C, 2 h, then r.t., 2 h; (b) NH2OH.HCl, H2O/MeOH 1:1, reflux, 5 h; (c) m-toluoyl chloride, NaH, anhydrous THF, r.t., 24 h.
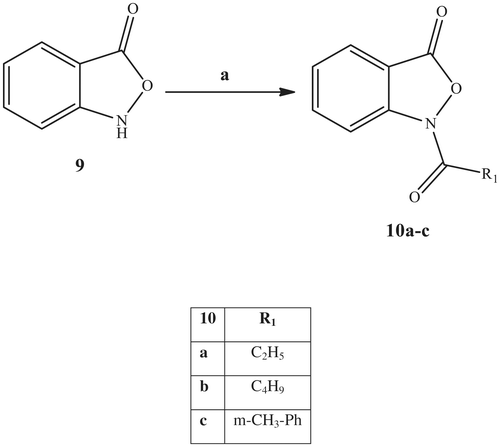
Scheme 3. Reagents and conditions: (a) R1-COCl, K2CO3, t-BuOH, 80 °C, 3 h.
Assignment of the isomer structures was first performed using the 1H NMR chemical shift value of the methyl at position 3. Compounds 4a–l (Scheme 1) had shifts of 2.8 ppm, which is characteristic of NCO derivatives. Likewise, we attributed the NCO-structure to compounds with a 2.8 ppm shift for 3-CH3 (7a–e) and the OCO-structure to compounds with a 2.4 ppm shift (8a,b,d,e) (see Supporting Information). In order to verify our findings, we performed additional techniques, such as IR spectroscopy and 2 D NMR (1H–13C HSQC, 1H–13C HMBC, and 1H–1H NOESY).
In Scheme 3, we show the synthetic pathways used to obtain the benzoisoxazolone derivatives 10a–c, which are an elaboration of the previous isoxazolone scaffold. The acylation of benzoisoxazolone intermediate 9Citation51 with the appropriate acyl chloride and potassium carbonate in t-BuOH led to final compounds 10a–c.
Biological evaluation and structure–activity relationship (SAR) analysis
All compounds were evaluated for their ability to inhibit HNE in comparison with Sivelestat, a reference HNE inhibitor, and the results presented in . PreviouslyCitation40, we examined the possibility of a two point of attack for Ser195, 2-NCO, and 5-CO, and docking studies confirmed that the endocyclic C = O at position 5 was involved in the catalysis process, whereas the amidic C = O group was important for anchoring to the sub-pocket of the binding site. We could expect a similar trend in this new series of isoxazolones.
Table 1. HNE inhibitory activity of isoxazolone derivatives 2, 3a–c, and 4a–u.
Table 2. HNE inhibitory activity of isoxazolone derivatives 7a–e and 8a,b,d,e.
Table 3. HNE inhibitory activity of benzoisoxazolone derivatives 10a–c.
Beginning analysis of the data with the 3-methyl-4-phenylisoxazol-5(2H)-one derivatives (compounds 4a–l), the results reported in suggest that, similar to our previous seriesCitation40, the best substituents at position N-2 are a m-methylbenzoyl or a cyclopropanecarbonyl fragment, which resulted in compounds 4a and 4b that were active in the nanomolar range (IC50 = 77 and 59 nM, respectively). Displacement of the methyl group from the meta to the para (4c) or ortho (4d) positions on the phenyl led to compounds with activity one order of magnitude lower than 4a, which is different than our previous seriesCitation40, where moving the methyl to the para position had no effect on HNE inhibitory activity. Substitution of m-methyl with other groups, such as trifluoromethyl (4e), cyano (4g,h), or methylsulfonyl (4f), which are found in other potent HNE inhibitors, was not favourable for activity, and only compound 4e exhibited activity in the submicromolar range (IC50 = 200 nM).
To evaluate the importance of the 2-CO amidic group in this series, we synthesised the alkyl derivative 2, which was completely inactive, suggesting that the carbonyl group is important for HNE inhibitory activity and also probably involved in catalysis. The insertion of the 3-methyl-4-phenylisoxazol-5(2H)-one scaffold of the sulfonamide fragment of the drug Sivelestat at position 2 led to compound 3b, which exhibited comparable activity to Sivelestat (IC50 = 59 nM, ). In contrast, its pivalamide derivative (3c) was completely inactive, suggesting that the possible point of attack of Ser195 is the CO of the pivalate function, which is also present in Sivelestat. These data were also confirmed by the inactivity of the hydrolysed derivative 3a. Moreover, we synthesised compounds 4l and 4i by substituting the SO2 group at position 2 (3b and 3c, respectively) with an amidic function. The amide 4l had activity (IC50 = 0.48 µM) that was one order of magnitude higher than the corresponding sulfonamide 3b, while the amide 4i had a higher potency (IC50 = 8.5 µM) than its corresponding sulfonamide 3c, which was completely inactive. These results suggest that the SO2 group at position 2 of 3b could also be important for anchoring the ligand to the sub-pocket of the binding site.
Keeping m-methylbenzoyl and ciclopropanecarbonyl at position N-2, we modified positions 3 and 4 of the isoxazolone (). Generally, we observed a collapse of activity. In particular, the 3,4-diphenylisoxazol-5(2H)-one derivatives (compounds 4m and 4n), the 4-methyl-3-phenylisoxazol-5(2H)-one derivatives (compounds 4o and 4p), and the 3-phenylisoxazol-5(2H)-one derivatives (compounds 4q,r and 4u) all had decreased activity (IC50 = 10–50 µM). The insertion in the para position of the phenyl ring at position 3 of compounds 4q,r with substituents that in the previous series gave good results (such as a nitro group or acetamide function) led to completely inactive (compounds 4s,t). Based on the results in , we can conclude that position 4 can bear bulky groups, such as an aromatic ring, when there is a methyl group in position 3, probably to fit constraints of the lipophilic pocket. Moving the phenyl ring to position 3 is not tolerated, either when there is a methyl or a hydrogen present in position 4.
In , we report the activity of the pair of isomers of type 7 and 8, the amidic and ester derivatives, respectively, which were obtained by introducing a substituent to the para position of the phenyl group at position 4. All N-benzoyl derivatives (7a–e), with the exception of the nitro compound 7c, exhibited good HNE inhibitory activity in the nanomolar range (IC50 = 20–70 nM). In addition, the activity seems to be independent of the electrophilic properties of the substituents (CH3, NO2, CN, NHCOR). The best compound of this series was the p-methyl derivative 7a, which had an IC50 of 20 nM. The O-benzoyl derivatives 8b,d,e, which were derived from tautomerism of the isoxazolone scaffold, surprisingly exhibited activity, although at one order of magnitude lower than the corresponding N-benzoyl type 7 derivatives (). Only the ester isomer of the potent 7a had a significant loss in activity (8a, IC50 = 10 µM). The unexpected data related to esters 8a,b,d,e could indicate a different interaction with the target, since the CO endocyclic implicated in the catalysis is missing, and the only carbonyl group presented in the molecule is an ester function at position 5. Further docking studies and kinetic experiments could help us to understand the interaction of these compounds with the HNE catalytic site and their mechanism of action.
shows the HNE inhibitory activity of benzoisoxazolone derivatives 10a–c, which are elaborations of the isoxazolone scaffold. In this case, only the m-methylbenzoyl derivative 10c had reasonable activity, with an IC50 of 638 nM, while the other compounds were less active (10a) or inactive (10b).
Molecular modelling
Several attempts were made using different solvents in order to obtain crystals of 7d and 8d. However, it was not possible to obtain crystals suitable for X-ray diffraction (probably in part due to their quite low melting points). As a consequence, the starting 3D geometry of these molecules was obtained by using as building blocks the solid state structures of 3,5-dicyano-4-(4-methoxyphenyl) isoxazole (QAQPON refcode)Citation68 found in the Cambridge Structural Database (CSD; v 5.37)Citation69 and that of the isoxazolone derivative 2j (3-ethyl-2–(3-methylbenzoyl)isoxazol-5(2H)-one)Citation40. The 3D arrangement of 7d and 8d was then roughly improved by an energy minimisation procedure, followed by molecular dynamics simulations (details in the Supplementary Material).
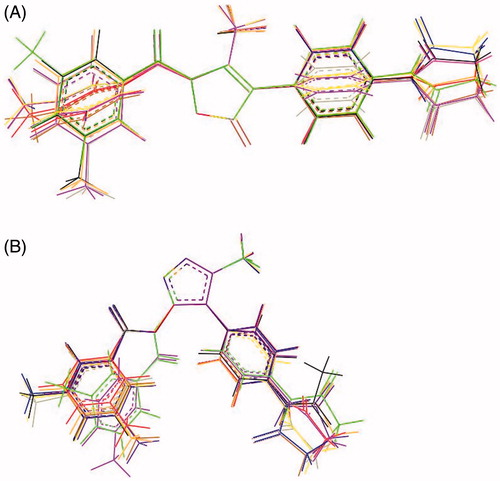
Preliminary molecular dynamics (MD) simulations at 300 and 600 K were performed on 7d and 8d to evaluate their flexibility, accessible conformational space, and preferred 3D arrangements (low-energy conformations). As expected, the overall shape of both ligands did not change significantly on changing the simulation medium, as roughly determined by comparison of the dihedral angles, which define the overall shape of the molecules (see Figure S1, Supplementary Material). For 7d, MD trajectories (300 and 600 K) showed that τ1 and τ2 adopted a trans conformation that is maintained throughout the simulations, while τ3–τ5 access different conformations (Figures S2 and S3, Supplementary Material). Overall, the molecule adopted an elongated cylindrical shape (Figure S4, Supplementary Material). In particular, the conformational behaviour of dihedrals τ1 and τ4 seems to be quite important (vide infra), given that the endocyclic C = O at position 5 appears to be involved in the catalytic process, while the closest C = O group could play an important role in anchoring to the sub-pocket of the binding siteCitation40. The superimposition (, panel A) of the minimised conformers of 7d extracted from the MD trajectory (T = 600 K, ε = 4r), which are comprised within about 2 kcal mol−1, exemplifies the conformational space accessible to the molecule through rotations about the τ3–τ5 dihedrals.
Figure 2. Panel (A): Superimposition of the minimised conformations of 7d extracted from the MD trajectory (T = 600 K, ε = 4r). Panel (B): Superimposition of the minimised conformations of 8d extracted from the MD trajectory (T = 600 K, ε = 4r).
Compound 8d appears to be a little bit more flexible compared to 7d, as suggested by the larger variability of the distance separating the centroids of the phenyl rings. While the τ1, τ2, and τ3 dihedrals were almost frozen irrespective of the simulation temperature and medium [they adopted cis, trans (with only few exceptions), and cis (with only few exceptions) conformations, respectively], increasing the temperature from 300 to 600 K made the τ5 and τ6 dihedrals freely rotate (Figures S5 and S6, Supplementary Material). Dihedral τ4 showed a preference for a gauche arrangement. Overall, 8d is V-shaped: in most cases both of the phenyl rings were rotated with respect to the mean plane defined by the heterocyclic ring (Figure S7, Supplementary Material). The superimposition (, panel B) of the minimised conformers of 8d extracted from MD simulations (T = 600 K, ε = 4r) revealed two possible orientations for the carbonyl group of the ester function at position 5, which is supposed to be involved in the catalytic process (energies are comprised within about 5 kcal mol−1).
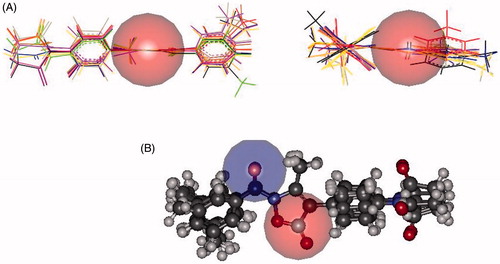
In both inhibitors, accessibility of the carbonyl group involved in the catalytic process was roughly estimated by the dimension of a sphere centred on the C = O carbon atom (r = 2.5 Å, in red in , panel A). Similarly a sphere (r = 2.5 Å, in blue in , panel B) centred on the oxygen atom, which could be involved in H-bond interactions with the receptor points, was used to assess the exposure to the environment of C = O in 7d. Panel A of suggests a more crowded region about the site of the nucleophilic attack of 8d, which could account for its lower activity compared to 7d (in 8d, the carbonyl group is directly bound to the phenyl ring and faces the pending arm at position 4). In addition, the almost cylindrical shape of 7d caused, at least in principle, the carbonyl carbon atom to be attacked from both the side of the plane defined by the 5-membered ring (Figure S8, Supplementary Material). In contrast, only one side of the ester group in 8d appeared to be easily accessible to the hydroxyl group of Ser195 (Figure S9, Supplementary Material). Finally, in both cases the terminal amide group appeared to be well exposed and thus prone for anchoring to the sub-pocket of the binding site.
Figure 3. Panel (A): Accessibility of the carbonyl grouping involved in the catalytic process estimated by the dimension of a sphere (in red, r = 2.5 Å) centred on the carbon atom of 7d (left) and 8d (right). Panel (B): Accessibilities of the carbonyl groups in 7d involved in the catalytic process and in H-bond interactions with the receptor points estimated by the dimension of a sphere centred on the carbon and oxygen atom (r = 2.5 Å, red and blue, respectively).
Two different approaches were used to gain an idea of the interaction of 7d and 8d with HNE. First, docking of the inhibitors (two different starting conformations were considered) into the active site of HNE using the CDocker protocol (in vacuum, target temperature 300 K, 20 poses retained) and secondly, MD simulations on each inhibitor-HNE complex (in vacuum, simulation time 5 ns, T = 300K, with the atoms outside the binding sphere constrained to fixed points in space in order to save computational time). The structure of HNE complexed with a peptide chloromethyl ketone inhibitor was used for the docking study and MD simulations (1HNECitation70 entry of the Protein Data Bank). The binding site of HNE was defined as a sphere with a 12 Å radius centred at the centroid of the five-membered ring of the peptide chloromethyl ketone inhibitor which covered all the active site amino acids of the HNE enzyme. All water molecules and bound inhibitor were removed from the macromolecule and hydrogen atoms were added.
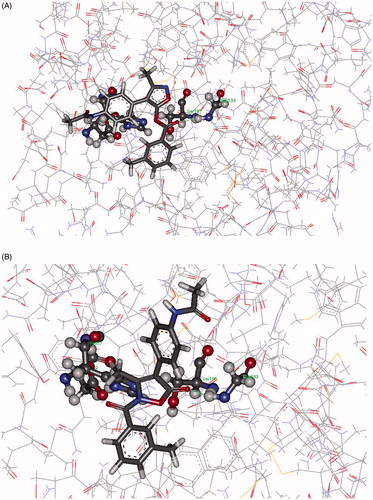
Interaction energies from docking protocols on 7d and 8d with HNE did not significantly differ, and the two inhibitors showed comparable binding modes. In particular for 8d, most of the saved poses featured Gly193 as the anchor group, and the H-bond interaction with this amino acid pushed the site of the nucleophilic attack quite distant from Ser195. However, a few poses show Gly193 involved as an H-bond donor to the ester function of 8d and, in these cases, the distance HO(Ser195)…CO ranged from 3.3 to 4.8 Å (, Panel A). The same amino acids are also involved in the interaction with 7d. Anchoring of the terminal NHCO group to Ser195 via H-bonds pushed the CO at position 5 distant from the nucleophilic –OH of Ser195, while H-bonds between 5-CO and Gly193 brought the two partners of the nucleophilic attack a little bit closer (distances range from 4.3 to 5.1 Å, , Panel B). However, the mean distance between the inhibitor-HNE reacting sites, as determined from the saved poses, was very long for both complexes (ca 7 Å).
Figure 4. Panel (A): Pose from the CDocker protocol showing Gly193 involved as H-bond donor to the carbonyl function of 8d. Panel (B): Pose from the CDocker protocol showing Gly193 involved as H-bond donor to 5-CO in 7d.
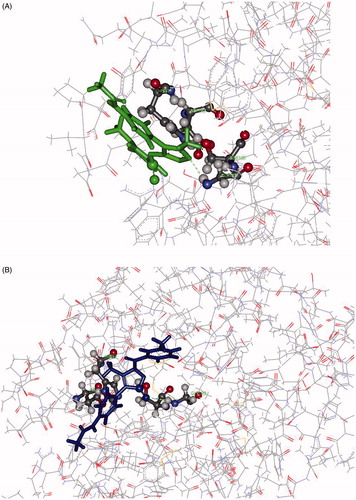
In summary, docking results do not help to explain the different activities of the two inhibitors towards HNE. The fact that the CDocker protocol keeps the receptor rigid, while the inhibitor is allowed to flex during the refinement, could however significantly affect the results, and this is the reason why the ligand–receptor binding affinity was also assessed by MD. On the other hand, MD simulations confirmed the leading role of Gly193 as an anchoring group for both inhibitors. However, in contrast with the docking results, MD shows the 5-CO grouping of 7d was significantly closer to the HNE reactive site (distance less than 4.5 Å in a large fraction of the snapshot conformations) with respect to the corresponding active site of 8d (d < 5 Å in few of the sampled conformations). In contrast, a comparison of the total number of the intermolecular DH…A contacts (distances less than 2.5 Å) shows a greater propensity for 8d to form H-bond interactions compared to 7d. In addition, while multiple intermolecular H-bonds were present for 8d, most of the snapshot conformations of 7d exhibited only one H-bond interaction. In other words, the net of intermolecular H-bonds that involves 8d keeps the molecule quite distant from Ser195. Finally, it is noteworthy that in the HNE complexes with 8d featuring closer distances between the active sites, the heteroatoms of the 5-membered ring acted as H-bond acceptors both towards the Ser195 –OH group and Gly193. As a result of these interactions, the relative 3 D arrangement of 8d in the HNE active site does not appear to be propitious for approach of the nucleophile of Ser195 (see for example , Panel A). In contrast, the H-bond between 5-CO and Gly193, which in most cases held together the 7d/HNE adduct, results in a favourable 3 D arrangement of the reacting partners (see for example , panel B) for nucleophilic attack.
Figure 5. Panel (A): View of the 8d-HNE adduct from MD showing the H-bond interaction between the nitrogen atom of the 5-membered ring acting as H-bond acceptor towards Gly193. Panel (B): View of the 7d-HNE adduct from MD showing the H-bond interaction between the oxygen of 5-CO acting as H-bond acceptor towards Gly193.
Stability and kinetic features
The most potent isoxazolones with IC50 < 100 nM and their ester analogs were further evaluated for chemical stability in aqueous buffer using spectrophotometry to detect compound hydrolysis. The compounds had t1/2 values from 2.9 to 9.6 h for spontaneous hydrolysis, indicating that the amides were more stable than the esters in the corresponding pairs of 7a/8a, 7d/8d, and 7e/8e (). In general, the isoxazolones were more stable than our previously described HNE inhibitors with cinnolinoneCitation39, N-benzoylindazoleCitation36,Citation37, and N-benzoylpyrazole scaffoldsCitation71.
Table 4. Half-life (t1/2) for the spontaneous hydrolysis of selected derivatives.
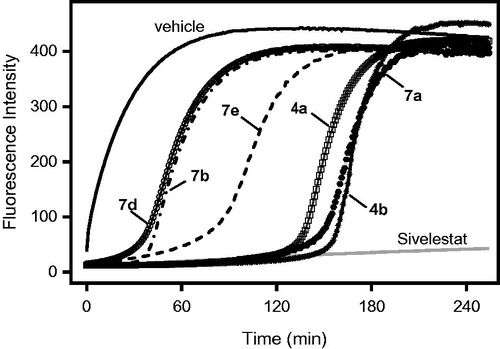
The most potent isoxazolones were also selected for evaluation of the reversibility of HNE inhibition over time. As shown in , HNE inhibition was rapidly (∼30 min) reversed for compounds 7d and 7b. The inhibition was maximal for up to 60 min with compound 7e and >120 min for the other tested compounds (4a, 4b, and 7a). However, inhibition by the compounds was eventually reversed, and full recovery of HNE activity was observed by 4 h after treatment with 8 µM of the compound (e.g. see ).
Figure 6. Evaluation of HNE inhibition by representative isoxazolones and Sivelestat over extended periods of time. HNE was incubated with the indicated compounds (8 µM), and kinetic curves monitoring substrate cleavage catalysed by HNE over time are shown. Representative curves are from two independent experiments.
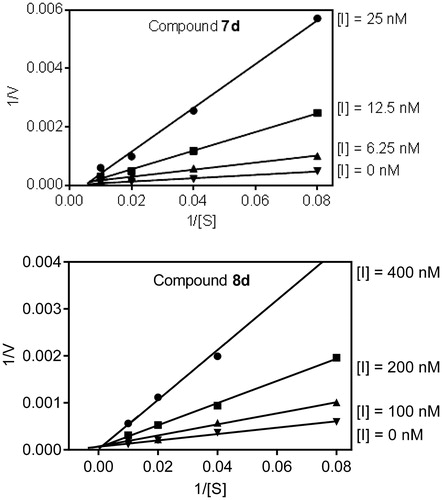
To better understand the mechanism of action of these isoxazolone HNE inhibitors, we performed kinetic experiments. As shown in , the representative double-reciprocal Lineweaver–Burk plot of fluorogenic substrate hydrolysis by HNE in the absence and presence of compounds 7d and 8d indicates that these compounds are competitive HNE inhibitors.
Figure 7. Kinetics of HNE inhibition by compounds 7d and 8d. Representative double reciprocal Lineweaver–Burk plots are shown from three independent experiments.
Conclusions
In the present study, we report a new series of isoxazolones as potent HNE inhibitors, confirming our previous resultsCitation40, which indicated this nucleus as an appropriate scaffold for this target. The most potent compounds had a methyl group at position 3 and a (substituted)phenyl ring at position 4, and the higher HNE inhibitory activity was found for compound 7a (IC50 = 20 nM). In addition to the 2-NCO derivatives, a number of 5-NCO compounds were obtained, although with lower activity than the corresponding amide. Studies of chemical stability in aqueous buffer indicated that amides were generally more stable than esters, while kinetic experiments confirmed that both amides and esters were competitive HNE inhibitors. Docking and molecular dynamics studies are the different approaches used to understand the interactions of the two isomers 7d and 8d with HNE. Both studies highlight the fundamental role of Gly193 as anchoring group for both the inhibitors, but the MD simulations help us to explain the difference in inhibitory activity between the inhibitors 7d and 8d. The ester isomer 8d shows a greater propensity to form H-bond interactions with respect to 7d and as a result the molecule is quite distant from the Ser195, the amino acid responsible for the nucleophilic attack. While the amide isomer 7b appears more mobile within the active site of HNE, being held in place by single H-bond interactions (vs. multiple in 8d), which leads the C = O at position 5 to obtain a favourable orientation for the nucleophilic attack by Ser195.
Supplemental Material
Download PDF (735.9 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Brocklehurst K, Willenbrock F, Salih E. In: Neuberger A, Brocklehurst K, eds. New comprehensive biochemistry. Amsterdam: Elsevier; 1987;16:39–158.
- Hedstrom L. Serine protease mechanism and specificity. Chem Rev 2002;102:4501–23.
- Rawling ND, Morton FR, Kok CY, et al. MEROPS: the peptidase database. Nucleic Acids Res 2007;36:D320–5.
- Perera NC, Schilling O, Kittel H, et al. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci USA 2012;109:6229–34.
- Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010;62:726–59.
- Heutinck KM, ten Berge IJ, Hack CE, et al. Serine proteases of the human immune system in health and disease. Mol Immunol 2010;47:1943–55.
- Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe 2014;15:526–36.
- Sinha S, Watorek W, Karr S, et al. Primary structure of human neutrophil elastase. Proc Natl Acad Sci USA 1987;84:2228–32.
- Chua F, Laurent GJ. Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc Am Thorac Soc 2006;3:424–7.
- Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 2006;6:541–50.
- Stapels DA, Geisbrecht BV, Rooijakkers SH. Neutrophil serine proteases in antibacterial defense. Curr Opin Microbiol 2015;23:42–8.
- Saitoh T, Komano J, Saitoh Y, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012;12:109–16.
- Fitch PM, Roghanian A, Howie SEM, Sallenave J-M. Human neutrophil elastase inhibitors in innate and adaptive immunity. Biochem Soc Trans 2006;34:279–82.
- Janciauskiene SM, Bals R, Koczulla R, et al. The discovery of α1-antitrypsin and its role in health and disease. Respir Med 2011;105:1129–39.
- Quabius ES, Görögh T, Fischer GS, et al. The antileukoprotease secretory leukocyte protease inhibitor (SLPI) and its role in the prevention of HPV-infections in head and neck squamous cell carcinoma. Cancer Lett 2015;357:339–45.
- Zhong QQ, Wang X, Li YF, et al. Secretory leukocyte protease inhibitor promising protective roles in obesity-associated atherosclerosis. Exp Biol Med 2017;242:250–7.
- Zani M-Z, Nobar SM, Lacour SA, et al. Kinetics of the inhibition of neutrophil proteinases by recombinant elafin and pre-elafin (trappin-2) expressed in Pichia pastoris. Eur J Biochem 2004;271:2370–8.
- Kawabata K, Hagio T, Matsuoka S. The role of neutrophil elastase in acute lung injury. Eur J Pharmacol 2002;451:1–10.
- Barnes PJ, Stockley RA. COPD: current therapeutic interventions and future approaches. Eur Respir J 2005;25:1084–106.
- Pandey KC, De S, Mishra PK. Role of proteases in chronic obstructive pulmonary disease. Front Pharmacol 2017;8:512–9.
- Kelly E, Greene CM, McElvaney NG. Targeting neutrophil elastase in cystic fibrosis. Expert Opin Ther Targets 2008;12:145–57.
- Wagner CJ, Schultz C, Mall MA. Neutrophil elastase and matrix metalloproteinase 12 in cystic fibrosis lung disease. Mol Cell Pediatr 2016;3:25.
- Hilbert N, Schiller J, Arnhold J, Arnold K. Cartilage degradation by stimulated human neutrophils: elastase is mainly responsible for cartilage damage. Bioorg Chem 2002;30:119–32.
- Henriksen PA, Sallenave J-M. Human neutrophil elastase: mediator and therapeutic target in atherosclerosis. Int J Biochem Cell Biol 2008;40:1095–100.
- Dhanrajani PJ. Papillon–Lefevre syndrome: clinical presentation and a brief review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2009;108:e1–7.
- Cools-Lartigue J, Spicer J, Najmeh S, Ferri L. Neutrophil extracellular traps in cancer progression. Cell Mol Life Sci 2014;71:4179–94.
- Lucas SD, Costa E, Guedes RC, Moreira R. Targeting COPD: advances on low-molecular-weight inhibitors of human neutrophil elastase. Med Res Rev 2013;33:E73–101.
- Tsai Y-F, Hwang T-L. Neutrophil elastase inhibitors: a patent review and potential applications for inflammatory lung diseases (2010–2014). Expert Opin Ther Pat 2015;25:1145–58.
- Tebbutt SJ. Technology evaluation: transgenic alpha-1-antitrypsin (AAT), PPL therapeutics. Curr Opin Mol Ther 2000;2:199–204.
- American Thoracic Society/European Respiratory Society Statement, Standards for the diagnosis and management of individual with alpha-1-antitrypsin deficiency. Am J Respir Crit Care Med 2003;168:818–900.
- Von Nussbaum F, Li VMJ. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: into clinical testing with pre-adaptive pharmacophores. Bioorg Med Chem 2015;25:4370–81.
- Ohbayashi H. Current synthetic inhibitors of human neutrophil elastase in 2005. Expert Opin Ther Pat 2005;15:759–71.
- Iwata K, Doi A, Ohji G, et al. Effect of neutrophil elastase inhibitor (Sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress (ARDS): a systemic review and meta-analysis. Intern Med 2010;49:2423–32.
- Stockley R, De Soyza A, Gunawardena K, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med 2013;107:524–33.
- Von Nussbaum F, Li VMJ, Allerheiligen S, et al. Freezing the bioactive conformation to boost potency: the identification of BAY 85-8501, a selective and potent inhibitor of human neutrophil elastase for pulmonary diseases. ChemMedChem 2015;10:1163–73.
- Crocetti L, Giovannoni MP, Schepetkin IA, et al. Design, synthesis and evaluation of N-benzoylindazole derivatives and analogues as inhibitors of human neutrophil elastase. Bioorg Med Chem 2011;19:4460–72.
- Crocetti L, Schepetkin IA, Cilibrizzi A, et al. Optimization of N-benzoylindazole derivatives as inhibitors of human neutrophil elastase. J Med Chem 2013;56:6259–72.
- Crocetti L, Schepetkin IA, Ciciani G, et al. Synthesis and pharmacological evaluation of indole derivatives as deaza analogues of potent human neutrophil elastase inhibitors. Drug Dev Res 2016;77:285–99.
- Giovannoni MP, Schepetkin IA, Crocetti L, et al. Cinnoline derivatives as human neutrophil elastase inhibitors. J Enzyme Inhib Med Chem 2016;31:628–39.
- Vergelli C, Schepetkin IA, Crocetti L, et al. Isoxazol-5(2H)-one: a new scaffold for potent human neutrophil elastase (HNE) inhibitors. J Enzyme Inhib Med Chem 2017;32:821–31.
- Beccalli EM, Marchesini A. The Vilsmeier–Haack reaction of isoxazoline-5-ones. Synthesis and reactivity of 2-(dialkylamino)-1,3-oxazin-6-ones. J Org Chem 1987;52:3426–34.
- Breslow T, Eicher A, Krebs RA, et al. Diphenylcyclopropenone. J Am Chem Soc 1965;87:1320–5.
- Boulton AJ, Katritzky AR. The tautomerism of heteroaromatic compounds with five-membered rings-I: 5-hydroxyisoxazoles-isoxazol-5-ones. Tetrahedron 1961;12:41–50.
- Batra S, Bhaduri AP. An account of the chemistry of 3,4-disubstituted 5-isoxazolones. J Indian Inst Sci 1994;74:213–26.
- Safi R, Rodriguez F, Hilal G, et al. Hemisynthesis, antitumoral effect, and molecular docking studies of ferutinin and its analogues. Chem Biol Drug Des 2016;87:382–97.
- Onoda A, Yamada Y, Nakayama Y, et al. Stabilization of calcium- and terbium-carboxylate bonds by NH····O hydrogen bonds in a mononuclear complex. A functional model of the active site of calcium-binding proteins. Inorg Chem 2004;43:4447–55.
- Belzecki C, Urbanski T. New antituberculosis agents. XXXVII. Thiosemicarbazones of oxo acids. 2. Thiosemicarbazones of aroyl fatty acids. Roczniki Chemii 1958;32:769–78.
- Cadieux JA, Zhang Z, Mattice M, et al. Synthesis and biological evaluation of substituted pyrazoles as blockers of divalent metal transporter 1 (DMT1). Bioorg Med Chem Lett 2012;22:90–5.
- Xie X, Cai G, Ma D. CuI/L-proline-catalyzed coupling reactions of aryl halides with activated methylene compounds. Org Lett 2005;7:4693–5.
- Monastyrskyi A, Namelikonda NK, Manetsch R. Metal-free arylation of ethyl acetoacetate with hypervalent diaryliodonium salts: an immediate access to diverse 3-aryl-4(1H)-quinolones. J Org Chem 2015;80:2513–20.
- Wierenga W, Evans BR, Zurenko G. Benzisoxazolones: antimicrobial and antileukemic activity. J Med Chem 1984;27:1212–5.
- Forist AA, Weber DJ. Kinetics of hydrolysis of hypoglycemic 1-acyl 3,5-dimethylpyrazoles. J Pharm Sci 1973;62:318–9.
- Accelrys Software Inc., San Diego, CA 92121, USA.
- Brooks BR, Bruccoleri RE, Olafson BD, et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 1983;4:187–217.
- Gaussian 09, Revision C.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ, Gaussian, Inc., Wallingford CT, 2010.12.
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev 1988;38:3098–100.
- Vosko SH, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys 1980;58:1200–11.
- Grimme SJ. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem 2006;27:1787–99.
- Hehre WJ, Ditchfield R, Pople JA. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys 1972;56:2257–61.
- Peng C, Ayala PY, Schlegel HB, Frisch MJ. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J Comput Chem 1996;17:49–56.
- Dassault Systèmes BIOVIA, Discovery Studio Visualizer, Release 4.5, San Diego: Dassault Systèmes; 2015.
- Katritzky AR, Barczynski P, Ostercamp DL, Yousaf TI. Mechanisms of heterocyclic ring formations. 4. A 13C-NMR study of the reaction of β-keto esters with hydroxylamine. J Org Chem 1986;51:4037–42.
- Laufer SA, Margutti S. Isoxazolone based inhibitors of p38 MAP kinases. J Med Chem 2008;51:2580–4.
- Franchini PF. Dipole moments and tautomerism of isoxazolin-5-ones. Corsie Sem Chim 1968;14:23–5.
- Cencioni R, Franchini PF, Orienti M. Dipole moments of 5-substitued isoxazoles. I. Dipole moments and tautomerism of isoxazolin-5-one. Tetrahedron 1968;24:151–66.
- Elguero J, Marzin C, Katritzky AR, Linda P, eds. Advances in heterocyclic chemistry, supplement 1, the tautomerism of heterocycles. New York: Academic Press; 1976:656.
- Frolund B, Jensen LS, Guandalini L, et al. Potent 4-aryl- or 4-arylalkyl-substituted 3-isoxazolol GABAA antagonists: synthesis, pharmacology, and molecular modeling. J Med Chem 2005;48:427–39.
- Nishiwaki N, Nogami T, Kawamura T, et al. One-pot synthesis of polyfunctionalised isoxazol(in)es. J Org Chem 1999;64:6476–8.
- Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B 2002;58:380–8.
- Navia MA, McKeever BM, Springer JP, et al. Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84 Å resolution. Proc Natl Acad Sci USA 1989;86:7–11.
- Schepetkin IA, Khlebnikov AI, Quinn MT. N-benzoylpyrazoles are novel small-molecule inhibitors of human neutrophil elastase. J Med Chem 2007;50:4928–38.