
Abstract
A series of dithiocarbamate esters of parthenolide (PTL) was designed, synthesised, and evaluated for their anti- acute myelogenous leukaemia (AML) activities. The most promising compound 7l showed greatly improved potency against AML progenitor cell line KG1a with IC50 value of 0.7 μM, and the efficacy was 8.7-folds comparing to that of PTL (IC50 = 6.1 μM). Compound 7l induced apoptosis of total primary human AML cells and leukaemia stem cell (LSCs) of primary AML cells while sparing normal cells. Furthermore, 7l suppressed the colony formation of primary human leukaemia cells. Moreover, compound 12, the salt form of 7l, prolonged the lifespan of mice in two patient-derived xenograft models and had no observable toxicity. The preliminary molecular mechanism study revealed that 7l-mediated apoptosis is associated with mitogen-activated protein kinase signal pathway. On the basis of these investigations, we propose that 12 might be a promising drug candidate for ultimate discovery of anti-LSCs drug.
Introduction
Acute myelogenous leukaemia (AML) is a malignant disease characterised by an aberrant accumulation of immature myeloid haematopoietic cellsCitation1. AML is the most common form of acute leukaemia in adults and constitutes approximately 80% of casesCitation2. Although current treatments could significantly improve the rate of remission in AML, more than 50% relapse with resistant disease; it is still a main challenge for AML chemotherapyCitation3. Leukaemia stem cells (LSCs) are a group of leukemic cells with self-renewal ability and capable of producing heterogeneous leukaemia cell populationsCitation4,Citation5. It has been considered to play significant role in the initiation and relapse of acute leukaemiaCitation6. Therefore, targeting LSCs is considered to be an effective strategy for treatment of AML and might cure AMLCitation7–10. However, LSCs are refractory to clinical used chemotherapy drugs, such as nucleoside analogues (e.g. cytosine arabinoside) and anthracyclines (e.g. idarubicin and daunorubicin)Citation11,Citation12. Therefore, effective agents that can selectively eradicate LSCs are urgently needed for the development of new therapies for treatment of leukaemia.
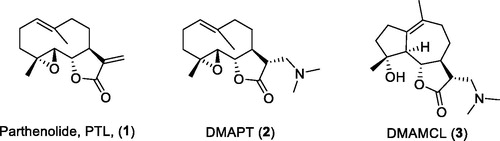
Parthenolide (PTL, 1, ), a sesquiterpene lactone originally separated from the shoots of Feverfew (Tanacetum parthenium), was reported to induce apoptosis of cancer stem cell (CSC), including LSCs, breast cancer stem cells, and prostate tumour-initiating cellsCitation13–17. PTL was shown to inhibit NF-κB, activate p53 and overturn the redox balance in LSCsCitation13,Citation18–23. However, the instability in both acidic and basic conditions and poor solubility limited the clinical application of PTLCitation24. DMAPT (2, ), a dimethylamine adduct of PTL, was in clinical trial for treatment of AML, acute lymphoblastic leukaemia, and chronic lymphocytic leukaemia in the United Kingdom in 2009Citation25–27. The other PTL derivative Dimethylaminomicheliolide (DMAMCL) (3) is conducting clinical trials in Australia for the treatment of gliomasCitation28.
Figure 1. Structures of PTL (1), DMAPT (2), and DMAMCL (3).
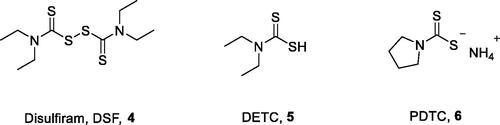
Dithiocarbamates have received considerable attention for their excellent biological activities, such as anti-fungal, anti-bacterial, and carbonic anhydrase inhibiting activitiesCitation29–32. It has been reported that many compounds containing the dithiocarbamate moiety exhibited anticancer activity in recent yearsCitation33–39. Disulphiram (DSF, 4, ) is used as an anti-alcoholism drug in clinical practiceCitation40. It was reported that DSF could inhibit NF-κB activity and enhance the anticancer activity of cytotoxic drugsCitation41,Citation42. DSF or DSF/copper complex exhibited inhibitory effect on a variety of cancer cellsCitation43–47. More importantly, they showed the ability to eliminate LSCs and breast cancer stem-like cellsCitation14,Citation46–48. Clinical trials of DSF for treatment of multiple malignant gliomas are ongoing in GreeceCitation49. DSF’s analogues, diethyldithiocarbamate (DETC, 5, ), and ammonium pyrrolidinedithiocarbamate (PDTC, 6) could inhibit breast cancer stem cells via NF-κB pathwayCitation14.
Figure 2. Dithiocarbamates that can selectively ablate CSCs.
Mitogen-activated protein kinase (MAPK) is a group of serine/threonine kinases in vivo, which can accelerate the proliferation of tumour cells and inhibit their apoptosis after being stimulated by external stimuliCitation50. MAPK signal pathway abnormality is one of the reasons that affect the emergence and development of tumour cells. The abnormal activation of the MAPK pathway is also involved in the production, development, and metastasis of cancer stem cells, including LSCsCitation51, breast cancer stem cellsCitation52, liver cancer stem cellsCitation53, prostate cancer stem cellsCitation54, colon cancer cellsCitation55, and glioblastoma multiforme stem cellsCitation56. Therefore, MAPK pathway is considered to be an important target for ablating CSCsCitation57–60.
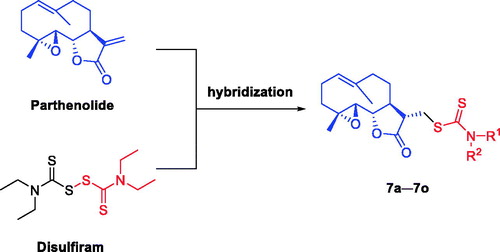
Inspired by the above-mentioned findings and in continuation with our previous efforts to find the new PTL-based anti-LSCs candidatesCitation61–65, herein, we report the design () and synthesis of a series of dithiocarbamate esters of PTL, and evaluation of their anti-AML activities in vitro and in vivo. The preliminary molecular mechanism of the most promising compound 7l was also investigated.
Figure 3. Design of compounds 7a–7o.
Materials and methods
Chemistry
Unless otherwise mentioned, all reactions were carried out under a nitrogen atmosphere with dry solvents under anhydrous conditions. The used solvents were purified and dried according to common procedures. Reactions were monitored by thin-layer chromatography carried out on 0.25 mm Tsingdao silica gel plates (60F-254). Visualisation was achieved using UV light, phosphomolybdic acid in ethanol or potassium permanganate in water, each followed by heating. Tsingdao silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography. Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. NMR spectra were recorded with a 400 MHz (1H: 400 MHz, 13C: 100 MHz) spectrometer and referenced to the solvent peak for CDCl3, CD3OD, and DMSO-d6. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartette, br. = broad, m = multiplet), coupling constants, and integration. All NMR copies are shown in Supplemental data. The purity of the final compounds was determined to be ≥95% by means of analytical high pressure liquid chromatography (HPLC) with an ODS-C18 column (4.6 × 150 mm, 5 μm) eluted at 1 ml/min with Milli-Q water and CH3CN.
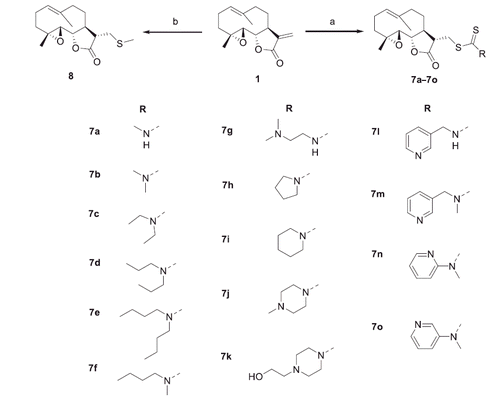
General procedure for the synthesis of compounds 7a–7m
After a mixture of corresponding amine or amine hydrochloride (1.2 eq), triethylamine (TEA) (1.1 eq) and CS2 (1.5 eq) in dichloromethane (DCM) and menthol (4/1) was stirred at 0 °C for 30 min, PTL (1 eq) was added. The reaction was stirred at room temperature for 2–8 h and quenched by adding water. The resulting mixture was extracted with DCM for three times. The combined organic layer was dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by column chromatography on silica gel to give compounds 7a–7m.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl methylcarbamodithioate (7a). White amorphous solid (yield: 71%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 4.2 Hz, 1H, N-H), 5.12 (d, J = 10.5 Hz, 1H, H-1), 3.84 (t, J = 8.8 Hz, 1H, H-6), 3.81–3.76 (m, 1H, H-13), 3.67 (dd, J = 14.5, 3.6 Hz, 1H, H-13), 3.18 (d, J = 4.5 Hz, 3H, N-CH3), 2.78 (dt, J = 11.9, 4.5 Hz, 1H, H-11), 2.73 (d, J = 8.9 Hz, 1H, H-5), 2.33 (dd, J = 12.8, 4.7 Hz, 1H, H-7), 2.25–2.02 (m, 6H, CH2), 1.64 (s, 3H, H-14), 1.59 (d, J = 9.1 Hz, 1H, CH2), 1.24 (d, J = 8.1 Hz, 3H, H-15), 1.22–1.13 (m, 1H, CH2). 13 C NMR (100 MHz, CDCl3) δ 197.4 (C-16), 176.4 (C-12), 134.8 (C-10), 124.9 (C-1), 82.7 (C-6), 66.1 (C-5), 62.1 (C-4), 48.4 (C-11), 47.0 (C-7), 41.0 (C-9), 36.5 (CH2), 34.4 (CH2), 32.6 (C-13), 29.9 (CH2), 24.0 (C-2), 17.2 (C-14), 16.9 (C-15). HRMS (ESI) calcd for C17H26NO3S2 [M + H] + 356.1349, found 356.1348.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl dimethylcarbamodithioate (7b). White amorphous solid (yield: 85%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 5.13 (d, J = 9.9 Hz, 1H, H-1), 3.88–3.70 (m, 3H, H-6, -13), 3.54 (s, 3H, N-CH3), 3.38 (s, 3H, N-CH3), 2.81 (ddd, J = 12.1, 6.1, 4.3 Hz, 1H, H-11), 2.69 (d, J = 8.9 Hz, 1H, H-5), 2.30–2.24(m, 3H, H-7, CH2), 2.18–2.03 (m, 4H, CH2), 1.66 (s, 3H, H-14), 1.66–1.58 (m, 1H, CH2), 1.26 (s, 3H, H-15), 1.17 (td, J = 13.0, 5.9 Hz, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 196.4 (C-16), 175.9 (C-12), 134.9 (C-10), 125.0 (C-1), 82.4 (C-6), 66.2 (C-5), 61.7 (C-4), 47.9 (N-CH3), 47.7 (N-CH3), 45.9 (C-11), 41.6 (C-7), 41.1 (C-9), 36.6 (CH2), 35.3 (C-13), 30.0 (CH2), 24.1 (CH2), 17.2 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C18H27NNaO3S2 [M + Na] + 392.1325, found 392.1323.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl diethylcarbamodithioate (7c). White amorphous solid (yield: 81%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 5.11 (d, J = 10.1 Hz, 1H, H-1), 4.07–3.97 (m, 2H, N-CH2), 3.89–3.72 (m, 5H, H-6, 13, N-CH2), 2.87–2.78 (m, 1H, H-11), 2.67 (d, J = 8.9 Hz, 1H, H-5), 2.43–2.22 (m, 3H, H-7, CH2), 2.19–2.05 (m, 4H, CH2), 1.67 (s, 3H, H-14), 1.66–1.58 (m, 1H, CH2), 1.32–1.23 (m, 9H, H-15, N-CH2CH3), 1.18 (td, J = 12.6, 5.5 Hz, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 194.8 (C-16), 175.9 (C-12), 135.0 (C-10), 124.9 (C-1), 82.4 (C-6), 66.4 (C-5), 61.7 (C-4), 50.1 (C-11), 48.0 (N-CH2), 47.9 (N-CH2), 47.0 (C-7), 41.2 (C-9), 36.7 (CH2), 34.9 (C-13), 30.1 (CH2), 24.2 (CH2), 17.3 (C-14), 17.0 (C-15), 12.7 (N-CH2CH3), 11.7 (N-CH2CH3); HRMS (ESI) calcd for C20H32NO3S2 [M + H] + 398.1818, found 398.1820.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl dipropylcarbamodithioate (7d). White amorphous solid (yield: 75%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 5.11 (d, J = 9.9 Hz, 1H, H-1), 3.97–3.86 (m, 2H, N-CH2), 3.83 (dt, J = 12.6, 6.0 Hz, 3H, H-6, -13), 3.70–3.61 (m, 2H, N-CH2), 2.82 (dt, J = 12.0, 5.1 Hz, 1H, H-11), 2.67 (d, J = 8.9 Hz, 1H, H-5), 2.44–2.22 (m, 3H, H-7, CH2), 2.11 (dt, J = 18.4, 11.1 Hz, 4H, CH2), 1.74 (dd, J = 14.8, 7.4 Hz, 4H, N-CH2CH2CH3), 1.67 (s, 3H, H-14), 1.66–1.58 (m, 1H, CH2), 1.27 (s, 3H, H-15), 1.24–1.13 (m, 1H, CH2), 0.94 (dd, J = 16.7, 7.5 Hz, 6H, N-CH2CH2CH3); 13 C NMR (100 MHz, CDCl3) δ 195.5 (C-16), 175.9 (C-12), 135.0 (C-10), 125.0 (C-1), 82.4 (C-6), 66.5 (C-5), 61.7 (C-4), 57.4 (C-11), 54.4 (C-7), 47.92 (N-CH2), 47.90 (N-CH2), 41.3 (C-9), 36.8 (CH2), 35.0 (C-13), 30.2 (CH2), 24.2 (CH2), 20.9 (N-CH2CH2CH3), 19.8 (N-CH2CH2CH3), 17.3 (C-14), 17.1 (C-15), 11.3 (2 C, N-CH2CH2CH3); HRMS (ESI) calcd for C22H36NO3S2 [M + H]+ 426.2131, found 426.2129.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl dibutylcarbamodithioate (7e). White amorphous solid (yield: 80%, purity: 97%). 1H NMR (400 MHz, CDCl3) δ 5.10 (d, J = 10.5 Hz, 1H, H-1), 4.01–3.90 (m, 2H, N-CH2), 3.89–3.76 (m, 3H, H-6, -13), 3.74–3.61 (m, 2H, N-CH2), 2.87–2.76 (m, 1H, H-11), 2.65 (d, J = 8.9 Hz, 1H, H-5), 2.42–2.28 (m, 2H, H-7, CH2), 2.28–2.20 (m, 1H, CH2), 2.18–2.03 (m, 4H, CH2), 1.73–1.57 (m, 8H, N-CH2CH2CH2CH3), 1.34 (dt, J = 14.3, 7.1 Hz, 4H, H-14, CH2), 1.26 (s, 3H, H-15), 1.17 (td, J = 13.0, 5.8 Hz, 1H, CH2), 0.93 (dd, J = 12.0, 7.1 Hz, 6H, N-CH2CH2CH2CH3); 13 C NMR (100 MHz, CDCl3) δ 195.2 (C-16), 175.9 (C-12), 135.0 (C-10), 124.9 (C-1), 82.4 (C-6), 66.4 (C-5), 61.6 (C-4), 55.5 (C-11), 52.6 (C-7), 47.9 (N-CH2), 47.8 (N-CH2), 41.2 (C-9), 36.7 (CH2), 34.9 (C-13), 30.2 (CH2), 29.6 (CH2), 28.5 (CH2), 24.1 (CH2), 20.1 (2 C, N-CH2CH2CH2CH3), 17.3 (C-14), 17.0 (C-15), 13.9 (N-CH2CH2CH2CH3), 13.8 (N-CH2CH2CH2CH3); HRMS (ESI) calcd for C24H40NO3S2 [M + H]+ 454.2444, found 454.2450.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl butyl(methyl)carbamodithioate (7f). White amorphous solid (yield: 89%, purity: 99%). 1H NMR (400 MHz, CDCl3, rotamer) δ 5.13 (d, J = 10.7 Hz, 1H, H-1), 4.12–4.00 (m, 1H, H-6), 3.89–3.69 (m, 4H, H-13, N-CH2), 3.49 (s, 1.5 H, N-CH3), 3.34 (s, 1.5 H, N-CH3), 2.82 (dt, J = 11.9, 5.1 Hz, 1H, H-11), 2.68 (d, J = 8.9 Hz, 1H, H-5), 2.45–2.21 (m, 3H, H-7, CH2), 2.18–2.02 (m, 4H, CH2), 1.71–1.61 (m, 6H, H-14, CH2), 1.43–1.31 (m, 2H, CH2), 1.27 (s, 3H, H-15), 1.19 (td, J = 13.0, 5.8 Hz, 1H, CH2), 0.95 (td, J = 7.3, 2.8 Hz, 3H, N-CH2CH2CH2CH3); 13 C NMR (100 MHz, CDCl3) δ 196.2 (C-16), 195.7 (C-16), 175.9 (C-12), 135.0 (C-10), 125.0 (C-1), 82.4 (C-6), 66.4 (C-5), 61.7 (C-4), 57.5 (C-11), 54.6 (C-7), 48.01 (N-CH2), 47.96 (N-CH2), 47.90 (N-CH3), 47.85 (N-CH3), 44.2 (CH2), 41.2 (CH2), 39.8 (CH2), 36.7 (N-CH2CH2CH2CH3), 35.1 (N-CH2CH2CH2CH3), 30.2 (CH2), 29.6 (CH2), 28.6 (CH2), 24.2 (CH2), 20.1 (CH2), 17.3 (C-14), 17.1 (C-15), 14.0 (N-CH2CH2CH2CH3), 13.9 (N-CH2CH2CH2CH3); HRMS (ESI) calcd for C21H34NO3S2 [M + H] + 412.1975, found 412.1981.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl (2-(dimethylamino)ethyl)carbamodithioate (7g). White amorphous solid (yield: 77%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 8.15 (br m, 1H, NH), 5.12 (d, J = 10.2 Hz, 1H, H-1), 3.84–3.46 (m, 5H, H-6, -13, N-CH2), 2.82–2.72 (m, 1H, H-11), 2.70 (d, J = 8.9 Hz, 1H, H-5), 2.52 (t, J = 6.2 Hz, 2H, (CH3)2NCH2) , 2.42–2.24 (m, 3H, H-7, CH2), 2.23 (s, 6H, N-CH3), 2.17–2.01 (m, 4H, CH2), 1.66 (s, 3H, H-14), 1.65–1.55 (m, 1H, CH2) , 1.26 (s, 3H, H-15), 1.17 (td, J = 12.9, 5.8 Hz, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 196.6 (C-16), 175.9 (C-12), 134.8 (C-10), 125.0 (C-1), 82.5 (C-6), 66.2 (C-5), 61.7 (C-4), 56.2 (CH3)2NCH2, 48.2 (C-11), 47.7 (C-7), 45.0 (N-CH3), 44.9 (N-CH3), 44.8 (N-CH2), 41.2 (CH2), 36.7 (CH2), 32.9 (C-13), 30.0 (CH2), 24.1 (CH2), 17.3 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C20H33N2O3S2 [M + H]+ 413.1927, found 413.1935.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl pyrrolidine-1-carbodithioate (7h). White amorphous solid (yield: 87%, purity: 96%). 1H NMR (400 MHz, CDCl3) δ 5.15 (d, J = 10.6 Hz, 1H, H-1), 3.93 (t, J = 6.9 Hz, 2H, N-CH2), 3.88–3.74 (m, 3H, H-6, -13), 3.74–3.63 (m, 2H, N-CH2), 2.86–2.76 (m, 1H, H-11), 2.71 (d, J = 8.9 Hz, 1H, H-5), 2.45–2.24 (m, 3H, H-7, CH2), 2.20–2.04 (m, 6H, CH2), 2.03–1.93 (m, 2H, CH2), 1.68 (s, 3H, H-14), 1.63 (s, 1H, CH2), 1.27 (s, 3H, H-15), 1.26–1.14 (m, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 192.1 (C-16), 175.9 (C-12), 135.0 (C-10), 125.1 (C-1), 82.5 (C-6), 66.3 (C-5), 61.7 (C-4), 55.6 (C-11), 50.8 (C-7), 48.0 (N-CH2), 48.0 (N-CH2), 41.2 (CH2), 36.8 (CH2), 34.1 (C-13), 30.1 (CH2), 26.1 (CH2), 24.3 (N-CH2CH2), 24.2 (N-CH2CH2), 17.3 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C20H30NO3S2 [M + H]+ 396.1662, found 396.1667.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl piperidine-1-carbodithioate (7i). White amorphous solid (yield: 83%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 5.12 (d, J = 9.9 Hz, 1H, H-1), 4.27 (d, J = 29.0 Hz, 2H, H-13), 3.98–3.75 (m, 5H, H-6, N-CH2), 2.82 (ddd, J = 12.0, 6.1, 4.3 Hz, 1H, H-11), 2.68 (d, J = 8.9 Hz, 1H, H-5), 2.39–2.22 (m, 3H, H-7, CH2), 2.11 (dq, J = 12.1, 8.3 Hz, 4H, N-CH2CH2), 1.73–1.62 (m, 10H, H-14, CH2), 1.27 (d, J = 6.1 Hz, 3H, H-15), 1.21–1.13 (m, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 194.9 (C-16), 175.9 (C-12), 135.0 (C-10), 125.0 (C-1), 82.4 (C-6), 66.4 (C-5), 61.7 (C-4), 53.7 (C-11), 51.7 (C-7), 48.1 (N-CH2), 47.9 (N-CH2), 41.3 (CH2), 36.8 (CH2), 34.9 (C-13), 30.1 (CH2), 26.3 (CH2), 25.7 (N-CH2CH2CH2), 24.4 (N-CH2CH2), 24.2 (N-CH2CH2), 17.3 (C-14), 17.1 (C-15); HRMS (ESI) calcd for C21H32NO3S2 [M + H]+ 410.1818, found 410.1817.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl 4-methylpiperazine-1-carbodithioate (7j). White amorphous solid (yield: 79%, purity: 97%). 1H NMR (400 MHz, CDCl3) δ 5.14 (d, J = 9.9 Hz, 1H, H-1), 4.35 (d, J = 31.6 Hz, 2H, N-CH2CH2NCH3), 4.00 (s, 2H, H-13), 3.83 (qd, J = 14.1, 5.2 Hz, 3H, H-6, N-CH2), 2.82 (ddd, J = 12.0, 6.4, 4.1 Hz, 1H, H-11), 2.69 (d, J = 8.9 Hz, 1H, H-5), 2.49 (s, 4H, H-7, CH2), 2.43–2.23 (m, 6H, N-CH2CH2NCH3, CH2), 2.11 (dt, J = 18.0, 11.0 Hz, 4H, N-CH3, CH2), 1.68 (s, 3H, H-14), 1.65–1.59 (m, 1H, CH2), 1.27 (s, 3H, H-15), 1.23–1.13 (m, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 196.3 (C-16), 175.8 (C-12), 134.9 (C-10), 125.1 (C-1), 82.5 (C-6), 66.4 (C-5), 61.7 (C-4), 54.6 (2 C, N-CH2CH2NCH3), 51.9 (C-11), 50.0 (C-7), 48.1 (N-CH2CH2NCH3), 47.8(N-CH2CH2NCH3), 45.7(N-CH3), 41.2 (CH2), 36.7 (CH2), 34.9 (C-13), 30.1 (CH2), 24.2 (CH2), 17.3 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C21H33N2O3S2 [M + H]+ 425.1927, found 425.1922.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl 4–(2-hydroxyethyl)piperazine-1-carbodithioate (7k). White amorphous solid (yield: 75%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 5.12 (d, J = 9.8 Hz, 1H, H-1), 4.30 (s, 2H, H-13), 4.02 (br m, 2H, N-CH2), 3.87–3.72 (m, 3H, H-6, N-CH2), 3.70–3.58 (m, 2H, CH2OH), 3.11 (s, 1H, O-H), 2.81 (ddd, J = 12.1, 6.4, 4.1 Hz, 1H, H-11), 2.68 (d, J = 8.9 Hz, 1H, H-5), 2.63–2.56 (m, 6H, (CH2)2NCH2), 2.40–2.24 (m, 3H, H-7, CH2), 2.19–2.06 (m, 4H, CH2), 1.66 (s, 3H, H-14), 1.65–1.57 (m, 1H, CH2), 1.25 (s, 3H, H-15), 1.17 (td, J = 13.0, 5.8 Hz, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 196.2 (C-16), 175.8 (C-12), 134.8 (C-10), 125.0 (C-1), 82.4 (C-6), 66.3 (C-5), 61.7 (C-4), 59.2 (CH2OH), 58.1 (NCH2CH2OH), 52.5 (2C, CH2NCH2CH2OH), 51.7 (C-11), 49.9 (C-7), 48.1 (N-CH2), 47.7 (N-CH2), 41.1 (CH2), 36.7 (CH2), 34.8 (CH2), 30.0 (CH2), 24.1 (CH2), 17.2 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C22H35N2O4S2 [M + H]+ 455.2033, found 455.2039.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl (pyridin-3-ylmethyl)carbamodithioate (7l). White amorphous solid (yield: 68%, purity: 95%). 1H NMR (400 MHz, CDCl3) δ 9.38 (s, 1H, N-H), 8.50 (s, 1H, H-2’), 8.37 (s, 1H, H-6’), 7.70 (d, J = 5.7 Hz, 1H, H-4’), 7.21 (s, 1H, H-5’), 5.00 (d, J = 11.3 Hz, 1H, H-1), 4.91 (s, 2H, H-7’), 3.87–3.73 (m, 2H, H-6, -13), 3.67 (d, J = 14.3 Hz, 1H, H-13), 2.75 (d, J = 8.7 Hz, 1H, H-11), 2.62 (d, J = 8.2 Hz, 1H, H-5), 2.38–2.24 (m, 1H, H-2), 2.21–1.92 (m, 6H, H-2, -3, -7, -8, -9), 1.62 (s, 4H, H-8, -14), 1.22 (s, 3H, H-15), 1.15 (d, J = 12.3 Hz, 1H, H-3).13C NMR (100 MHz, CDCl3) δ 198.0 (C-16), 176.3 (C-12), 149.4 (C-2’), 148.6 (C-6’), 136.4 (C-4’), 134.7 (C-10), 132.7 (C-3’), 125.0 (C-1), 123.7 (C-5’), 82.6 (C-6), 66.1 (C-5), 62.1 (C-4), 48.4 (C-7’), 48.3 (C-11), 47.1 (C-7), 41.1 (C-9), 36.5 (C-3), 32.6 (C-13), 30.0 (C-8), 24.1 (C-2), 17.2 (C-14), 16.9 (C-15); HRMS (ESI) calcd for C22H29N2O3S2 [M + H]+ 433.1614, found 433.1608.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl methyl(pyridin-3-ylmethyl)carbamodithioate (7m). White amorphous solid (yield: 77%, purity: 97%). 1H NMR (400 MHz, CDCl3, rotamer) δ 8.55–8.50 (m, 2H, Ar-H), 7.67–7.59 (m, 1H, Ar-H), 7.28–7.26 (m, 1H, Ar-H), 5.37 (s, 1.5 H, Ar-CH2), 5.14 (d, J = 11.1 Hz, 1H, H-1), 5.05 (br s, 0.5 H, Ar-CH2), 3.92–3.72 (m, 3H, H-6, -13), 3.50 (s, 1H, N-CH3), 3.34 (s, 2H, N-CH3), 2.84 (d, J = 6.2 Hz, 1H, H-11), 2.70 (d, J = 8.8 Hz, 1H, H-5), 2.47–2.23 (m, 3H, H-7, CH2), 2.22–2.04 (m, 4H, CH2), 1.68 (s, 3H, H-14), 1.67–1.60 (s, 1H, CH2), 1.28 (s, 3H, H-15), 1.24–1.14 (m, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 198.8 (C-16), 175.8 (C-12), 149.5 (Ar-C), 149.3 (Ar-C), 135.5 (Ar-C), 134.8 (C-10), 131.4 (Ar-C), 125.1 (C-1), 123.8 (Ar-C), 82.5 (C-6), 66.3 (C-5), 61.7 (C-4), 57.6 (Ar-CH2), 48.2 (C-11), 47.7 (C-7), 41.3 (C-9), 39.4 (N-CH3), 36.7 (C-3), 35.6 (C-13), 30.1 (C-8), 24.2 (C-2), 17.3 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C23H31N2O3S2 [M + H]+ 447.1771, found 447.1765.
General procedure for the synthesis of compounds 7n and 7o
To a mixture of corresponding amine (1.2 eq) in tetrahydrofurane (THF) was added n-BuLi (1.2 eq) at 0 °C, the mixture was stirred for 1 h at 0 °C, CS2 was added. After 2 h, PTL (1 eq) dissolved in THF was added to the mixture. The reaction was stirred overnight at room temperature. The reaction was quenched by adding saturated aqueous ammonium chloride solution, extracted with ethyl acetate three times, organic phase was washed with water and saturated brine, the combined organic layer was dried over anhydrous Na2SO4, concentrated under reduced pressure and purified by column chromatography on silica gel to give compounds 7n and 7o.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl methyl(pyridin-2-yl)carbamodithioate (7n). White amorphous solid (yield: 9%, purity: 98%). 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 3.9 Hz, 1H, Ar-H), 7.87–7.78 (m, 1H, Ar-H), 7.37–7.32 (m, 2H, Ar-H), 5.16 (d, J = 10.2 Hz, 1H, H-1), 3.86–3.79 (m, 2H, H-6, -13), 3.78 (s, 3H, N-CH3), 3.73 (dd, J = 14.3, 4.0 Hz, 1H, H-13), 2.82–2.74 (m, 1H, H-11), 2.71 (d, J = 8.9 Hz, 1H, H-5), 2.39–2.24 (m, 3H, CH2), 2.20–2.07 (m, 4H, H-7, CH2), 1.67 (s, 3H, H-14), 1.65–1.56 (m, 1H, CH2), 1.26 (s, 3H, H-15), 1.24–1.16 (m, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 198.8 (C-16), 175.7 (C-12), 156.4 (Ar-C), 150.0 (Ar-C), 138.8 (Ar-C), 134.9 (C-10), 125.1 (C-1), 124.0 (Ar-C), 122.5 (Ar-C), 82.5 (C-6), 66.3 (C-5), 61.7 (C-4), 47.8 (C-11), 47.7 (C-7), 44.2 (N-CH3), 41.2 (C-9), 36.7 (C-3), 35.4 (C-13), 30.2 (C-8), 24.2 (C-2), 17.3 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C22H29N2O3S2 [M + H]+ 433.1614, found 433.1606.
((3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10]cyclodeca[1,2-b]furan-3-yl)methyl methyl(pyridin-3-yl)carbamodithioate (7o). White amorphous solid (yield: 18%, purity: 96%). 1H NMR (400 MHz, CDCl3) δ 8.64 (d, J = 4.3 Hz, 1H, Ar-H), 8.51 (s, 1H, Ar-H), 7.60 (d, J = 8.1 Hz, 1H, Ar-H), 7.41 (dd, J = 8.0, 4.8 Hz, 1H, Ar-H), 5.14 (d, J = 10.3 Hz, 1H, H-1), 3.79 (t, J = 9.2 Hz, 1H, H-6), 3.76 (s, 3H, N-CH3), 3.74–3.60 (m, 2H, H-6, -13), 2.81–2.71 (m, 1H, H-11), 2.66 (d, J = 8.9 Hz, 1H, H-5), 2.41–2.24 (m, 3H, CH2), 2.16–2.03 (m, 4H, H-7, CH2), 1.67 (s, 3H, H-14), 1.65–1.58 (m, 1H, CH2), 1.25 (s, 3H, H-15), 1.23–1.12 (m, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 199.5 (C-16), 175.5 (C-12), 150.0 (Ar-C), 148.2 (Ar-C), 134.8 (2 C, Ar-C, C-10), 125.1 (C-1), 124.4 (Ar-C), 82.4 (C-6), 66.2 (C-5), 61.7 (C-4), 48.3 (C-11), 47.3 (2 C, N-CH3, C-7), 41.1 (C-9), 36.6 (C-3), 36.0 (C-13), 30.1 (C-8), 24.1 (C-2), 17.2 (C-14), 17.0 (C-15); HRMS (ESI) calcd for C22H29N2O3S2 [M + H]+ 433.1614, found 433.1609.
(3S,3aS,9aR,10aS,10bS,E)-6,9a-dimethyl-3-((methylthio)methyl)-3a,4,5,8,9,9a,10a,10b-octahydrooxireno[2',3':9,10] cyclodeca[1,2-b] furan-2(3H)-one (8). To a solution of PTL (248 mg, 1.0 mmol) and NaH2PO4·2H2O (378 mg, 1.05 mmol) in THF (2 ml), triethylamine (0.15 ml), and 15% aqueous sodium methanethiolate solution (0.5 ml, 1.0 mmol) were added sequentially. After stirred for 1 h, the reaction was quenched by adding saturated aqueous ammonium chloride solution, extracted with ethyl acetate (3 × 15 ml). Organic phase was washed with water and saturated brine. The combined organic was dried over anhydrous Na2SO4, concentrated under reduced pressure and purified by column chromatography on silica gel (ethyl acetate:hexane =60:40) to give compound 8 as a white amorphous solid (144 mg, yield: 49%, purity: 98%). 1H NMR (400 MHz, CDCl3) δ 5.20 (d, J = 10.9 Hz, 1H, H-1), 3.84 (t, J = 9.0 Hz, 1H, H-6), 2.95 (dd, J = 14.0, 4.6 Hz, 1H, H-13), 2.89 (dd, J = 14.0, 4.6 Hz, 1H, H-13), 2.74 (d, J = 9.0 Hz, 1H, H-11), 2.61 (dt, J = 11.7, 4.6 Hz, 1H, H-5), 2.43–2.33 (m, 2H, CH2), 2.25 (dd, J = 13.2, 6.3 Hz, 1H, H-7), 2.16 (s, 3H, S-CH3), 2.15–2.03 (m, 3H, CH2), 1.94 (dd, J = 15.0, 6.3 Hz, 1H, CH2), 1.73–1.61 (m, 1H, CH2), 1.68 (s, 3H, H-14), 1.27 (s, 3H, H-15), 1.21 (td, J = 13.0, 6.0 Hz, 1H, CH2); 13 C NMR (100 MHz, CDCl3) δ 175.7 (C-12), 134.6 (C-10), 125.2 (C-1), 82.4 (C-6), 66.4 (C-5), 61.6 (C-4), 47.9 (C-11), 47.6 (C-7), 41.1 (C-9), 36.7 (CH2), 32.5 (C-13), 30.1 (CH2), 24.2 (CH2), 17.3 (C-14), 17.2 (C-15), 17.0 (S-CH3); HRMS (ESI) calcd for C16H25O3S [M + H]+ 297.1519, found 297.1522.
Potassium (pyridin-3-ylmethyl) carbamodithioate (10). 3-(aminomethyl) pyridine (9) (265 mg, 2.45 mmol) in methanol (10 ml) was mixed with CS2 (1.03 ml, 17.04 mmol) and KOH (137 mg, 2.44 mmol), then stirred at 0 °C for 2 h, concentrated under reduced pressure and recrystallisation in ethanol to afford compound 10 as a white amorphous solid (377 mg, yield: 69%, purity: 96%). 1H NMR (400 MHz, CD3OD) δ 8.55 (d, J = 1.4 Hz, 1H, Ar-H), 8.44–8.37 (m, 1H, Ar-H), 7.87 (d, J = 7.9 Hz, 1H, Ar-H), 7.45–7.33 (m, 1H, Ar-H), 4.90 (s, 2H, CH2); 13 C NMR (100 MHz, CD3OD) δ 216.4 (S = C), 149.6 (Ar-C), 148.4 (Ar-C), 137.8 (Ar-C), 137.1 (Ar-C), 125.1 (Ar-C), 68.2 (CH2); HRMS (ESI-MS) calcd for C7H9N2S2 [M + H] + 185.0202, found 185.0202.
Methyl (pyridin-3-ylmethyl) carbamodithioate (11). A mixture of TEA (0.3 ml, 2.15 mmol), CS2 (0.18 ml, 2.98 mmol), and 3-(aminomethyl) pyridine (9) (212 mg, 1.96 mmol) was dissolved in THF (5 ml), the mixture was stirred at 0 °C for 10 min. MeI (134 µl, 2.16 mmol) was added to the reaction and stirred for 4 h. Water (10 ml) was added to quench the reaction. The resulting mixture was extracted with ethyl acetate (3 × 25 ml). Organic phase was washed with water and saturated brine. The combined organic layer was dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by column chromatography on silica gel (ethyl acetate:hexane =50:50) to give compound 11 as a white amorphous solid (366 mg, yield: 94%, purity: 97%). 1H NMR (400 MHz, CDCl3) δ 8.43 (t, J = 19.0 Hz, 3H, Ar-H, N-H), 7.70 (d, J = 7.6 Hz, 1H, Ar-H), 7.26 (t, J = 6.1 Hz, 1H, Ar-H), 4.95 (d, J = 5.2 Hz, 2H, CH2), 2.65 (s, 3H, CH3); 13 C NMR (100 MHz, CDCl3) δ 200.1 (S = C), 149.1 (Ar-C), 148.9 (Ar-C), 136.2 (Ar-C), 132.5 (Ar-C), 123.7 (Ar-C), 48.1 (CH2), 18.3 (CH3); HRMS (ESI) calcd for C8H11N2S2 [M + H] + 199.0358, found 199.0359.
((3S,3aS,9aR,10aS,10bS, E)-6,9a-dimethyl-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2',3':9,10] cyclodeca[1,2-b] furan-3-yl) methyl (pyridin-3-ylmethyl) carbamodithioate oxalate (12). To a solution of compound 7l (42 mg, 0.097 mmol) in methanol (1 ml), oxalic acid (8.7 mg, 0.097 mmol) was added. The mixture was stirred for 0.5 h and concentrated under vacuum. The residue was washed three times with ethyl acetate to afford compound 12 as a white amorphous solid (49.9 mg, yield 98%). 1H NMR (400 MHz, DMSO-d6) δ 12.82 (s, 2H, COOH), 10.67 (t, J = 5.3 Hz, 1H, N-H), 8.65–8.37 (m, 2H, Ar-H), 7.73 (d, J = 6.7 Hz, 1H, Ar-H), 7.49–7.29 (m, 1H, Ar-H), 5.01 (d, J = 10.2 Hz, 1H, H-1), 4.87 (ddd, J = 39.4, 15.0, 5.6 Hz, 2H, H-7’), 4.10–3.93 (m, 1H, H-6), 3.81–3.71 (m, 1H, H-13), 3.60 (dd, J = 14.3, 4.3 Hz, 1H, H-13), 2.99–2.88 (m, 1H, H-11), 2.64 (d, J = 9.1 Hz, 1H, H-5), 2.33 (ddd, J = 25.6, 12.9, 5.1 Hz, 1H, CH2), 2.11–2.00 (m, 5H, H-7, CH2), 1.90 (t, J = 12.3 Hz, 1H, CH2), 1.75–1.65 (m, 1H, CH2), 1.61 (s, 3H, H-14), 1.18 (d, J = 3.5 Hz, 3H, H-15), 1.12–1.04 (m, 1H, CH2). 13 C NMR (100 MHz, DMSO-d6) δ 197.0 (C-16), 176.0 (C-12), 161.1 (COOH), 148.7 (C-2’), 148.2 (C-6’), 135.8 (C-4’), 134.4 (C-10), 133.0 (C-3’), 124.4 (C-5’), 123.6 (C-1), 81.5 (C-6), 65.5 (C-5), 61.2 (C-4), 59.7 (C-7’), 47.4 (C-11), 46.7 (C-7), 40.5 (C-9), 36.0 (C-3), 32.3 (C-13), 29.1 (C-8), 23.6 (C-2), 16.8 (C-14), 16.6 (C-15). HRMS (ESI) calcd for C22H29N2O3S2 [M + H] + 433.1614, found 433.1613.
Materials
Cell culture medium (1640) and foetal bovine serum were purchased from Gibco (NY, USA). H4434 culture medium was purchased from stem cell. MTT, cremophor EL, and DMSO were purchased from Sigma Chemical Company (St. Louis, MA, USA). Cell lysis buffer was purchased from Beyotime Institute of Biotechnology (Beijing, China). AnnexinV-fluorescein isothiocyanate (FITC) and propidium iodide (PI) apoptosis detection kit, human CD34-APC, and human CD38-PE.cy7 antibody were purchased from BD (BD, USA). Rabbit polyclonal anti-human p65, XIAP, Bax, Bcl-2, JNK, p-JNK, ERK1/2, p-ERK1/2, p38, p-p38, c-Jun, p-c-Jun, c-Fos, c-Myc, PARP, caspase-3, caspase-9, and β-actin antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). ECL-Plus Kit was purchased from Thermo Scientific (Rockford, IL, USA). Kunming mice and NOD/SCID mice were purchased from Chinese Academy of Sciences (Shanghai, China).
Cell isolation and culture
Human leukaemia cell lines HL-60, HL-60/adriamycin (ADR), THP-1, K562, and KG1a were cultured in 1640 containing supplements (10% foetal bovine serum, penicillin/streptomycin, and L-glutamine) at 37 °C, 5% CO2. Primary human AML samples were obtained from Yuhuangding Hospital (Yantai, Shandong).
Cytotoxicity assay
Leukaemia cells in exponential growth were seeded in 96 well plates (1 × 104 each well). After 24 h different concentrations of the compounds were added into each well. After 72 h, 20 μl MTT (5 mg/ml) were added and incubated for 4 h. The cells were centrifuged by 2000 rpm for 20 min in follow, then the supernatant were removed and 200 μl DMSO was added to measure the absorbance at 570 nm. The IC50 was calculated by GraphPad Prism 5.
Apoptosis assay
Leukaemia cells treated with different concentrations of the compound 7l for 48 h (for cultured cells) or 24 h (for leukaemia cells from primary specimens) were collected and re-suspended by 100 μl × loading buffer. Annexin-V FITC and PI were added for 15 min according to the manufacturer’s protocol. Then the cells were analysed by flow cytometry.
Methylcellulose colony-forming cell assay
Mononuclear cells from primary AML specimens were incubated in serum-free Iscove's modified dulbecco medium (IMDM) in the presence or absence of 0.5, 1, and 2 μM 7l for 24 h. After then 2 × 105 cells were plated into 24 well plates in Methocult H4434. The number of colonies was recorded after 10 days.
Acute toxicity assay in Kunming mice
To verify the toxicity of 7l, Kunming mice were administrated orally with compound 12 (salt form of 7l) or vehicle control with a dose of 500 mg/kg. The body weight was recorded every day. After 22 days, the blood samples were collected and assayed by blood routine analysis. Meanwhile, the viscus tissues included liver, spleen, lung, kidney, and brain were collected for immune-histochemical analysis.
Patient-derived xenograft model assay
To further evaluate the anti-AML effect of 12, patient derived xenograft model of human AML was established. First, the mice were irradiated with 200 centi-gray (cGy). After 8 h, 5 × 106 primary AML mononuclear cells were injected by tail vein at a final volume of 200 μl. After 30 days, the mice were administrated orally with 12 with a dose of 100 mg/kg every other day. The survival rate was analysed and graphed by Kaplan-Meier plot.
Microarray transcriptional profiling
KG1a cells were treated with 2 µM 7l for 24 h. Gene chip assay was performed by Genergy Biotechnology Company (Shanghai, China). Each sample was performed in triplicate. These data are available at National Center for Biotechnology Information Gene Expression Omnibus with accession number GSE103717.
Western blotting assay
KG1a cells were collected after being treated with 7l at different concentrations for 24 h. The cells were re-suspended by 200 μl RIPA lysis for 30 min on ice to extract the total protein. Equal amounts of protein extract sample (50 μg) was separated by SDS-PAGE in a 12% gel and then transferred to a polyvinylidene fluoride (PVDF) membrane. After blocked in 5% skim milk the membranes were incubated with primary antibodies at 4 °C overnight. Then the membrane was washed with phosphate buffered saline+Tween-20 (PBST) and incubated with secondary antibodies at room temperature for 2 h. Finally, bound antibodies were assayed by ECL-Plus Kit.
Statistical analysis
Each experiment was performed in triplicate and all results were repeated for three times. Student’s t-test was performed to analyse the significance level by GraphPad Prism 5 software. A p-value of less than 0.05 was considered to be statistically significant.
Results and discussion
Chemistry
The synthesis of compounds 7a–7o was shown in Scheme 1. The desired compounds 7a–7m could be obtained by one-pot reaction of corresponding amine, carbon disulphide, and PTL using TEA as base with yields from 73% to 89%. For synthesis of compounds 7n–7o, n-butyllithium was used as a base in tetrahydrofuran solution. Michael addition of PTL with sodium methanethiolate provided compound 8. As shown in Scheme 2, treatment of 3-(aminomethyl) pyridine (9) and carbon disulphide with KOH or iodomethane yielded compounds 10 and 11, respectively. Reaction of compound 7l and oxalic acid in methanol gives salt 12.
Scheme 1. Synthesis of compounds 7a–7o and 8a. aReagents and conditions: (a) for 7a–7m: amine, CS2, TEA, DCM-MeOH, 0 °C to rt, 68–89%; for 7n and 7o: amine, CS2, n-BuLi, THF, 0 °C to rt, 7n: 9%, 7o: 18%; (b) MeSNa, NaH2PO4, TEA, THF, H2O, rt, 49%.
Scheme 2. Synthesis of Analogues 10–12a. aReagents and conditions: (a) CS2, KOH, MeOH, 0 °C to rt, 69%; (b) CS2, TEA, MeI, DCM, 0 °C to rt, 94%; (c) Oxalic acid, MeOH, rt, 98%.
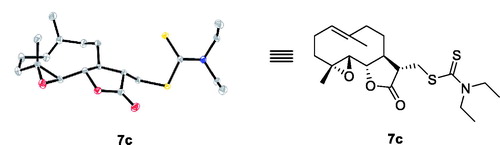
To determine the absolute stereospecificity of the PTL Michael addition products, X-ray analysis of compound 7c was performed (). The details of the synthetic procedures and structural characterisations are described in the Experimental Section. The purity of all analogues was confirmed to be ≥95% by HPLC.
Figure 4. X-ray structure of compound 7c.
Biological activities against AML cell lines
Compounds 7a–7o and 8, 10, 11 were evaluated for their effects on viability of the AML cell lines KG1a and HL-60. In addition, ADR was introduced as a positive control, and the natural product, PTL (1), was also included for comparison. KG1a, a human AML cell line, showed high multidrug resistance and self-renewal potential. KG1a cells have characteristics of LSCs, a large portion of cells bearing a CD34+CD38– immunophenotype. KG1a was considered as a type of AML progenitor cell lineCitation66–68. The results were shown in . The natural parent compound PTL (1) exhibited moderate potency against the KG1a cells (IC50 = 6.1 μM) and HL-60 cells (IC50 = 3.8 μM). Introduction of dithiocarbamate moieties with linear aliphatic amino (7a–7g with IC50 values of 6.5–24.7 μM) or with cyclic aliphatic amino (7h–7k with IC50 values of 4.4–7.7 μM) led to comparable or decreased activities against the HL-60 cell line. For KG1a cell line, most of these compounds showed declined potencies with IC50 values of 6.9–50 μM, except that compound 7 b (IC50 = 4.8 μM) exhibited slightly improved activity compared with that of PTL (IC50 = 6.1 μM).
Table 1. Inhibitory effects of dithiocarbamate esters of parthenolide against KG1a and HL-60 cells.
To further explore the influence of dithiocarbamate moiety for the anti-AML activity, different patterns of heterocycle substitution were introduced (7l–7o). To our surprise, a significant advance was achieved when pyridinylmethylamino group was installed to PTL, which is compound 7l. Compound 7l exhibited more potent anti-AML activity than PTL. Compound 7l showed greatly increased activities against KG1a and HL-60 with IC50 values of 0.7 and 1.7 μM, respectively, and the activities against KG1a and HL-60 were 8.7- and 2.2-folds comparing to those of PTL, respectively. It is worth noting that 7l was more active against AML progenitor cell line KG1a (IC50 = 0.7 μM) comparing to sensitive cell line HL-60 (IC50 = 1.7 μM). In contrast, ADR, a clinically used drug, showed 34-folds drop of activity against KG1a (IC50 = 0.75 μM) than that against HL-60 (IC50 = 0.022 μM). Compound 7l may present superior physico/chemical properties to penetrate the cellular biomembranes when compared to the parent PTL with tricyclic scaffold. With introduction of a methyl group to 7l (7m) or replacement of pyridinylmethylamino group with pyridinylamino group (7n and 7o), the anti-AML activities were significantly decreased.
To investigate the role of dithiocarbamate moiety in anti-AML activity of 7l, we synthesised the derivatives 8, 10, and 11 for comparison. Compound 8 lost anti-AML activity (IC50 > 50 μM). Compounds 10 and 11 or combination of 10 and PTL (1:1) exhibited moderate anti-AML activity, which were less potent than 7l. These results suggest that the anti-AML activity of 7l may be attributed to the synergic effects of both moieties of PTL and dithiocarbamate.
Compound 7l inhibited the proliferation of different cultured leukaemia cells
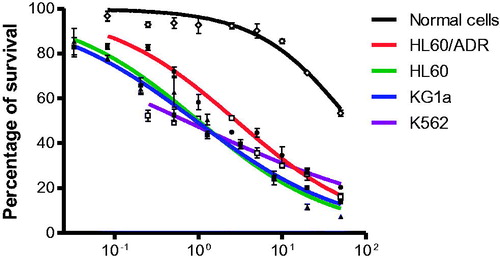
Compound 7l was further evaluated for inhibitory effects on other different leukaemia cells by MTT assay using leukaemia cell lines K562 and the ADR-resistant cell line HL-60/ADR. All leukaemia cells were treated with 7l for 72 h. From the results ( and ), 7l showed strong inhibitory effects on K562 cells with the IC50 value of 1.3 μM. Moreover, 7l showed similar inhibitory effects on ADR-resistant cell line HL-60/ADR (IC50 = 2.2 μM) with sensitive cell line HL-60 (IC50 = 1.7 μM).
Figure 5. Compound 7l inhibited the proliferation of different cultured leukaemia cells while sparing normal cells.
Compound 7l selectively inhibited AML cells while sparing normal cells
The most potent 7l was selected for further characterisation to evaluate its selectivity against AML over normal cells. For the study, normal cells were obtained from health donors. As shown in and , compound 7l did not significantly affect the viability of normal cells, which indicates that 7l could selectively eliminate AML cells (IC50 = 0.7 and 1.7 µM towards KG1a and HL-60, respectively) with relatively low toxicity against normal cells (IC50 = 59.8 µM). The selectivity indexes of compound 7l for AML cells KG1a and HL-60 were 85.4 and 35.2, respectively.
Compound 7l induced the apoptosis of diverse cultured leukaemia cells
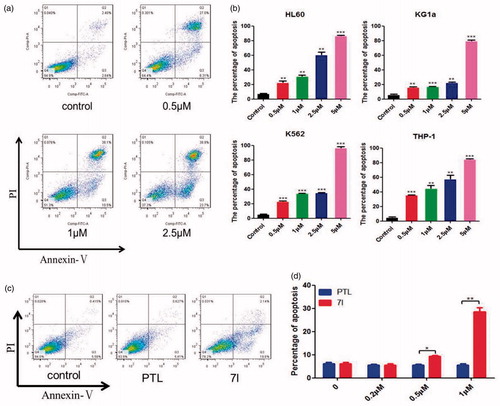
The apoptosis induced by 7l on leukaemia cells were detected by flow cytometry using AnnexinV/PI double staining (). As shown in 7l significantly induced the apoptosis of leukaemia cells HL-60, K562, THP-1, and especially KG1a which was considered to be a leukaemia stem-like cell line. Moreover, the percentages of apoptosis in the KG1a cells after the treatment with 7l at a concentration of 1 μM were remarkably higher than those of PTL (). These results indicated that the cytotoxicity of 7l to leukaemia cells was accomplished through inducing apoptosis of leukaemia cells.
Figure 6. Compound 7l induced the apoptosis of diverse cultured leukaemia cells. (a) The representative picture of apoptosis induced by 7l in KG1a cells. (b) Apoptosis of THP-1, HL-60/ADR, K562, KG1a cells after being exposed to different concentrations of 7l for 48 h. The percentages of apoptosis were determined by flow cytometry using Annexin V/PI. (c) The representative picture of apoptosis induced by 7l and PTL in KG1a cells at 1 µM. (d) Apoptosis of KG1a cells after being exposed to 0.2, 0.5, 1 µM of 7l or PTL for 48 h. These experiments were performed for three times. Analysis results represented mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001.
Compound 7l induced the apoptosis of total primary leukaemia cells and CD34+CD38– cells from clinical AML patients
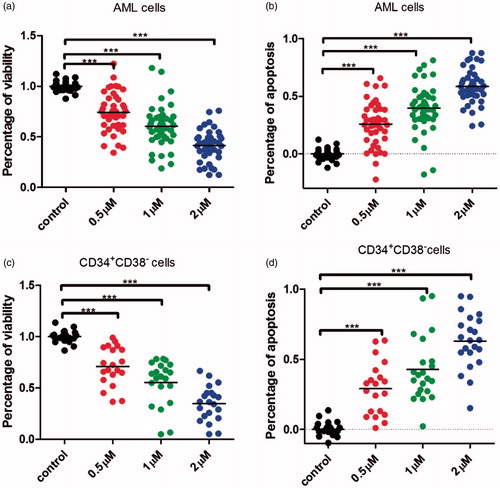
To further identify the effects of 7l on leukaemia cells and LSCs, 18 primary AML specimens were collected. Cells bearing a CD34+CD38– immune-phenotype were considered as LSCs. The apoptosis of total leukaemia cells and CD34+CD38– LSCs were assayed after being treated with 7l for 24 h. The result showed that 7l could greatly ablate total leukaemia cells and CD34+CD38– primary LSCs with a dose-dependent manner ().
Figure 7. Compound 7l induced the apoptosis of primary leukaemia cells and CD34+CD38– cells from AML patients. (a) Compound 7l reduced the percentage of viability with a dose-dependent manner in primary AML cells after being treated for 24 h. (b) Compound 7l induced apoptosis with a dose-dependent manner in primary AML cells after being treated for 24 h. (c) Compound 7l reduced the percentage of viability with a dose-dependent manner in primary AML CD34+CD38– cells after being treated for 24 h. (d) Compound 7l induced apoptosis with a dose-dependent manner in primary AML CD34+CD38– cells after being treated for 24 h. ***p < 0.001.
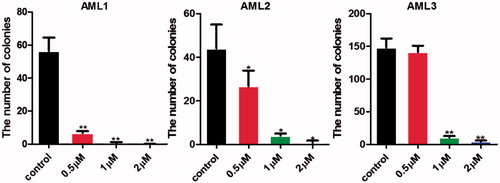
Compound 7l suppressed the colony formation of primary human leukaemia cells
Colony formation was an important characteristic of LSCs when cultured in methocult H4434 medium. To determine the effect of 7l on LSCs, colony formation assay was performed. From the result in , the numbers of colony-forming units were significantly reduced after being treated with 7l for 10 days with a dose-dependent manner.
Figure 8. Compound 7l suppressed the colony formation of primary human leukaemia cells. *p < 0.05, **p < 0.01.
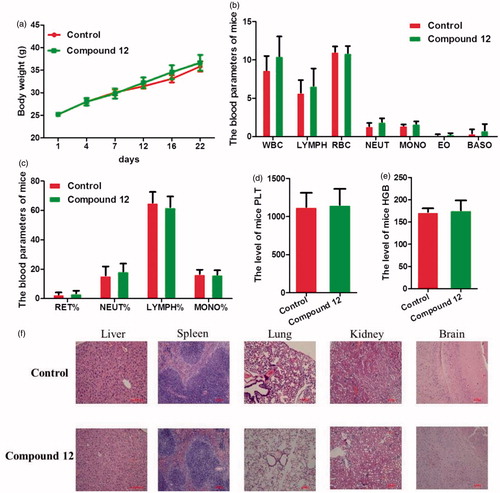
Compound 12 displayed no observable toxicity in Kunming mice
Taking account of the significant anti-AML activity of 7l, we planned to evaluate its toxicity to mice by oral administration. However, 7l showed low solubility in water. Therefore, 7l was converted to its salt form, which is compound 12. To explore the safety of 12 to haematopoietic system and main organs, acute toxicity assay was performed. Kunming mice were treated with 12 (500 mg/kg) or vehicle control for 22 days. At the end of the experiments, blood samples, liver, spleen, lung, kidney, and brain were collected and detected. The body weights did not significantly reduce after being treated with 12 (). Furthermore, from the results of complete blood counts, all the parameters of routine blood test including red blood cell count, lymphocyte count, neutrophil cell count, monocyte cell count, eosinophil cell count, basophil cell count, platelet count, and the level of haemoglobin were not changed apparently compared with vehicle control. No pathologic changes were apparent in the examined tissues (). These results suggested that 12 was safe to mice.
Figure 9. Compound 12 displayed no observable toxicity to Kunming mice. (a) The body weights did not significantly reduced after being treated with 12 compared with vehicle control. (b) The parameters of routine blood test included white blood cell count, lymphocyte count, neutrophil cell count, monocyte cell count, eosinophil cell count, and basophil cell count were not changed apparently after treatment of 12 compared with vehicle control. (c) The blood parameters included the percentage of reticulocyte cell, neutrophil cell, lymphocyte cell, and monocyte cell were not changed significantly after being treated with 12 compared with vehicle control. (d) The level of platelet was not changed significantly after treatment of 12 compared with vehicle control. (e) The level of haemoglobin was not changed clearly compared with vehicle control. (f) The representative pictures of haematoxylin and eosin (H&E) staining of the liver, spleen, lung, kidney, and brain.
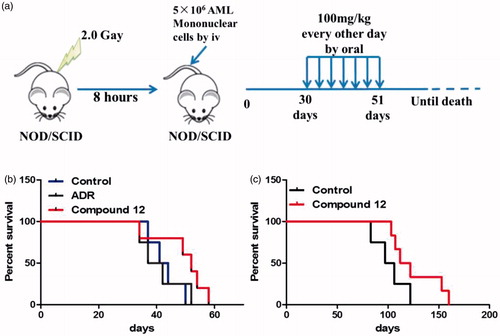
Compound 12 prolonged the lifespan in patient-derived xenograft model assay
Compound 7l showed significant cytotoxicity against cultured leukaemia cells, total primary leukaemia cells, and LSCs in vitro, which prompted us to further investigate the anti-AML effect in vivo. Patient-derived xenograft model was established with primary human AML mononuclear cells. Two clinical AML samples were taken to establish nonobesediabetic/severe combined immunodeficiency (NOD/SCID) patient-derived xenograft model. After injecting AML mononuclear cells from primary specimens by tail vein for 30 days, compound 12, salt form of 7l, was administrated orally with a dose of 100 mg/kg for 7 times every other day (). Meanwhile, the survival of human AML mice was calculated. From the result (), the lifespan of patient-derived xenograft mice which was administrated with 12 was improved compared to the control group which was administered with PBS in two different patient-derived xenograft models (). These results suggested that treatment of 12 improved the survival of patient-derived xenograft mice. Therefore, 12 might be considered as a potential promising drug candidate for the treatment of AML.
Figure 10. Compound 12 prolonged the lifespan of mice in two patient-derived xenograft mice models. (a) The flowchart of establishing patient-derived xenograft mice model. (b) The lifespan of the first AML patient-derived xenograft mice model which was orally administrated with 12 with a dose of 100 mg/kg was extended compared to the control group and ADR group. (c) The lifespan of the second AML patient-derived xenograft mice model which was administrated with 12 was improved compared to the control group.
Compound 7l induced apoptosis of leukaemia stem and progenitor cells through MAPK signal pathway
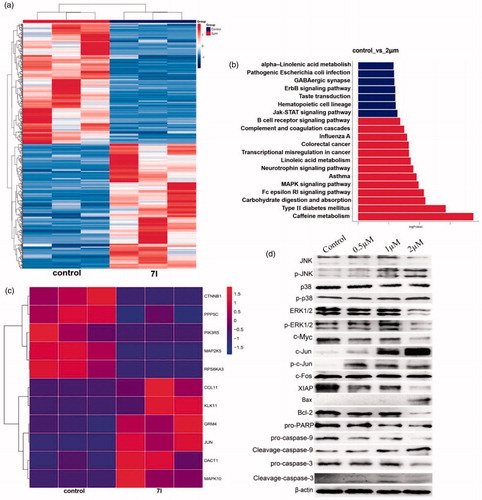
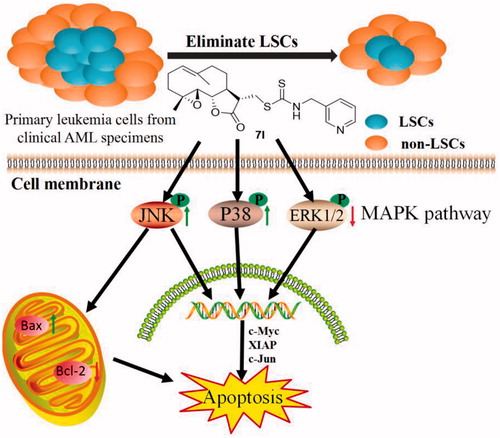
To investigate the mechanism of 7l, microarray gene expression profiling was performed. From the results of microarray gene expression profiling, MAPK signal pathway in KG1a cells was clearly changed after treatment of 7l for 24 h (). ERK1/2, p38 and JNK played significant roles in MAPK signal pathway which was very important for CSC survival. Activation of ERK1/2 activity contributes to inhibition of apoptosis and rising activities of p38 and JNK promotes apoptosis. From the results of western blot assay in KG1a cells, 7l activated p38, JNK by phosphorylation and inhibited ERK1/2. Furthermore, the level of apoptosis-related protein Bax and c-Jun were clearly up-regulated. Meanwhile, the level of anti-apoptosis protein c-Myc, XIAP, and Bcl-2 were significantly down-regulated. The cleavage of proteins caspase-3, caspase-9, and PARP that are associated with activating apoptosis was increased significantly after the treatment of 7l. These data prompted us to propose that 7l might induce apoptosis of leukaemia stem and progenitor cells through MAPK signal pathway.
Figure 11. Preliminary mechanism study of 7l. (a) Heat map analysis of microarray data between control group and 7l-treated group in KG1a cells. (b) The KEGG enrichment analysis of microarray data between control group and 7l-treated group in KG1a cells. (c) Heat map analysis of microarray data of MAPK pathway after treatment of 7l at a concentration of 2 µM. (d) Western blot analysis of MAPK pathway related proteins and apoptosis mediated proteins after exposing to different concentrations of compound 7l for 24 h in KG1a cells.
Conclusion
In summary, a series of dithiocarbamate esters of PTL was synthesised and evaluated for their anti-AML activity, which led to discovery of the most potent compound 7l. Compound 7l exhibited enhanced activities against KG1a and HL-60 with IC50 values of 0.7 and 1.7 μM, respectively, and the activities against KG1a and HL-60 were 8.7- and 2.2-folds comparing to those of PTL, respectively. It is worth noting that compound 7l was more active against AML progenitor cell line KG1a comparing to sensitive cell line HL-60 (). Importantly, 7l could induce apoptosis of total primary leukaemia cells and CD34+CD38– primary LSCs from AML patients with a dose-dependent manner () while sparing normal cells from healthy donors ( and ). Compound 7l significantly suppressed the colony formation of primary human leukaemia cells in dose-dependent manner (). Moreover, compound 12, salt form of 7l, showed no observable toxicity with a dose of 500 mg/kg by oral ().
These encouraging in vitro results and low acute toxicity encouraged us to further evaluate its anti-AML efficacy in vivo. The lifespan of patient-derived xenograft mice in 12-group was improved compared to the control PBS-group in two NOD/SCID patient-derived xenograft models ().
Microarray assay indicated that 7l might mediate MAPK pathway. Western blot analysis showed that 7l activated p38, JNK by phosphorylation and inhibited ERK1/2. After treatment of 7l, western blot assay demonstrated up-regulation of apoptosis-related proteins (Bax and c-Jun), down-regulation of anti-apoptosis proteins (c-Myc, XIAP, and Bcl-2), and increase in the cleaved caspase-3, caspase-9, and PARP proteins associated with activation of apoptosis (). These data showed that molecular mechanism of 7l-mediated apoptosis is associated with MAPK signal pathway ().
Figure 12 Compound 7l induced apoptosis of leukaemia stem and progenitor cells through MAPK signal pathway.
On the basis of these studies, we propose that 12 might be considered as a promising drug candidate deserving to be further developed for ultimate discovery of anti-LSCs drug.
Supplemental Material
Download PDF (1.3 MB)Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
Related Research Data
References
- Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 2005;105:4163–9.
- Siveen KS, Uddin S, Mohammad RM. Targeting acute myeloid leukemia stem cell signaling by natural products. Mol Cancer 2017;16:13.
- Sarkozy C, Gardin C, Gachard N, et al. Outcome of older patients with acute myeloid leukemia in first relapse. Am J Hematol 2013;88:758–64.
- Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645–8.
- Sarry J-E, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2R gamma c-deficient mice. J Clin Invest 2011;121:384–95.
- Stiehl T, Baran N, Ho AD, Marciniak-Czochra A. Cell division patterns in acute myeloid leukemia stem-like cells determine clinical course: a model to predict patient survival. Cancer Res 2015;75:940–9.
- Ho T-C, LaMere M, Stevens BM, et al. Evolution of acute myelogenous leukemia stem cell properties following treatment and progression. Blood 2016;128:1671–8.
- Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017;547:104–8.
- Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017;129:1577–85.
- Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 2011;17:1086–91.
- Chan WI, Huntly BJ. Leukemia stem cells in acute myeloid leukemia. Semin Oncol 2008;35:326–35.
- Jin L, Hope KJ, Zhai Q, et al. Targeting of CD44 eradicates human acute myeloid leukemia stem cells. Nat Med 2006;12:1167–74.
- Guzman ML, Rossi RM, Neelakantan S, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 2007;110:4427–35.
- Zhou J, Zhang H, Gu P, et al. NF-kappa B pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res Treat 2008;111:419–27.
- Huynh DT, Iannotti MJ, Gunn EJ, et al. Parthenolide and structurally related natural products as anti-cancer stem cell agents: a new era in treatment of multiple myeloma. Cancer Res 2010;70:4292.
- Kawasaki BT, Hurt EM, Kalathur M, et al. Effects of the sesquiterpene lactone parthenolide on prostate tumor-initiating cells: an integrated molecular profiling approach. Prostate 2009;69:827–37.
- Ghantous A, Sinjab A, Herceg Z, Darwiche N. Parthenolide: from plant shoots to cancer roots. Drug Discov Today 2013;18:894–905.
- Dell'Agli M, Galli GV, Bosisio E, D'Ambrosio M. Inhibition of NF-kB and metalloproteinase-9 expression and secretion by parthenolide derivatives. Bioorg Med Chem Lett 2009;19:1858–60.
- Gopal YV, Arora TS, Van Dyke MW. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem Biol 2007;14:813–23.
- Kim YJ, Choi M-H, Hong S-T, Bae YM. Resistance of cholangiocarcinoma cells to parthenolide-induced apoptosis by the excretory-secretory products of Clonorchis sinensis. Parasitol Res 2009;104:1011–6.
- Riganti C, Doublier S, Viarisio D, et al. Artemisinin induces doxorubicin resistance in human colon cancer cells via calcium-dependent activation of HIF-1 alpha and P-glycoprotein overexpression. Br J Pharmacol 2009;156:1054–66.
- Hassane DC, Guzman ML, Corbett C, et al. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood 2008;111:5654–62.
- Kim YR, Eom JI, Kim SJ, et al. Myeloperoxidase expression as a potential determinant of parthenolide-induced apoptosis in leukemia bulk and leukemia stem cells. J Pharmacol Exp Ther 2010;335:389–400.
- Nasim S, Crooks PA. Antileukemic activity of aminoparthenolide analogs. Bioorg Med Chem Lett 2008;18:3870–3.
- Nakshatri H, Appaiah HN, Anjanappa M, et al. NF-kappa B-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT. Cell Death Dis 2015;6:e1608.
- Neelakantan S, Nasim S, Guzman ML, et al. Aminoparthenolides as novel anti-leukemic agents: discovery of the NF-kappaB inhibitor, DMAPT (LC-1). Bioorg Med Chem Lett 2009;19:4346–9.
- Peese K. New agents for the treatment of leukemia: discovery of DMAPT (LC-1). Drug Discov Today 2010;15:322.
- Lickliter J. A phase 1 dose-escalation study to evaluate the safety, tolerability and pharmacokinetics of ACT001 in patients with advanced solid tumors. Availabel from: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12616000228482p.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun 2012;48:1868–70.
- Imamura H, Ohtake N, Jona H, et al. Dicationic dithiocarbamate carbapenems with anti-MRSA activity. Bioorg Med Chem 2001;9:1571–8.
- Len C, Boulogne-Merlot A-S, Postel D, et al. Synthesis and antifungal activity of novel bis(dithiocarbamate) derivatives of glycerol. J Agric Food Chem 1996;44:2856–8.
- Dehmel F, Weinbrenner S, Julius H, et al. Trithiocarbonates as a novel class of HDAC inhibitors: SAR studies, isoenzyme selectivity, and pharmacological profiles. J Med Chem 2008;51:3985–4001.
- Ding PP, Gao M, Mao BB, et al. Synthesis and biological evaluation of quinazolin-4(3H)-one derivatives bearing dithiocarbamate side chain at C2-position as potential antitumor agents. Eur J Med Chem 2016;108:364–73.
- Li B, Zhou S, Wang S, et al. Efficient synthesis of organic sulfonic acid derivatives containing dithiocarbamate side chains. Tetrahedron 2016;72:3885–9.
- Li R-D, Wang H-L, Li Y-B, et al. Discovery and optimization of novel dual dithiocarbamates as potent anticancer agents. Eur J Med Chem 2015;93:381–91.
- Qian Y, Ma G-Y, Yang Y, et al. Synthesis, molecular modeling and biological evaluation of dithiocarbamates as novel antitubulin agents. Bioorg Med Chem 2010;18:4310–6.
- Zahran MAH, Salem TAR, Samaka RM, et al. Design, synthesis and antitumor evaluation of novel thalidomide dithiocarbamate and dithioate analogs against Ehrlich ascites carcinoma-induced solid tumor in Swiss albino mice. Bioorg Med Chem 2008;16:9708–18.
- Zhang Y, Liu B, Wu X, et al. New pyridin-3-ylmethyl carbamodithioic esters activate pyruvate kinase M2 and potential anticancer lead compounds. Bioorg Med Chem 2015;23:4815–23.
- Johansson B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl 1992;369:15–26.
- Guo X, Xu B, Pandey S, et al. Disulfiram/copper complex inhibiting NF kappa B activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett 2010;290:104–13.
- Jivan R, Damelin LH, Birkhead M, et al. Disulfiram/copper-disulfiram damages multiple protein degradation and turnover pathways and cytotoxicity is enhanced by metformin in oesophageal squamous cell carcinoma cell lines. J Cell Biochem 2015;116:2334–43.
- Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res 2006;66:10425–33.
- Dastjerdi MN, Babazadeh Z, Salehi M, et al. Comparison of the anti-cancer effect of disulfiram and 5-Aza-CdR on pancreatic cancer cell line PANC-1. Adv Biomed Res 2014;3:156.
- Lin J, Haffner MC, Zhang Y, et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 2011;71:333–43.
- Liu P, Kumar IS, Brown S, et al. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br J Cancer 2013;109:1876–85.
- Liu P, Brown S, Goktug T, et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer 2012;107:1488–97.
- Yip NC, Fombon IS, Liu P, et al. Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer 2011;104:1564–74.
- Clinical trials. Gov identifier NCT 01777919.
- Shehzad A, Lee J, Lee YS. Autocrine prostaglandin E2 signaling promotes promonocytic leukemia cell survival via COX-2 expression and MAPK pathway. BMB Rep 2015;48:109–14.
- Konig H, Copland M, Chu S, et al. Effects of dasatinib on Src kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res 2008;68:9624–33.
- Balko JM, Schwarz LJ, Bhola NE, et al. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res 2013;73:6346–58.
- Piao LS, Hur W, Kim T-K, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett 2012;315:129–37.
- Mulholland DJ, Kobayashi N, Ruscetti M, et al. Pten Loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res 2012;72:1878–89.
- Wang YK, Zhu YL, Qiu FM, et al. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+primary colon cancer cells. Carcinogenesis 2010;31:1376–80.
- Karsy M, Albert L, Tobias ME, et al. All-trans retinoic acid modulates cancer stem cells of glioblastoma multiforme in an MAPK-dependent manner. Anticancer Res 2010;30:4915–20.
- Kumar D, Kumar S, Gorain M, et al. Notch1-MAPK signaling axis regulates CD133+ cancer stem cell-mediated melanoma growth and angiogenesis. J Invest Dermatol 2016;136:2462–74.
- Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011;19:387–400.
- Bharti R, Dey G, Mandal M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: a snapshot of IL-6 mediated involvement. Cancer Lett 2016;375:51–61.
- Dreesen O, Brivanlou AH. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev 2007;3:7–17.
- Long J, Ding Y-H, Wang P-P, et al. Protection-group-free semisyntheses of parthenolide and its cyclopropyl analogue. J Org Chem 2013;78:10512–8.
- Long J, Zhang S-F, Wang P-P, et al. Total syntheses of parthenolide and its analogues with macrocyclic stereocontrol. J Med Chem 2014;57:7098–112.
- Yang Z, Kuang B, Kang N, et al. Synthesis and anti-acute myeloid leukemia activity of C-14 modified parthenolide derivatives. Eur J Med Chem 2017;127:296–304.
- Yang Z-J, Ge W-Z, Li Q-Y, et al. Syntheses and biological evaluation of costunolide, parthenolide, and their fluorinated analogues. J Med Chem 2015;58:7007–20.
- Zhang Q, Lu Y, Ding Y, et al. Guaianolide sesquiterpene lactones, a source to discover agents that selectively inhibit acute myelogenous leukemia stem and progenitor cells. J Med Chem 2012;55:8757–69.
- Fuchs D, Daniel V, Sadeghi M, et al. Salinomycin overcomes ABC transporter-mediated multidrug and apoptosis resistance in human leukemia stem cell-like KG-1a cells. Biochem Biophys Res Commun 2010;394:1098–104.
- Liu Y, Chen F, Wang S, et al. Low-dose triptolide in combination with idarubicin induces apoptosis in AML leukemic stem-like KG1a cell line by modulation of the intrinsic and extrinsic factors. Cell Death Dis 2013;4:e948
- Weng G, Zeng Y, Huang J, et al. Curcumin enhanced busulfan-induced apoptosis through downregulating the expression of survivin in leukemia stem-like KG1a cells. Biomed Res Int 2015;2015:1.