
Abstract
A series of eighteen pyrrolo[3,2-c]pyridine derivatives were tested for inhibitory effect against FMS kinase. Compounds 1e and 1r were the most potent among all the other tested analogues (IC50 = 60 nM and 30 nM, respectively). They were 1.6 and 3.2 times, respectively, more potent than our lead compound, KIST101029 (IC50 = 96 nM). Compound 1r was tested over a panel of 40 kinases including FMS, and exerted selectivity against FMS kinase. It was further tested against bone marrow-derived macrophages (BMDM) and its IC50 was 84 nM (2.32-fold more potent than KIST101029 (IC50 = 195 nM)). Compound 1r was also tested for antiproliferative activity against a panel of six ovarian, two prostate, and five breast cancer cell lines, and its IC50 values ranged from 0.15–1.78 µM. It possesses also the merit of selectivity towards cancer cells than normal fibroblasts.
Introduction
Colony-stimulating factor-1 receptor (CSF-1R), or FMS, kinase is a member of type III receptor tyrosine kinases family. It interacts with CSF-1 or Il-34, and the produced signal transduction results in proliferation and survival of the monocyte/macrophage lineage cellsCitation1,Citation2. FMS kinase is over-expressed in different cancer types (e.g. ovarianCitation3, prostateCitation4, and breastCitation5–7) as well as inflammatory disorders (e.g. rheumatoid arthritis)Citation8. So FMS inhibitors could be useful drug candidates for treatment of these disorders.
Several FMS kinase inhibitors of different chemical classes have been recently reviewed by our groupCitation9. Among them, diarylamide derivatives have been reportedCitation10–15. We have previously reported a diarylamide derivative (KIST101029, ) as a potent and selective FMS kinase inhibitorCitation16. It exerted also high potency against ovarian, prostate, and breast cancer cell linesCitation17.
Figure 1. Structure of the lead compound, KIST101029.
In the present study, we tested a series of diarylamides and diarylureas possessing pyrrolo[3,2-c]pyridine scaffold against FMS kinase. Many of them were previously reported as antiproliferative agentsCitation18–20. The most potent compound against FMS kinase was tested against a panel of 40 kinases including FMS, tested against a panel of 13 cancer cell lines of 3 different cancer types and HS 27 fibroblasts, and against bone marrow-derived macrophages (BMDM) in order to investigate its biological effects. The results were compared with those of the lead compound KIST101029. The results, structure-activity relationship (SAR) studies, and relevant discussions are presented in details.
Experimental
Synthesis of the target molecules 1a–q
Compounds 1f, 1m–o, and 1q are new, while the other 12 compounds are previously reportedCitation18–20. In addition, compound 1r is also new. The synthetic procedure utilised for its synthesis as well as its spectral data are reported in the next section. The synthetic procedures, purification methods, and the spectral analysis data for compounds 1a–q have been provided in the supplementary file.
Synthesis of compound 1r
A mixture of compound 8b (24 mg, 0.1 mmol), 4-morpholino-3-(trifluoromethyl)benzoic acid (57 mg, 0.2 mmol), HOBt (31 mg, 0.22 mmol), and EDCI (50 mg, 0.26 mmol) in dry DMF (2.0 ml) was cooled to 0 °C in inert atmosphere. TEA (0.04 ml, 0.02 mmol) was added thereto at the same temperature. The mixture was then stirred at 80 °C for 12 h. The reaction mixture was cooled and then partitioned between water (10 ml) and ethyl acetate (15 ml), and the organic layer was separated. The aqueous layer was then extracted with ethyl acetate (2 × 10 ml) and the combined organic phase was washed with brine and dried over anhydrous sodium sulphate. The organic solvent was evaporated, and the residue was purified by column chromatography (silica gel, hexane-ethyl acetate 4:1 v/v followed by 1:1 v/v) to get the pure compound. Yield 7.5%; 1H NMR (400 MHz, CDCl3): δ 8.56 (s, 1H), 8.39 (s, 1H), 8.17–8.07 (m, 2H), 7.71–7.53 (m, 2H), 7.39 (brd, 2H, J = 8.6 Hz), 7.34 (t, 1H, J = 3.2 Hz), 7.30 (brd, 1H, J = 8.0 Hz), 7.13 (brs, 2H), 6.83 (brd, 1H, J = 7.7 Hz), 6.74 (dd, 1H, J = 1.9 Hz and 1.5 Hz), 3.86 (t, 4H, J = 2.6 Hz), 3.02 (t, 4H, J = 2.1 Hz); LC-MS: 482.1 (M+ +1), 481.1 (M+); elemental analysis: Calculated: C: 62.36%, H: 4.61%, N: 14.55%, Found: C: 62.23%, H: 4.50%, N: 14.68%.
Bioactivity protocol
In vitro kinase screening
Reaction Biology Corp. Kinase HotSpotSM service [http://www.reactionbiology.com] was utilised for screening of the target compounds as per the protocol reported in the literatureCitation21. In order to calculate the IC50 values and the inhibition percentages at different concentrations, each compound was tested in 10-dose duplicate assay mode starting with 81 µM and threefold serial dilution.
Antiproliferative screening against cancer cell lines
Compounds 1r and KIST101029 were tested at the National Cancer Institute (NCI, Bethesda, MD) following their standard protocol [https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm].
Antiproliferative screening against HS 27 fibroblasts
It was conducted following the previously reported protocolCitation17.
BMDM assay
The macrophages were isolated from C57BL6 murine bone marrow using Miltenyi Biotec MACS beads (CD11b). The macrophages (30,000 cells/well) were cultured in RPMI 1640 medium containing 10% foetal bovine serum, 2-mercaptoethanol (50 mM), and the test compound for 1 h at 37 °C in the presence of 5% CO2. CSF-1 (20 ng/mL) was then added and the system was incubated for 5 days. Each compound was tested in a 10-dose testing mode, threefold serial dilution starting with 1 µM concentration. The inhibitory effects of the tested compounds against CSF-1-induced BMDM growth were determined using Alamar BlueTM (Serotec).
Results and discussion
Chemistry
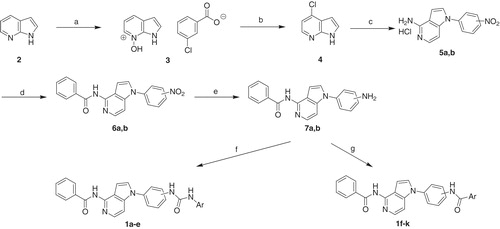
The 4-benzamidopyrrolo[3,2-c]pyridine derivatives 1a–k were synthesised as per the pathway illustrated in Scheme 1. Pyrrolo[2,3-b]pyridine (2) was treated with m-chloroperoxybenzoic acid to produce the m-chlorobenzoate salt 3. Heating compound 3 with phosphorus oxychloride yielded 4-chloropyrrolo[2,3-b]pyridine (4)Citation22. Fusion of compound 4 with the appropriate nitroaniline yielded 1-aryl-4-aminopyrrolo[3,2-c]pyridine HCl salts (5a,b) after ring rearrangementCitation23. The amines 5a,b were treated with benzoyl chloride in the presence of diisopropylamine to produce the corresponding benzamido analogues 6a,b. The nitro group of compounds 6a,b was reduced using hydrogen gas and Pd/C to yield the corresponding aniline derivatives 7a,b. In order to get the diarylurea products 1a–e, compounds 7a,b were reacted with appropriate aryl isocyanate at room temperature. On the other hand, heating the amines 7a,b with the appropriate benzoic acid derivative in the presence of EDCI, HOBt, and triethylamine led to condensation reaction and formation of the bisamide products 1f–kCitation18–20.
Scheme 1. Reagents and conditions: (a) 3-chloroperoxybenzoic acid, DME:heptane (1:2), rt, 2.5 h; (b) POCl3, 55 °C then rt then 85–90 °C, 18 h; (c) appropriate nitroaniline, 180 °C, 2–5 h; (d) benzoyl chloride, diisopropylamine, CH3CN, rt, 8 h; (e) Pd/C, H2, THF, rt, 2 h; (f) aryl isocyanate, THF, rt, 8 h; (g) benzoic acid derivative, HOBt, EDCI, TEA, DMF, 80 °C, 12 h.
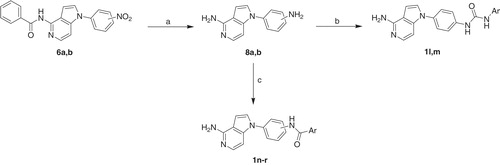
Reaction of compounds 6a,b using stannous chloride refluxing ethanol led to reduction of the nitro group and hydrolysis of the benzamido moiety to yield the diamine derivatives 8a,b. The aniline amino group is more reactive in the next reactions than the other amino attached to the heterocyclic nucleus due to higher electron density and higher nucleophilicity. Formation of the urea products 1l,m and the amide derivatives 1n–r (Scheme 2) was carried out by the same methods described for synthesis of compounds 1a–e and 1f–k, respectively (Scheme 1).
Scheme 2. Reagents and conditions: (a) SnCl2.H2O, EtOH, reflux, 4 h; (b) aryl isocyanate, THF, rt, overnight; (c) benzoic acid derivative, HOBt, EDCI, TEA, DMF, 80 °C, 12 h.
Biology
Kinase screening results and discussion
Compounds 1a–r were tested in a 10-dose testing mode starting with 81 µM, threefold serial dilutions, against FMS kinase in a cell-free enzyme assay. The IC50 values are summarised in . Upon comparing the structures and potencies, it was found that compounds 1c, 1e, and 1g possessing benzamido moiety at position 4 of the pyrrolopyridine nucleus were more potent than the corresponding primary amino analogues 1 l, 1m, and 1o. This can be due to occupancy of a hydrophobic pocket with that additional benzoyl moiety and/or formation of additional hydrogen bond by the carbonyl oxygen. On the contrary, the amino analogues 1p and 1r were more potent than the corresponding benzamido derivatives 1j and KIST101029.
Table 1. Structures of the target compounds and their IC50 values against FMS kinase. 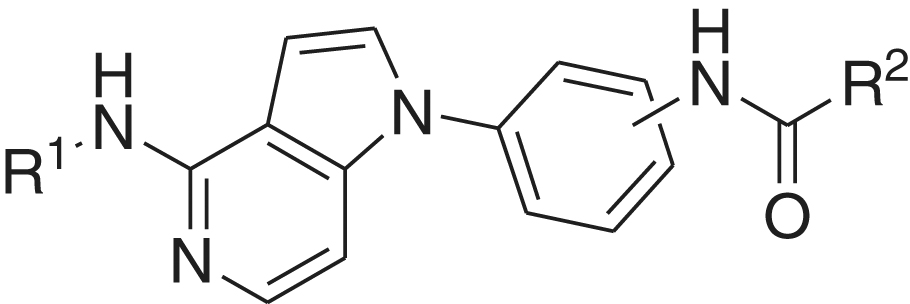
The central phenyl ring attached to N1 atom of the pyrrolopyridine nucleus was para-disubstituted in some derivatives or meta-disubstituted in others. The results showed that para-disubstituted compounds 1a, 1c, and 1h were more potent than the corresponding meta-disubstituted derivatives 1b, 1d, and 1i. On the other hand, the meta-disubstituted compounds 1g, 1o, and 1r showed stronger inhibitory effect against FMS kinase than the para-disubstituted positional isomers 1f, 1n, and 1p. These differences in the substitution position can affect proper orientation and fitting at the active site.
The urea derivatives 1a, 1b, 1e, and 1m were generally more potent than the corresponding analogues with amide linker 1h, 1i, 1k, and 1q. The urea linker is longer and can be optimal for best fitting at the binding site. The urea spacer contains also additional NH, compared with the amide. This terminal NH could contribute to stronger affinity by additional hydrogen bonding. Any or both of these effects can account for stronger enzyme inhibition effect of the urea derivatives.
Upon investigating the terminal aryl ring effect on the activity, it was found that 3′,5′-bis(trifluoromethyl)phenyl and 4′-morpholino-3′-(trifluoromethyl)phenyl are the best rings in diarylureas and diarylamides, respectively. These rings may contribute to stronger affinity with the enzyme. The morpholino moiety is also a polar ring, which enhances the solvent exposure and aqueous solubility of the molecule.
Among all the tested compounds, 1e and 1r were more potent than the lead compound KIST101029. Their potencies were 1.6 and 3.2 times, respectively, higher than that of KIST101029. Compound 1r possessing primary amino, meta-disubstituted central phenyl, amide linker, and 4′-morpholino-3′-(trifluoromethyl)phenyl terminal ring was the most potent among this series of compounds. So we decided to consider it for further biological investigations.
Compound 1r was tested against a panel of 40 kinases including FMS. The inhibition percentages at 1 µM concentration are summarised in . It showed selectivity against FMS kinase with 81% inhibition. Its inhibition percentages against FLT3 (D835Y) and c-MET kinases were 42% and 40%, respectively. So its IC50 values against both of them will be in micromolar scale. Compared with its IC50 values against FMS kinase (30 nM, ), it can be concluded that compound 1r is more than 33 times more selective towards FMS than the other kinases. The inhibition percentages exerted by compound 1r against the other 37 tested kinases were less than 28%.
Table 2. Inhibition percentages exerted by compounds 1r and KIST101029 over 40-kinase panel including FMS kinase.
Cell-based biological assays of compounds 1r and KIST101029
Due to over-expression of FMS kinase in certain types of tumours such as ovarianCitation3, prostateCitation4, and breastCitation5–7 cancers, we decided to test the antiproliferative activity of compound 1r against a panel of six ovarian, two prostate, and five breast cancer cell lines. It was also tested against HS 27 fibroblasts in order to investigate the selectivity indexes. The results were compared with those of KIST101029 as a reference standard. The IC50 values are presented in , and the dose-response curves of compound 1r are shown in .
Figure 2. Dose-response curves of compound 1r over the ovarian, prostate, and breast cancer subpanels.
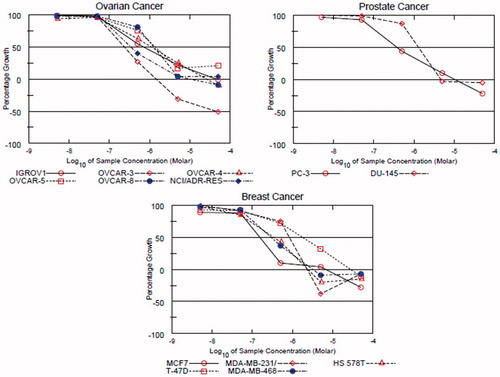
Table 3. IC50 and TGI values in μM of compound 1r and KIST101029 over ovarian, prostate, and breast cancer cell lines.
At 10 µM concentration, compound 1r was more active than KIST101029 against four ovarian, two prostate, and two breast cancer cell lines (Supplementary file). 1r was also more potent than KIST101029 against HS 578 T breast cancer cell line. TGI values reflect the efficacy of the molecule. Compound 1r was more efficacious than KIST101029 against MCF7, MDA-MB-231/ATCC, and HS 578 T breast cancer cell lines. But in general, KIST101029 was more potent than 1r against most of the tested cell lines. The structural difference, benzamido in KIST101029 compared with amino in 1r, is responsible for this potency difference. The benzamido moiety is more hydrophobic and is expected to allow greater ability of the molecule to cross the cell membrane to inside the cell. So the exposure of the intracellular components to compound KIST101029 is expected to be higher. Furthermore, the benzamido moiety may form additional interactions with the receptor site, which can contribute to stronger affinity and potency. However, the IC50 values of compound 1r were in the range of 0.15–1.78 µM, which is an excellent range of potency. In addition, compound 1r has another merit over KIST101029. Its IC50 value against HS 27 fibroblasts was higher, i.e. greater selectivity towards cancer cells than normal cells. Upon dividing its IC50 value against HS 27 fibroblasts by IC50 range over cancer cells, we find that the selectivity range of compound 1r ranges from 3.21 to 38.13 times towards cancer cells than normal cells.
BMDM assay
FMS over-expression contributes to inflammatory disorders as explained in the Introduction section. So compounds 1r and KIST101029 were tested for their ability to inhibit CSF-1-induced BMDM growth. The IC50 values are summarised in . The more potent FMS kinase inhibitor 1r exerted 2.32-fold superior potency than KIST101029 in this assay. So, the compound 1r can be a promising candidate for future development of anti-arthritic drugs.
Table 4. IC50 values of the tested compounds against bone marrow-derived macrophages (BMDM).
Each compound was tested in a 10-dose testing mode, threefold serial dilution starting with 1 µM concentration.
Conclusions
Our previous report on the potent and selective FMS kinase inhibitor, KIST101029, encouraged us to investigate the FMS kinase inhibitory effects of analogues of that lead compound. Eighteen compounds were tested, and this study led to discovery of compound 1r as a more potent and selective FMS kinase inhibitor (3.2 times more potent than KIST101029). It showed strong potency against the tested ovarian, prostate, and breast cancer cell lines with IC50 values ranged from 0.15 to 1.78 µM. It selectivity index towards cancer cells than normal fibroblasts ranged from 3.21 to 38.13. In addition, compound 1r showed potential anti-inflammatory effect against bone marrow-derived macrophages. This compound is a promising candidate for anticancer and anti-arthritic drug development. Further lead optimisation and biological investigations are required.
Supplemental Material
Download PDF (272.9 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Sherr CJ. The fms oncogene. Biochim Biophys Acta 1988;948:225–43.
- Woolford J, Rothwell V, Rohrschneider L. Characterization of the human c-fms gene product and its expression in cells of the monocyte-macrophage lineage. Mol Cell Biol 1985;5:3458–66.
- Kacinski BM, Carter D, Mittal K, et al. Ovarian adenocarcinomas express fms-complementary transcripts and fms antigen, often with coexpression of CSF-1. Am J Pathol 1990;137:135–47.
- Zhu P, Baek SH, Bourk EM, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 2006;124:615–29.
- Morandi A, Barbetti V, Riverso M, et al. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PLoS One 2011;6:e27450.
- Swierczak A, Cook A, Lenzo J, et al. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol Res 2014;2:765–76.
- Sullivan AR, Pixley FJ. CSF-1R signaling in health and disease: a focus on the mammary gland. J Mammary Gland Biol Neoplasia 2014;19:149–59.
- El-Gamal MI, Anbar HS, Yoo KH, et al. FMS kinase inhibitors: current status and future prospects. Med Res Rev 2013;33:599–636.
- El-Gamal MI, Al-Ameen SK, Al-Koumi DM, et al. Recent advances of colony-stimulating factor-1 receptor (CSF-1R) kinase and its inhibitors. J Med Chem 2018;61:5450–466.
- Genovese MC, Hsia E, Belkowski SM, et al. Results from a phase IIA parallel group study of JNJ-40346527, an oral CSF-1R inhibitor, in patients with active rheumatoid arthritis despite disease-modifying antirheumatic drug therapy. J Rheumatol 2015;42:1752–61.
- Li P, He K, Li J, et al. The role of Kupffer cells in hepatic diseases. Mol Immunol 2017;85:222–9.
- von Tresckow B, Morschhauser F, Ribrag V, et al. An open-label, multicenter, phase I/II study of JNJ-40346527, a CSF-1R inhibitor, in patients with relapsed or refractory Hodgkin lymphoma. Clin Cancer Res 2015;21:1843–50.
- Tresckow BV, Morschhauser F, Ribrag V, et al. A phase 1 study of JNJ-40346527, a colony stimulating factor-1 receptor (CSF-1R) inhibitor, in patients with relapsed or refractory Hodgkin lymphoma. Blood 2013;122:1812.
- Meegalla SK, Wall MJ, Chen J, et al. Structure-based optimization of a potent class of arylamide FMS inhibitors. Bioorg Med Chem Lett 2008;18:3632–7.
- Illig CR, Manthey CL, Meegalla SK, et al. Enhancement of kinase selectivity in a potent class of arylamide FMS inhibitors. Bioorg Med Chem Lett 2013;23:6363–9.
- El-Gamal MI, Jung M-H, Oh C-H. Discovery of a new potent bisamide FMS kinase inhibitor. Bioorg Med Chem Lett 2010;20:3216–8.
- El-Gamal MI, Abdel-Maksoud MS, Gamal El-Din MM, et al. Cell-based biological evaluation of a new bisamide FMS kinase inhibitor possessing pyrrolo[3,2-c]pyridine scaffold. Arch Pharm 2014;347:635–41.
- El-Gamal MI, Jung M-H, Lee WS, et al. Design, synthesis, and antiproliferative activity of new 1H-pyrrolo[3,2-c]pyridine derivatives against melanoma cell lines. Eur J Med Chem 2011;46:3218–26.
- Jung M-H, El-Gamal MI, Abdel-Maksoud MS, et al. Design, synthesis, and antiproliferative activity of new 1H-pyrrolo[3,2-c]pyridine derivatives against melanoma cell lines. Part 2. Bioorg Med Chem Lett 2012;22:4362–7.
- Khan MA, El-Gamal MI, Abdel-Maksoud MS, et al. Broad-spectrum antiproliferative activity of diarylureas and diarylamides possessing pyrrolo[3,2-c]pyridine scaffold. J Pharm Pharmacol 2014;2:157–69.
- Gamal El-Din MM, El-Gamal MI, Abdel-Maksoud MS, et al. Design, synthesis, broad-spectrum antiproliferative activity, and kinase inhibitory effect of triarylpyrazole derivatives possessing arylamides or arylureas moieties. Eur J Med Chem 2016;119:122–31.
- Wang X, Zhi B, Baum J, et al. A Practical Synthesis of 2-((1H-Pyrrolo[2,3-b]pyridine-4-yl)methylamino)-5-fluoronicotinic Acid. J Org Chem 2006;71:4021–3.
- Girgis NS, Larson SB, Robins RK, et al. The synthesis of 5‐azaindoles by substitution‐rearrangement of 7‐azaindoles upon treatment with certain primary amines. J Heterocycl Chem 1989;26:317–25.