
Abstract
In our endeavour towards the development of effective anticancer therapeutics, a novel series of isoxazole-piperazine hybrids were synthesized and evaluated for their cytotoxic activities against human liver (Huh7 and Mahlavu) and breast (MCF-7) cancer cell lines. Within series, compounds 5l-o showed the most potent cytotoxicity on all cell lines with IC50 values in the range of 0.3–3.7 μM. To explore the mechanistic aspects fundamental to the observed activity, further biological studies with 5m and 5o in liver cancer cells were carried out. We have demonstrated that 5m and 5o induce oxidative stress in PTEN adequate Huh7 and PTEN deficient Mahlavu human liver cancer cells leading to apoptosis and cell cycle arrest at different phases. Further analysis of the proteins involved in apoptosis and cell cycle revealed that 5m and 5o caused an inhibition of cell survival pathway through Akt hyperphosphorylation and apoptosis and cell cycle arrest through p53 protein activation.
Introduction
Cancer is one of the leading cause of deaths globally, and can be classified as a multifactorial disease, which is diligently orchestrated by a combination of genetic, epigenetic, and environmental factors working together towards the progression of tumoursCitation1,Citation2. Hepatocellular carcinoma (HCC) is the most common type of liver cancer accounting for about 75% of all primary liver cancers and is the 6th most frequent and the 2nd deadly cancer worldwideCitation3. Etiological factors, which are associated with HCC are chronic infection by hepatitis B virus (HBV) and hepatitis C virus (HCV), alcohol consumption, obesity, and aflatoxin exposureCitation4,Citation5. Primary liver cancer is extremely resistant to conventional chemotherapeutics, only 7% of patients have five-year survivalCitation6. Sorafenib and Regorafenib are the only FDA approved agents for advanced liver cancer cases, which extend patient survival approximately 3–7 monthsCitation7,Citation8. One of the biggest challenge in anticancer drug development is that HCC cells are reported to display high levels of cellular heterogeneityCitation9, which hampers significantly the development of new cancer therapeutics and causes failures in clinical studies since many candidate drugs do not produce clinical benefit in the overall populationCitation10. Consequently, an endless effort has continuously been devoted to the discovery and development of new and more effective anticancer agents that are capable to intervene with this complex disease.
Diverse azaheterocyclic ring systems have been in the centre of medicinal chemists, and used as versatile tools and building blocks for the synthesis of small molecule cancer therapeuticsCitation11. Among them, one of the most constantly growing area was the investigation of the antitumor properties of compounds containing isoxazole core structureCitation12. For example, a series of resorcinylic 4,5-diarylisoxazole amides have been developed as potent heat shock protein (HSP90) inhibitors, exemplified by NVP-AUY922 (Luminespib)Citation13, which was active against a variety of tumor xenografts and has been evaluated in phase II clinical trialsCitation14. Currently marketed immunosuppresory drug Leflunomide, which has recently been identified as a potential anticancer drugCitation15, is also an isoxazole derivative. Isoxazole derivatives as comberastatin A-4 analogues are also successfully described as tubulin polymerisation inhibitors with antiproliferative activities towards various cell linesCitation16,Citation17. Hewings and others reported the 3,5-dimethylisoxazole moiety as an effective acetylated lysine (KAc) mimic, which was used for developing bromodomain inhibitors with anticancer activityCitation18–21. In addition, a naturally occurring diarylisoxazole derivative has recently been reported as a new chemical tool with efficacy against AR-expressing breast cancer cellsCitation22.
Another widely occurring structural fragment in anticancer compounds is the piperazine moiety and a large number of compounds have appeared in the literature having piperazine motif with cytotoxicity against various cancer cellsCitation23–27. For instance, studies on an arylpiperazine derivative naftopidilCitation28,Citation29, a well-known α1-adrenergic receptor antagonist, and several other arylpiperazines have shown significant cytotoxicity against prostate cancer cellsCitation30–32. A series of piperazine derivatives have also been demonstrated to bear potent antiproliferative activities against various cancer cells including colon, prostate, breast, lung, and leukaemia as well as to suppress experimental tumours in small animal models by a mechanism involving inhibition of microtubule synthesis, inhibition of cell-cycle progression and angiogenesisCitation33,Citation34. Recently, incorporation of arylpiperazine moiety in purine nucleoside analogues resulted in compounds with senescence-induced cell death in liver cancer cellsCitation26. Other recent progress on anticancer piperazine derivatives can be found elsewhereCitation35.
In the course of our ongoing research interest concerning bioactive heterocyclesCitation36–40, we have relied on the aforementioned data for the design, synthesis, and biological evaluation of novel isoxazole derivatives containing in their structural framework an aryl piperazine residue. Since primary liver cancer incidence is expected to increase due to the obesity associated non-alcoholic fatty liver diseaseCitation4, we think that the synthesis of novel anticancer agents as reported herein will contribute not only the mechanistic bioactivity analysis of these isoxazole-piperazine hybrids, but also the future treatment options for HCC. In this context, we hereby report the straightforward synthesis of the novel isoxazole-piperazine hybrids, which were evaluated for their antitumor activities.
Experimental
Chemistry
Starting materials were purchased from commercial suppliers and used without further purification. 1H and 13C NMR spectra were recorded in CDCl3 or DMSO-d6 on a Varian Mercury 400 MHz spectrometer (Agilent Technologies, Santa Clara, CA, USA) using tetramethylsilane as the internal standard. All chemical shifts were recorded as δ (ppm), coupling constants are reported as Hertz. High resolution mass spectra data (HRMS) were collected using Waters LCT Premier XE Mass Spectrometer (high sensitivity orthogonal acceleration time-of-flight instrument) operating in ESI (+) or ESI (−) method, also coupled to an AQUITY Ultra Performance Liquid Chromatography system (Waters Corporation, Milford, MA, USA) using a UV detector monitoring at 254 nm. Purity for all final compounds were >95%, according to the UPLC-MS method using (A) water + 0.1% formic acid and (B) acetonitrile + 0.1% Formic Acid; flow rate = 0.3 mL/min, Column: Aquity BEH C18 column (2.1 × 100 mm, 1.7 mm; Waters Corporation, Milford, MA, USA). Flash chromatography on silica gel was performed on RediSep prepacked disposable silica gel columns using Teledyne Isco Combiflash. Preparative liquid chromatography was performed on Vydac Denali C18 Column (150 × 20 mm, 5 µ; Grace, Columbia, MD, USA) using Reveleris PREP purification system. All microwave irradiation experiments were carried out in a Biotage Initiator + microwave apparatus with Biotage sealed microvawe vials. Melting points of the synthesized compounds were determined by SMP50 automatic melting point apparatus and uncorrected. Experimental data for all intermediate compounds can be found in Supporting Information.
Synthesis of Compounds 5a-o
The mixture of the appropriate bromide derivative (4a-o) (0.5 mmol, 1 eq), 4-trifluoromethylbenzylpiperazine (0.6 mmol, 1.2 eq) and DIEA (1 mmol, 2 eq) in DMF (2 ml) was heated by microwave irradiation at 80 °C for 20 min. Then, it was poured into ice-water and formed precipitate was filtrated. The crude product was purified by flash chromatography.
5-Phenyl-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5a)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 87.0%; mp 112.6–113.9 °C. 1H NMR (CDCl3): δ 2.51 (4H, bs), 2.59 (4H, bs), 3.57 (2H, s), 3.66 (2H, s), 6.55 (1H, s), 7.42–7.48 (5H, m), 7.56 (2H, d, J = 8.0 Hz), 7.76–7.78 (2H, m). 13C NMR (CDCl3) δ 52.88, 52.98, 53.29, 62.26, 99.60, 124.20 (q, 1JC-F = 270.0 Hz), 125.15 (q, 3JC-F = 3.9 Hz), 125.77, 127.51, 128.92, 129.18, 129.44 (q, 2JC-F = 30.0 Hz), 130.08, 142.26, 161.68, 169.97. HRMS (m/z) [M + H]+ calcd for C22H23F3N3O: 402.1793, found, 402.1794.
5-(4-Fluorophenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5b)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 74.0%; mp 110.1–111.0 °C. 1H NMR (CDCl3): δ 2.60–2.72 (8H, m), 3.64 (2H, s), 3.72 (2H, s), 6.58 (1H, s), 7.12–7.18 (2H, m), 7.49–7.52 (2H, m), 7.58 (2H, d, J = 8.4 Hz), 7.74–7.79 (2H, m). 13C NMR (CDCl3) δ 53.06, 53.19, 53.46, 62.47, 99.60, 116.37 (d, 2JC-F = 21.8 Hz), 124.05 (d, 4JC-F = 3.2 Hz), 124.40 (q, 1JC-F = 270.5 Hz), 125.38 (q, 3JC-F = 3.8 Hz), 128.04 (d, 3JC-F = 8.4 Hz), 129.41, 142.46, 162.05, 163.90 (d, 1JC-F = 250.0 Hz), 169.20. HRMS (m/z) [M + H]+ calcd for C22H22F4N3O: 420.1699, found, 420.1686.
5-(4-Chlorophenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5c)
Purified by flash column chromatography (0% → 20% EA in DCM). Yield 68.1%; mp 126.2–126.9 °C. 1H NMR (CDCl3): δ 2.52 (4H, bs), 2.59 (4H, bs), 3.57 (2H, s), 3.66 (2H, s), 6.55 (1H, s), 7.41–7.45 (4H, m), 7.56 (2H, d, J = 7.6 Hz), 7.69 (2H, d, J = 8.4 Hz). 13C NMR (CDCl3) δ 52.80, 52.94, 53.21, 62.23, 99.95, 124.19 (q, 1JC-F = 270.6 Hz), 125.15 (q, 3JC-F = 3.8 Hz), 125.91, 127.03, 129.24, 129.28, 136.18, 141.95, 161.79, 168.86. HRMS (m/z) [M + H]+ calcd for C22H22ClF3N3O: 436.1404, found, 436.1402.
5-(p-Tolyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)-isoxazole (5d)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 84.0%; mp 141.0–142.3 °C. 1H NMR (CDCl3): δ 2.40 (3H. s), 2.61 (4H, bs), 2.69 (4H, bs), 3.63 (2H, s), 3.71 (2H, s), 6.57 (1H, s), 7.27 (2H, d, J = 8.0 Hz), 7.48 (2H, d, J = 7.6 Hz), 7.58 (2H, d, J = 7.6 Hz), 7.66 (2H, d, J = 8.0 Hz). 13C NMR (CDCl3) δ 21.66, 53.07, 53.17, 53.52, 62.48, 99.22, 124.41 (q, 1JC-F = 269.8 Hz), 124.98, 125.37 (q, 3JC-F = 3.8 Hz), 125.91, 129.41, 129.82, 140.60, 142.50, 161.81, 170.36. HRMS (m/z) [M + H]+ calcd for C23H25F3N3O: 416.1950, found, 416.1948.
3-((4-(4-(Trifluoromethyl)benzyl)piperazin-1-yl)methyl)-5-(4-(trifluoromethyl) phenyl)isoxazole (5e)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 44.0%; mp 110.1–111.5 °C. 1H NMR (CDCl3): δ 2.53 (4H, bs), 2.61 (4H, bs), 3.58 (2H, s), 3.68 (2H, s), 6.67 (1H, s), 7.45 (2H, d, J = 8.2 Hz), 7.56 (2H, d, J = 8.2 Hz), 7.72 (2H, d, J = 7.8 Hz), 7.88 (2H, d, J = 7.8 Hz). 13C NMR (CDCl3) δ 52.83, 52.99, 53.19, 62.23, 101.08, 123.71(q, 1JC-F = 270.6 Hz), 124.19 (q, 1JC-F = 270.5 Hz), 125.17 (q, 3JC-F = 3.9 Hz), 125.99 (q, 3JC-F = 3.8 Hz), 126.03, 129.19, 129.50 (q, 2JC-F = 31.0 Hz), 130.58, 131.84 (q, 2JC-F = 32.7 Hz), 142.18, 162.01, 168.33. HRMS (m/z) [M + H]+ calcd for C23H22F6N3O: 470.1667, found, 470.1667.
5-(4-Isopropylphenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl) isoxazole (5f)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 67.0%; mp 111.8–112.2 °C. 1H NMR (CDCl3): δ 1.27 (6H, d, J = 6.8 Hz), 2.51 (4H, bs), 2.58 (4H, bs), 2.93–2.96 (1H, m), 3.57 (2H, s), 3.65 (2H, s), 6.51 (1H, s), 7.31 (2H, d, J = 8.2 Hz), 7.44 (2H, d, J = 7.8 Hz), 7.56 (2H, d, J = 7.8 Hz), 7.69 (2H, d, J = 8.2 Hz). 13C NMR (CDCl3) δ 23.76, 34.08, 52.83, 52.94, 53.30, 62.25, 99.06, 124.21 (q, 1JC-F = 270.0 Hz), 125.10, 125.18 (q, 3JC-F = 3.8 Hz), 125.84, 127.04, 129.23, 129.40 (q, 2JC-F = 34.0 Hz), 142.15, 151.30, 161.53, 170.19. HRMS (m/z) [M + H]+ calcd for C25H29F3N3O: 444.2263, found, 444.2265.
5-(4-(Trifluoromethoxy)phenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5g)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 85.0%; mp 103.2–103.9 °C. 1H NMR (CDCl3): δ 2.59 (4H, bs), 2.67 (4H, bs), 3.63 (2H, s), 3.71 (2H, s), 6.62 (1H, s), 7.31 (2H, d, J = 8.4 Hz), 7.47 (2H, d, J = 8.0 Hz), 7.58 (2H, d, J = 8.0 Hz), 7.81 (2H, d, J = 8.4 Hz). 13C NMR (CDCl3) δ 53.05, 53.21, 53.44, 62.46, 100.30, 120.55 (q, 1JC-F = 257.0 Hz), 121.53, 124.40 (q, 1JC-F = 270.5 Hz), 125.39 (q, 3JC-F = 3.8 Hz), 126.28, 127.61, 129.41, 142.50, 150.50, 162.15, 168.76. HRMS (m/z) [M + H]+ calcd for C23H22F6N3O2: 486.1616, found, 486.1600.
5-(4-(Methylsulfonyl)phenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5h)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 84.0%; mp 163.5–164.1 °C. 1H NMR (CDCl3): δ 2.51 (4H, bs), 2.59 (4H, bs), 3.08 (3H, s), 3.57 (2H, s), 3.68 (2H, s), 6.73 (1H, s), 7.44 (2H, d, J = 7.8 Hz), 7.56 (2H, d, J = 7.8 Hz), 7.96 (2H, d, J = 8.8 Hz), 8.04 (2H, d, J = 8.8 Hz). 13C NMR (CDCl3) δ 44.43, 52.83, 53.00, 53.17, 62.24, 101.94, 124.19 (q, 1JC-F = 269.8 Hz), 125.20 (q, 3JC-F = 3.6 Hz), 126.52, 128.20, 129.23, 132.11, 141.55, 142.09, 162.21, 167.69. HRMS (m/z) [M + H]+ calcd for C23H25F3N3O3S: 480.1569, found, 480.1567.
4-(3-((4-(4-(Trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazol-5-yl)phenol (5i)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 71.6%; mp 154.2–156.0 °C. 1H NMR (CDCl3): δ 2.56 (4H, bs), 2.66 (4H, bs), 3.57 (2H, s), 3.67 (2H, s), 6.22 (1H, s), 6.70 (2H, d, J = 8.4 Hz), 7.42–7.47 (4H, m), 7.56 (2H, d, J = 8.4 Hz). 13C NMR (CDCl3): δ 52.83, 53.18, 62.47, 98.64, 116.31, 119.78, 124.20 (q, 1JC-F = 270.0 Hz), 125.46 (q, 3JC-F = 3.1 Hz), 127.65, 129.62, 142.10, 158.19, 160.44, 170.43. HRMS (m/z) [M + H]+ calcd for C22H23F3N3O2: 418.1742; found, 418.1736.
5-(4-Methoxyphenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5j)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 59.4%; mp 117.9–118.2 °C. 1H NMR (CDCl3): δ 2.49 (4H, bs), 2.57 (4H, bs), 3.55 (2H, s), 3.63 (2H, s), 3.85 (3H, s), 6.42 (1H, s), 6.96 (2H, d, J = 9.2 Hz), 7.43 (2H, d, J = 7.8 Hz), 7.55 (2H, d, J = 7.8 Hz), 7.70 (2H, d, J = 9.2 Hz). 13C NMR (CDCl3) δ 52.94, 53.05, 53.37, 55.37, 62.33, 98.24, 114.34, 120.35, 124.23 (q, 1JC-F = 270.0 Hz), 125.14 (q, 3JC-F = 3.8 Hz), 127.35, 129.17, 129.32 (q, 2JC-F = 32.0 Hz), 142.40, 161.03, 161.72, 169.90. HRMS (m/z) [M + H]+ calcd for C23H25F3N3O2: 432.1899, found, 432.1891.
5-(4-Propoxyphenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5k)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 79.0%; mp 103.0–103.5 °C. 1H NMR (CDCl3): δ 1.05 (3H, t, J = 7.4 Hz), 1.80–1.85 (2H, m), 2.51 (4H, bs), 2.58 (4H, bs), 3.56 (2H, s), 3.64 (2H, s), 3.96 (2H, t, J = 6.4 Hz), 6.42 (1H, s), 6.95 (2H, d, J = 8.8 Hz), 7.43 (2H, d, J = 7.8 Hz), 7.56 (2H, d, J = 7.8 Hz), 7.68 (2H, d, J = 8.8 Hz). 13C NMR (CDCl3) δ 10.46, 22.48, 52.82, 52.92, 53.30, 62.25, 69.64, 98.17, 114.85, 120.07, 124.21 (q, 1JC-F = 270.6 Hz), 125.18 (q, 3JC-F = 3.8 Hz), 127.33, 129.24, 129.40 (q, 2JC-F = 32.7 Hz), 142.10, 160.66, 161.48, 170.07. HRMS (m/z) [M + H]+ calcd for C25H29F3N3O2: 460.2212, found, 460.2213.
5-(4-(Allyloxy)phenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5l)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 78.5%; mp 107.1–107.4 °C. 1H NMR (CDCl3): δ 2.51 (4H, bs), 2.58 (4H, bs), 3.56 (2H, s), 3.64 (2H, s), 4.58 (2H, dt, J = 5.6 Hz, 1.6 Hz), 5.33 (1H, dq, J = 10.4, 1.4 Hz), 5.44 (1H, dq, J = 17.2, 1.6 Hz), 6.01-6.10 (1H, m), 6.43 (1H, s), 6.97 (2H, d, J = 9.0 Hz), 7.44 (2H, d, J = 7.8 Hz), 7.56 (2H, d, J = 7.8 Hz), 7.69 (2H, d, J = 9.0 Hz). 13C NMR (CDCl3) δ 52.66, 52.78, 53.20, 62.14, 68.86, 98.35, 115.12, 118.05, 120.38, 124.18 (q, 1JC-F = 270.6 Hz), 125.23 (q, 3JC-F = 3.8 Hz), 127.36, 129.32, 132.70, 142.02, 160.07, 161.25, 170.04. HRMS (m/z) [M + H]+ calcd for C25H27F3N3O2: 458.2055, found, 458.2061.
5-(4-((3-Methylbut-2-en-1-yl)oxy)phenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5m)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 66.8%; mp 100.0–100.3 °C. 1H NMR (CDCl3): δ 1.76 (3H, s), 1.81 (3H, s), 2.53 (4H, bs), 2.60 (4H, bs), 3.58 (2H, s), 3.66 (2H, s), 4.55 (2H, d, J = 6.8 Hz), 5.47–5.51 (1H, m), 6.44 (1H, s), 6.97 (2H, d, J = 8.8 Hz), 7.45 (2H, d, J = 8.0 Hz), 7.56 (2H, d, J = 8.0 Hz), 7.69 (2H, d, J = 8.8 Hz). 13C NMR (CDCl3) δ 18.23, 25.81, 52.86, 52.95, 53.31, 62.26, 64.93, 98.21, 115.05, 119.15, 120.18, 124.10 (q, 1JC-F = 270.6 Hz), 125.18 (q, 3JC-F = 3.2 Hz), 127.33, 129.23, 138.72, 160.38, 170.04. HRMS (m/z) [M + H]+ calcd for C27H31F3N3O2: 486.2368, found, 486.2362.
3,5-Dimethyl-4-((4-(3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazol-5-yl)phenoxy)methyl)isoxazole hydrochloride (5n)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 58.4%; mp 200.4–201.2 °C (decomp). 1H NMR (DMSO-d6): δ 2.23 (3H, s), 2.28 (3H, s), 2.43 (3H, s), 3.40 (8H, bs), 4.33 (2H, s), 4.47 (2H, s), 5.02 (2H, s), 7.09 (1H, s), 7.19 (2H, d, J = 8.8 Hz), 7.80–7.85 (4H, m), 7.90 (2H, d, J = 8.0 Hz). 13C NMR (DMSO-d6) δ 9.70, 10.65, 47.99, 48.01, 49.68, 57.38, 59.23, 100.13, 110.08, 115.70, 119.50, 124.00 (q, 1JC-F = 243.6 Hz), 125.59 (q, 3JC-F = 3.8 Hz), 127.36, 129.61, 130.08 (q, 2JC-F = 32.1 Hz), 132.25, 159.54, 159.92, 167.60, 169.81. HRMS (m/z) [M + H]+ calcd for C28H30ClF3N4O3: 527.2270, found, 527.2275.
5-(4-((1,3-Dimethyl-1H-pyrazol-4-yl)methoxy)phenyl)-3-((4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methyl)isoxazole (5o)
Purified by flash column chromatography (0% → 10% MeOH in DCM). Yield 58.0%; mp 131.4–131.9 °C. 1H NMR (CDCl3): δ 2.52 (3H, s), 2.51 (4H, bs), 2.58 (4H, bs), 3.57 (2H, s), 3.64 (2H, s), 3.84 (3H, s), 5.03 (2H, s), 6.11 (1H, s), 6.45 (1H, s), 7.02 (2H, d, J = 8.8 Hz), 7.44 (2H, d, J = 7.8 Hz), 7.56 (2H, d, J = 7.8 Hz), 7.71 (2H, d, J = 8.8 Hz). 13C NMR (CDCl3) δ 13.38, 36.46, 52.40, 52.56, 53.05, 60.62, 61.98, 98.71, 106.88, 115.18, 120.99, 124.13 (q, 1JC-F = 270.6 Hz), 125.31 (q, 3JC-F = 3.8 Hz), 127.50, 129.45, 137.26, 147.38, 159.50, 160.95, 169.91. HRMS (m/z) [M + H]+ calcd for C28H31F3N5O2: 526.2430, found, 526.2429.
Biology
Cell culture
Huh7 (epithelial-like) and Mahlavu (mesenchymal-like) human hepatocellular cancer cell lines and MCF7 human breast cancer carcinoma cells were grown in Dulbecco’s Modified Eagles Medium(DMEM) supplemented with %10 fetal bovine serum (Gibco, Invitrogen, Carlsbad, CA, USA), 1% non-essential amino acids (Gibco, Invitrogen) and 100 units/ml penicillin/streptomycin (Gibco, Invitrogen). Cells were maintained at 37 °C in a humidified incubator under 5% CO2.
NCI-60 sulforhodamine B assay
Huh7, MCF7 (2500 cell/well in 150 µl/well) and Mahlavu (1000 cell/well in 150 µl/well) cells were plated in 96-well plates and were grown in incubator for 24 hours. The compounds were dissolved in DMSO (Sigma, St Louis, MO, USA) as 20 mM stock solution. The compounds which were below 2.5 µM were tested in a concentration range of starting from 2.5 µM to 0.015 µM. Cells were fixed using 10% (v/v) trichloroacetic acid (Sigma ) for an hour after the end of 72 h incubation time. The fixed plates were dried and fixed cells were stained with sulforhodamine B (SRB) solution (Sigma) (50 µl of a 0.4% (m/v) of SRB in 1% acetic acid solution (Sigma)) for 10 min. In order to remove unbound SRB dye, cells were washed with 1% acetic acid three times and left for air-drying. The protein bound SRB dye was dissolved in 10 mM Tris-base (Sigma) and absorbance was measured with 96-well plate reader at 515 nm. The IC50 values were calculated and the cells treated with DMSO alone were used as control. All experiments were done in triplicate. Data with R2 values >0.9 was considered significant.
Real-time cell growth surveillance by cell electronic sensing (RT-CES)
Real-time cell growth analysis was performed using the xCELLigence System (Roche Applied Sciences, Penzberg, Germany). The Huh7, MCF7 (2500 cell/well) and Mahlavu (1000 cell/well) cells were seeded in E-Plates 96. In proliferation step, the cellular growth was analysed with cell index measurements in every 30 min for 24 h. After 24 h of incubation, when cells reached the log growth phase, they were treated with 5m and 5o starting from 10 µM and 1/2 folds’ serial dilutions three times. The cell index values (CI) were initially monitored every 10 min for 24 h and then CI were recorded in 30 min intervals. After 72 h of incubation, the cellular growth ratios were calculated by CIdrug/CIDMSO.
Oxidative stress assay
Mahlavu (35000 cells/well) and Huh7 (50000 cells/well) cells were inoculated into 6-wells plate for 24 h. Then cells were treated with 5o (1 µM for Huh7 and 4 µM for Mahlavu) and 5m (1 µM for Huh7 and Mahlavu). One group of cells did not receive the compounds, but they were grown in selenium-deficient serum-free medium as positive control for oxidative stressCitation41. Fourth group was treated with DMSO as negative control. At the end of 24 h, 48 h, and 72 h incubation period, the cells were collected and analysed by MUSE Oxidative Stress Kit (MCH100111, Merck Millipore, Burlington, MA, USA), which uses dihydroethium to monitor superoxide production in the cellsCitation42. Compound 5o impaired Mahlavu cells a lot, so 10,000 events were analysed for treated cells, 2000 events could be done in 5o treated Mahlavu cells. In parallel, Huh7 cells were plated into six-well plates for 24 h followed by treatment with 5o (1 µM) or 5m (1 µM) or DMSO or selenium deficient serum-free medium. After 24 h, 48 h, 72 h treatment periods, samples were washed three times with 1 × PBS, then they were incubated with dichloro-dihydro-fluorescein diacetate (DCFH-DA) solution (10 mM glucose, 0.5 µM DCFH-DA, 10 mM HEPES dissolved in 1 × PBS) in order to detect ROS (particularly H2O2) in the cells for 15 min in humidified chamber in dark at 37 °C. The solution was aspirated and cells were washed with PBS two times. The staining was analysed in situ with fluorescence microscope.
Flow cytometry for cell cycle analysis
Huh7 and Mahlavu cells were seeded onto 100 mm culture dishes. After 24 h, cells were treated with 5o (1 µM for Huh7 and 4 µM for Mahlavu) or 5m (1 µM for Huh7 and Mahlavu) or DMSO as a negative control. The end of 24 h, 48 h, and 72 h of incubation period, cells were fixed with ice-cold 70% ethanol for 3 h at −20 °C. Cell cycle analysis was carried out by PI (propidium iodide) staining using MUSE Cell Analyzer according to the manufacturer’s recommendations (Millipore).
Immunofluorescence staining
Huh7 (50,000 cells/well) and Mahlavu (35,000 cells/well) cells were inoculated on cover slides in 6-well plates after 24 h, cells were treated with 5o (1 µM for Huh7 and 4 µM for Mahlavu) or 5m (1 µM for Huh7 and Mahlavu) or DMSO control for 24 h, 48 h, and 72 h. After incubation time periods, the cells were washed three times with 1 × PBS and fixed with %100 ice-cold methanol. Then, the cells were stained with 1 µg/ml Hoechst (#33258, Sigma). Finally, the cells were analysed under a fluorescent microscope.
Western blot analysis
Cells were treated with the 5o (1 µM for Huh7 and 4 µM for Mahlavu), 5m (1 µM for Huh7 and Mahlavu) and with DMSO as control for 72 h. After 72 h incubation, the cells were collected with scraper, their total proteins were isolated and protein concentrations were calculated with Bradford assay. Bio-Rad protein electrophoresis (Mini-PROTEAN® TetraCellSystems and TGX™ precast gels, Bio-Rad, Hercules, CA, USA) and transfer system (Trans-Blot® TurboTransfer System, Bio-Rad, Hercules, CA, USA) were used according to the manufacturer’s protocol for all the Western blotting analyses. About 20–40 µg of protein were used per well. Proteins were transferred to a PVDF membrane. For immunoblotting, PARP (#9532S, Cell Signaling), p21/WAF1/Cip1 (#05-345, Millipore), p53 (#05-224, Millipore), phospho-p53Ser15 (#9286S, Cell Signaling), Rb (#9309, Cell Signaling), and phospho-RbSer807/811 (#9308S, Cell Signaling), α-phospho-AktSer473 (Cell Signaling, #9271), and AKT (#9272, Cell Signaling) antibodies were used in 1:100 to 1:500 5% BSA-TBS-T. β-actin (#A5441, Sigma) antibody was used in 1:1000 concentration for equal loading control. Proteins were visualized using a C-Digit® imaging system (Ll-COR)
Results and discussion
Chemistry
Compounds 5a–o was prepared following the reaction sequence illustrated in Schemes 1 and 2 using the known general methods. Hence, diethyloxalate has been treated with substituted acetophenones in the presence of a base to obtain β-ketoesters 1a–j. These intermediates (1a–j) were subsequently cyclized with hydroxylamine hydrochloride to provide isoxazole esters 2a–j. Reduction of 2a–j with LAH or NaBH4 followed by bromination with CBr4/PPh3 provided isoxazole methylbromides (4a–j). Finally, these intermediate alkyl bromides were treated with 4-trifluoromethylbenzylpiperazine to achieve target compounds 5a–j. For the synthesis of compounds 5k–o, alkylation of phenolic hydroxyl of the intermediate 3i with appropriate alkyl bromides was first accomplished, and then used to produce desired final compounds 5k–o following the reaction sequence shown in Scheme 2. All compounds were purified by automated flash chromatography and checked for purity by TLC and UPLC before being tested in biological assays (purity was 97% based on the peak area percentage of UPLC analysis). The structure of synthesized compounds was confirmed by means of 1H NMR, 13C NMR and high-resolution mass spectrometry (HRMS).
Scheme 1. Synthesis of compounds 5a-j. Reagents and conditions: i. NaOEt, EtOH ii. NH2OH.HCl, EtOH iii. LiAlH4, THF or NaBH4, THF/MeOH iv. CBr4/PPh3, DCM v. 4- (trifluoromethyl)benzylpiperazine, DIEA, DMF.
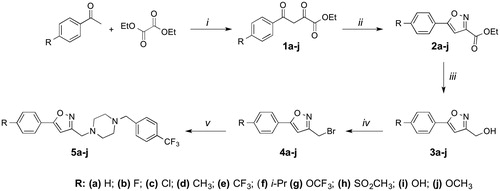
Scheme 2. Synthesis of compounds 5k-o. Reagents and conditions: i. R-Br, K2CO3 ii. CBr4/PPh3, DCM iii. 4- (trifluoromethyl)benzylpiperazine, DIEA, DMF.
Biological evaluation
Cytotoxicity of isoxazole-piperazine hybrids 5a–o in liver and breast cancer cells
The newly synthesized compounds (5a–o) were evaluated for their antitumor activities against human liver (Huh7 and Mahlavu), and breast (MCF7) carcinoma cell lines using the sulforhodamine B (SRB) assayCitation43. Data are expressed as IC50 values, defined as the half maximal inhibitory concentration, and are shown in .
Table 1. In vitro cytotoxic activities of 5a-o with 72 h of treatment.
Among the isoxazole analogues, compound having a non-substituted phenyl attachment at 5-position of the isoxazole nucleus (5a) showed the least potent cytotoxic activity for all the three cell lines (IC50 = 14.1 – 19.9 µM). However, para fluorine substitution improved the cytotoxic activity, and 5b appeared to be equally efficient in both liver cancer cells (IC50 = 5.3 µM for Huh7 and 6.2 µM for Mahlavu) but was less efficient in breast cancer cells (IC50 = 11.9 µM for MCF7), while the chlorine substituted derivative 5c was 4.3-fold less potent in Mahlavu as compared to 5b (). Compounds 5d–f having p-alkyl substituents showed only moderate cytotoxic activity against MCF7 breast cancer cells with IC50 values of 9.4–9.8 µM but proved to be less potent in liver cancer cells such as Huh7 (IC50 values of 19.9–24 µM) and Mahlavu (IC50 >40 µM). Introducing polarity to p-phenyl (5g–j) clearly improved the cytotoxic potency for both Huh7 (IC50 = 3.6 – 5.4 µM) and MCF7 cells (IC50 = 2.9 – 10.2 µM) along with a slight improvement in Mahlavu cells (IC50 = 7.5 – 26 µM). Next, O-alkylated analogues of compound 5i were evaluated for their anti-proliferative activity. As compared to the compounds 5g–j, cytotoxic potency significantly increased by allyl (5l), prenyl (5m), 3,5-dimethylisoxazol-4-ylmethyl (5n) and 1,3-dimethylpyrazol-5-ylmethyl (5o) substitutions with IC50 values between 0.3 and 3.7 µM, depending on the cell line (). Based on the promising cytotoxic activities, 5m and 5o were selected for further biological studies in order to understand the underlying mechanisms of their anticancer activities.
Real-time cellular response of cancer cells upon treatment with compounds 5m and 5o
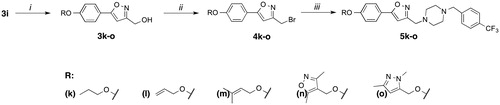
Time-dependent cytotoxic activities of 5m and 5o were scrutinized with real time cell electronic sensing (RT-CES)Citation44 by monitoring dynamic cell proliferation of Huh7, Mahlavu, and MCF7 cells. RT-CES assay revealed that 5m/5o significantly reduced the growth rate of cells as compared to DMSO control. This real-time growth pattern confirmed that 5m and 5o displayed time and dose-dependent growth inhibitory effects in all cells (). Cytotoxic effects of 5m and 5o on all three cell lines could be observed after 24 h of compound treatment, and reached to its highest values upon 72 h. As a result of cell cycle arrest or oxidative stress, this real-time growth pattern proposed growth inhibition in which the cells were neither proliferating nor dying, while the DMSO treated cells sustained to increase their number until they reached confluenceCitation45. In general, the RT-CES results were consistent with values obtained with the SRB assay. In light of this information, the molecular mechanisms underlying the cytotoxic activities of these isoxazole-piperazine hybrids were further investigated in detail with PTEN adequate epithelial like Huh7 cells and PTEN deficient mesenchymal like Mahlavu cellsCitation46.
Figure 1. RT-CES analysis of liver cancer cell lines (Huh7 and Mahlavu) and breast cancer cell line (MCF7) treated with compounds 5o (dark red, red and orange for 10, 5, and 2.5 µM, respectively) and 5m (dark blue, blue and turquoise for 10, 5 and 2.5 µM, respectively) with DMSO control (0.1%) at different concentrations (black, dark grey and grey for 10, 5 and 2.5 µM, respectively) for 120 h. The experiment was conducted in triplicate and was normalized to DMSO controls.
Oxidative stress induced by compounds 5m and 5o
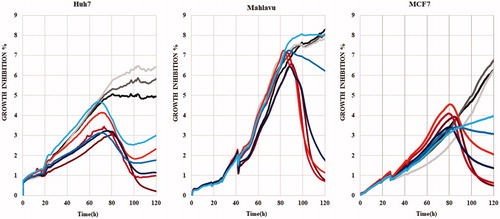
Reactive oxygen species (ROS), depending on their dose, can alter cellular pathways and promote cell cycle arrest and apoptosis in liver cancer cellsCitation41. Therefore, induction of oxidative stress by compounds 5m and 5o was analyzed in HCC cells (Mahlavu and Huh7) at different time periods (). Selenium-deficient serum free medium grown cells were used as experimental positive control for ROS generationCitation41. It is shown that while PTEN deficient Mahlavu cells can tolerate selenium deficient serum free medium, PTEN adequate Huh7 cells are strongly affectedCitation41. For visualization of in situ presence of oxidative stress, dichloro-dihydro fluorescein diacetate (DCFH-DA) assay was performed on these cells, which were treated with 5m/5o for 24 h, 48 h and 72 h (). In the presence of oxidative stress, DCFH-DA dye was oxidized to a green fluorescent molecule, DCF. Fluorescent microscopy images represented that oxidative stress was triggered by compounds 5m and 5o. While compounds 5m and 5o started to affect Mahlavu cells after 24 h, 5o and 5 m treated Huh7 cells displayed a raise in ROS (+) cells at 24 h (), which were in parallel to cell death as determined by RT-CES assay. We illustrated that 5o leads to an increase in ROS (+) cells with 40% and 13% for 48 h and 85% and 15% for 72 h in Mahlavu and Huh7 cells, respectively, when compared to DMSO controls (). In addition, compound 5m increased ROS (+) cells with 16% for 48 h and 25% for 72 h in Huh7, and it also caused a rise in ROS (+) cells with 32% in Mahlavu cells for 48 h ().
Figure 2. Oxidative stress induced by 5o and 5m liver cancer cells, which were treated with 5o (1 µM for Huh7 and 4 µM for Mahlavu) and 5m (1 µM for Huh7 and Mahlavu) or DMSO control for 24 h, 48 h, and 72 h. Selenium deficient serum-free medium was used as a positive control of ROS induction. A. DCFH-DA staining of the cells under oxidative stress with fluorescent microscope (20×) for 24 h, 48 h, and 72 h. (B) Cytometric analysis of oxidative stress induction. ROS positive cells are indicated with orange and ROS negative cells are shown in blue. (C) AKT and phospho-AKT in Mahlavu and Huh7 cells treated with 5o and 5m for 72h. Actin was used for equal loading.
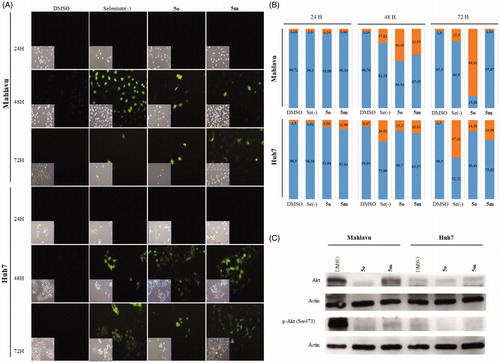
Metabolic stress induces cell death through ROS-induced apoptosis and Akt is one of the primary effectors in response to metabolic stressCitation47. Akt protein, which is hyperactivated in many tumours, plays a major role in both cell survival and resistance to tumor therapyCitation48. While Akt pathway is hyperactive in Mahlavu cells due to PTEN deletion, the pathway is normoactive in PTEN adequate Huh7 cellsCitation49. Therefore, the poorly differentiated Mahlavu cells are considered as aggressive HCC phenotype. In addition, Akt signaling was illustrated to be involved in the oxidative stress induced cellular pathwaysCitation50. Based on the findings that the compounds 5m and 5o caused ROS generation in HCC cells, Akt and phospho(p)-Akt protein levels comparatively analyzed in PTEN deficient Mahlavu and PTEN adequate Huh7 cells (). Significant decrease in the levels of Akt and p-Akt proteins was observed in 5m/5o treated Mahlavu cells as compared to DMSO control. However, compounds treated Huh7 cells did not result in a significant decrease in Akt protein levels. Although Mahlavu cells have hyperactive Akt signal pathway due to the deletion of PTEN gene, which leads to more drug resistant phenotype, more than 80% of Mahlavu cells were ROS positive upon 5o treatment (). Under normal conditions Mahlavu cells exhibit cryptic resistance to intrinsic oxidative stress-induced apoptosis due to selenium deficiency. However, treatment with 5m and 5o clearly induces oxidative stress along with cell death that the latter being more effective. Hence, we analyzed the nature of cell death induced with these compounds in liver cancer cells.
5m and 5o induce apoptosis
Compounds 5m and 5o induced apoptotic morphological changes as observed by nuclear staining with Hoechst in Huh7 and Mahlavu cells. Following treatment with 5m/5o for 24 h, 48 h, and 72 h, cells were analyzed under fluorescent microscopy. As shown in and S1, typical morphological changes such as chromatin condensation, nuclear fragmentation and apoptotic bodies were detected in treated cells for different time periods.
Figure 3. Characterization of cell death using fluorescent microscopy and Western blotting. (A) Hoechst staining of 5o and 5m treated Mahlavu and Huh7 cells with apoptotic nuclei at 24 h and 72 h. (B) PARP in Mahlavu and Huh7 cells treated with 5o and 5m for 72 h. Actin was used for equal loading. (C) The bar graphs representing relative band intensities of PARP and cleaved-PARP, which were normalized with their actin loading controls.
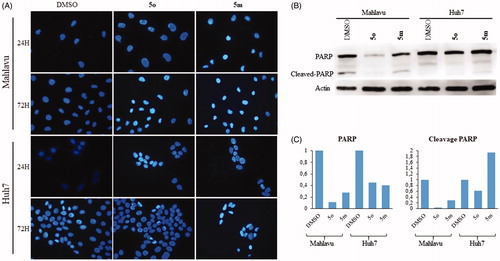
The activation of apoptotic pathways through 5m and 5o treatments were further confirmed with apoptosis-associated PARP protein levels. There was a significant decrease in total PARP protein in 5m/5o treated Mahlavu and Huh7 cells while an increase in PARP cleavage in the 5m treated Huh7 cells was identified at 72 h. These results supported the increased cytotoxic effects of the compounds on liver cancer cells (). Our data indicates that the 5m/5o-induced ROS leading to the cell death characterized with apoptosis. We then analyzed the proteins involved in apoptosis and cell cycle with the aim of further characterization of cell death mechanism.
Induction of cell cycle arrest and analysis of cellular pathways targeted by 5m and 5o
Initially, cell cycle arrest was analysed by flow cytometry analysis using propidium iodide (PI) staining of DNA. Huh7 cells treated with 5m and 5o showed an increase in entry to G2/M and G1 phases in 48 h () and 72 h (), respectively. While no arrest was observed in Mahlavu cells treated with 5m and 5o for 48 h (), the cells treated with 5m and 5o represented a higher cell population in S and G2/M phases for 72 h (). Quantitative results of cell cycle analysis were also revealed in Table S1.
Figure 4. Detection of cell cycle arrest. Cell cycle analysis of Mahlavu for (A) 48 h (B) 72 h and Huh7 for (C) 48 h and (D) 72 h after treatment with compounds 5o and 5m, and DMSO controls following 48 h and 72 h of treatment. Orange, blue, yellow and black show GO/S1, S, G2/M, and Sub-G1, respectively. (E) Rb, p53, phospho-Rb, and phospho-p53 in Mahlavu and Huh7 cells with 5o and 5m for 72h. Actin was used for equal loading.
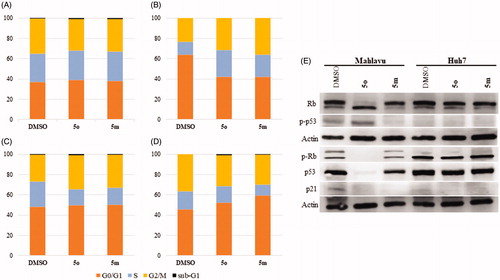
Based on the finding that 5m/5o caused ROS accumulation, cell cycle arrest and apoptosis, several targets involved in these pathways at the protein level were further investigated by Western blot analyses. Proteins p53, p21, Rb are inhibitory regulators of the cell cycle, and are effectors of responses to cellular stress leading to apoptosis and cell cycle arrest. Since 5m and 5o induce cellular stress-associated cell death, we examined the levels of these proteins upon compound treatment. Compound 5o treated Mahlavu cells exhibited significant alteration in protein levels of Rb, phosphorylated form of Rb (Ser807/811), p53 and phosphorylated form of p53 (Ser15) while p21 protein keeps its expression practically unchanged (). The phosphorylated from of p53 tumor-suppressor protein has critical roles for cell cycle arrest in response to DNA injury. It is a known fact that reactive oxygen species damage several complex biomolecues in the cell incuding DNA. Upon DNA damage, p53 is stabilized by phosphorylation and activates the expression of target genes. Primary among these is p21Citation51, which provides a direct link between p53 and pRb for cell cycle arrest. The p21 retains pRb in a hypophosphorylated state that inhibits E2F-1 activity and thereby S-phase entry. Our results with 5o treated Mahlavu cells clearly demonstrate that phosphorylation of p53 is associated with hypophoshorylation of pRb and arest in S phase.
Conclusion
We synthesized a series of isoxazole-piperazine hybrids and evaluated their cytotoxic activities against human cancer cell lines in comparison to DMSO control. The majority of derivatives showed moderate to significant cytotoxicity in the tested cell lines. As a general conclusion, we observed that the substitution pattern on the phenyl group linked to isoxazole at 5-position has a significant impact on the cytotoxicity and the selectivity of the compounds within the series. Therefore, compounds 5m and 5o, the most effective derivatives with respect to antiproliferative activity against hepatocellular cancer cells, were selected for detailed mechanistic studies. By further investigating their molecular effects, we showed that compounds 5m and 5o caused generation of ROS, induction of apoptotic cell death, and cell cycle arrest at different phases in HCC cells. Particularly, the bioactivities of 5o in poorly differentiated aggressive Mahlavu cells were prominent. Mahlavu cells were reported to be resistant to the ROS-induced cell death and drug resistant phenotypeCitation41. In this study, we clearly demonstated that 5o induces ROS and inhibits Akt cell survival pathway in Mahlavu cells. Decrease in levels of Akt and phosphorylated form of Akt (Ser473) upon treatment and the status of cell cycle proteins were worth exploring since it provided further information about mechanism of action of the compounds on cancer cells.
In conclusion, we were able to demostrate that our novel compounds induces chemically-induced extrinsic ROS, cell survival pathway inhibition through Akt hyperphosphorylation and apoptosis and cell cycle arrest through p53 protein activation. Future studies may evaluate the detailed cellular networks that are affected by the use of high throughput genomic screening methods such as transcriptome analysis with next generation sequencing in the presence of selected compounds. This may lead to identify molecular targets involved in induction of reactive oxygen species and cell cycle for eventual drug design and development against cancer. Therefore, we think that the synthesis of further derivatives as potent anticancer agents will be promising in terms of elucidating alternative mechanistic effects of isoxazole-piperazine hybrids as well as providing future treatment approaches for HCC associated with obesity non-alcoholic fatty liver disease.
Supplemental Material
Download PDF (871.9 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Wallace TA, Martin DN, Ambs S. Interactions among genes, tumor biology and the environment in cancer health disparities: examining the evidence on a national and global scale. Carcinogenesis 2011;32:1107–21.
- Stewart BW, Wild CP. World Cancer Report, Lyon, France: IARC Publications; ; 2014.
- Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol 2012;56:1384–91.
- Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol 2012;56:704–13.
- Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–86.
- Carr BI, Cavallini A, Lippolis C, et al. Fluoro-Sorafenib (Regorafenib) effects on hepatoma cells: growth inhibition, quiescence, and recovery. J Cell Physiol 2013;228:292–7.
- Kissel M, Berndt S, Fiebig L, et al. Antitumor effects of regorafenib and sorafenib in preclinical models of hepatocellular carcinoma. Oncotarget 2017;8:107096–108.
- Unsal H, Yakicier C, Marcais C, et al. Genetic heterogeneity of hepatocellular carcinoma. Proc Natl Acad Sci USA 1994;91:822–6.
- De Sousa EMF, Vermeulen L, Fessler E, et al. Cancer heterogeneity-a multifaceted view. EMBO Rep 2013;14:686–95.
- Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol 2012;6:155–76.
- Sysak A, Obminska-Mrukowicz B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur J Med Chem 2017;137:292–309.
- Eccles SA, Massey A, Raynaud FI, et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res 2008;68:2850–60.
- Felip E, Barlesi F, Besse B, et al. Brief report: phase 2 study of the HSP-90 inhibitor AUY922 in previously treated and molecularly defined patients with advanced non-small cell lung cancer. J Thorac Oncol 2018;13(4):576–84.
- Zhang C, Chu M. Leflunomide: a promising drug with good antitumor potential. Biochem Biophys Res Commun 2018;496(2):726–30.
- Kaffy J, Pontikis R, Carrez D, et al. Isoxazole-type derivatives related to combretastatin A-4, synthesis and biological evaluation. Bioorg Med Chem 2006;14:4067–77.
- Kamal A, Bharathi EV, Reddy JS, et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur J Med Chem 2011;46:691–703.
- Bamborough P, Diallo H, Goodacre JD, et al. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J Med Chem 2012;55:587–96.
- Chung CW, Dean AW, Woolven JM, et al. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J Med Chem 2012;55:576–86.
- Hewings DS, Fedorov O, Filippakopoulos P, et al. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. J Med Chem 2013;56:3217–27.
- Hewings DS, Wang M, Philpott M, et al. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J Med Chem 2011;54:6761–70.
- Robles AJ, McCowen S, Cai S, et al. Structure-activity relationships of new natural product-based diaryloxazoles with selective activity against androgen receptor-positive breast cancer cells. J Med Chem 2017;60:9275–89.
- Akkoc MK, Yuksel MY, Durmaz I, et al. Design, synthesis, and biological evaluation of indole-based 1,4-disubstituted piperazines as cytotoxic agents. Turk J Chem 2012;36:515–25.
- Mao ZW, Zheng X, Qi Y, et al. Synthesis and biological evaluation of novel hybrid compounds between chalcone and piperazine as potential antitumor agents. Rsc Adv 2016;6:7723–7.
- Saab AM, Dobmeier M, Koenig B, et al. Antiproliferative and erythroid differentiation of piperazine and triphenyl derivatives against k-562 human chronic myelogenous leukemia. Anticancer Res 2013;33:3027–32.
- Tuncbilek M, Guven EB, Onder T, et al. Synthesis of novel 6-(4-substituted piperazine-1-yl)-9-(beta-D-ribofuranosyl)purine derivatives, which lead to senescence-induced cell death in liver cancer cells. J Med Chem 2012;55:3058–65.
- Waszkielewicz AM, Gunia A, Szkaradek N, et al. Synthesis and evaluation of pharmacological properties of some new xanthone derivatives with piperazine moiety. Bioorg Med Chem Lett 2013;23:4419–23.
- Hori Y, Ishii K, Kanda H, et al. Naftopidil, a selective {alpha}1-adrenoceptor antagonist, suppresses human prostate tumor growth by altering interactions between tumor cells and stroma. Cancer Prev Res (Phila) 2011;4:87–96.
- Kanda H, Ishii K, Ogura Y, et al. Naftopidil, a selective alpha-1 adrenoceptor antagonist, inhibits growth of human prostate cancer cells by G1 cell cycle arrest. Int J Cancer 2008;122:444–51.
- Chen H, Liang X, Xu F, et al. Synthesis and cytotoxic activity evaluation of novel arylpiperazine derivatives on human prostate cancer cell lines. Molecules 2014;19:12048–64.
- Chen H, Xu BB, Sun T, et al. Synthesis and antitumor activity of novel arylpiperazine derivatives containing the saccharin moiety. Molecules 2017;22(11):1857.
- Chen H, Xu F, Liang X, et al. Design, synthesis and biological evaluation of novel arylpiperazine derivatives on human prostate cancer cell lines. Bioorg Med Chem Lett 2015;25:285–7.
- Cho EH, Chung SG, Lee SH, et al. Piperazine derivatives and process for the preparation thereof, Patent WO1998000402, A1;1998.
- Lee JH, Kang DW, Kwon HS, et al. Microtubule inhibitory effects of various SJ compounds on tissue culture cells. Arch Pharm Res 2004;27:436–41.
- Rathi AK, Syed R, Shin HS, et al. Piperazine derivatives for therapeutic use: a patent review (2010-present). Expert Opin Ther Pat 2016;26:777–97.
- Banoglu E, Caliskan B, Luderer S, et al. Identification of novel benzimidazole derivatives as inhibitors of leukotriene biosynthesis by virtual screening targeting 5-lipoxygenase-activating protein (FLAP). Bioorg Med Chem 2012;20:3728–41.
- Banoglu E, Celikoglu E, Volker S, et al. 4,5-Diarylisoxazol-3-carboxylic acids: A new class of leukotriene biosynthesis inhibitors potentially targeting 5-lipoxygenase-activating protein (FLAP). Eur J Med Chem 2016;113:1–10.
- Baytas SN, Inceler N, Yilmaz A, et al. Synthesis, biological evaluation and molecular docking studies of trans-indole-3-acrylamide derivatives, a new class of tubulin polymerization inhibitors. Bioorg Med Chem 2014;22:3096–104.
- Caliskan B, Yilmaz A, Evren I, et al. Synthesis and evaluation of analgesic, anti-inflammatory, and anticancer activities of new pyrazole-3(5)-carboxylic acid derivatives. Med Chem Res 2013;22:782–93.
- Cankara Pirol S, Caliskan B, Durmaz I, et al. Synthesis and preliminary mechanistic evaluation of 5-(p-tolyl)-1-(quinolin-2-yl)pyrazole-3-carboxylic acid amides with potent antiproliferative activity on human cancer cell lines. Eur J Med Chem 2014;87:140–9.
- Irmak MB, Ince G, Ozturk M, et al. Acquired tolerance of hepatocellular carcinoma cells to selenium deficiency: a selective survival mechanism? Cancer Res 2003;63:6707–15.
- Bindokas VP, Jordan J, Lee CC, et al. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci 1996;16:1324–36.
- Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–12.
- Solly K, Wang X, Xu X, et al. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev Technol 2004;2:363–72.
- System Xcelligence. Real-time and dynamic monitoring of cell proliferation and viability for adherent cells. Application Note 1. San Diego, CA: Acea Biosciences Inc.; 2013.
- Durmaz I, Guven EB, Ersahin T, et al. Liver cancer cells are sensitive to lanatoside C induced cell death independent of their PTEN status. Phytomedicine 2016;23:42–51.
- Zhao Y, Hu X, Liu Y, et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer 2017;16:79.
- Nogueira V, Park Y, Chen CC, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008;14:458–70.
- Buontempo F, Ersahin T, Missiroli S, et al. Inhibition of Akt signaling in hepatoma cells induces apoptotic cell death independent of Akt activation status. Invest New Drugs 2011;29:1303–13.
- Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J 2010;277:2–21.
- Sheahan S, Bellamy CO, Treanor L, et al. Additive effect of p53, p21 and Rb deletion in triple knockout primary hepatocytes. Oncogene 2004;23:1489–97.