
Abstract
To identify anticancer agents with high potency and low toxicity, a series of (Z)-styrylbenzene derivatives were synthesised and evaluated for anticancer activities using a panel of nine cancer cell lines and two noncancerous cell lines. Most derivatives exhibited significant anti-proliferative activities against five cancer cell lines, including MGC-803 and BEL-7402. (Z)-3-(p-Tolyl)-2-(3,4,5-trimethoxyphenyl)acrylonitrile (6h) showed a strong inhibitory effect on MGC-803 cells (IC50 < 0.01 µM) and exhibited stronger anti-proliferative activity than taxol (IC50 < 0.06 ± 0.01 µM). The IC50 value of 6h in L-02 cells was 10,000-fold higher than in MGC-803 cells. Compound 6h inhibited proliferation of BEL-7402 cells by arresting at the G2/M phase through up-regulation of cyclin B1 expression, down-regulation of cyclin A and D1 expression, and induction of apoptosis. In addition, 6h inhibited the migration of BEL-7402 cells and the formation of cell colonies.
Introduction
Cancer is becoming an increasingly important disease and is a leading cause of death worldwideCitation1. Although a variety of anticancer agents are currently available, none is able to eradicate cancer cells without having toxic effects on healthy tissuesCitation2. Therefore, finding new anticancer agents with higher potency and lower toxicity is a great challengeCitation3,Citation4. More than 60% of anticancer drugs used in the clinic originate from natural sourcesCitation5. Agents containing a styrylbenzene structure, such as resveratrol ((E)-3,5,4′-trihydroxystilbene)Citation6 and CA-4 ((Z)-2-methoxy-5–(3,4,5-trimethoxystyryl) phenol)Citation7, are naturally present in medicinal plants () and have been shown to exert anticancer effects with high toxicity to normal cellsCitation6–8.
Figure 1. The structures of resveratrol, compound 3, CA-4, colchicine, and the design of target compounds 6a-h, 7a-c, 13a-d, and 14a-b.
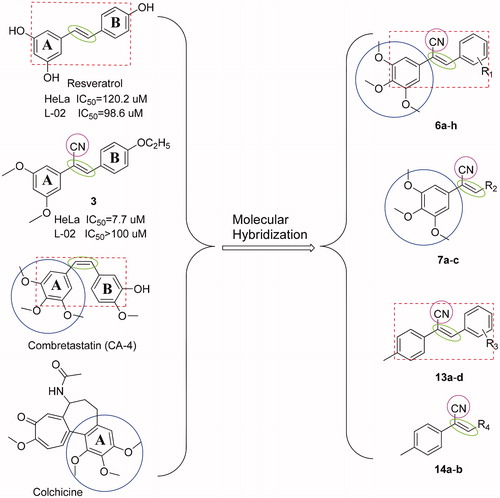
The microtubule system of eukaryotic cells is an important target for the development of anticancer agentsCitation9. Recently, much attention has been devoted to studying microtubule colchicine-binding sites of antimitotic agents, such as CA-4 and its analogsCitation10–13. Compounds with a structure similar to that of colchicine and having trimethoxy groups on the A benzene ring () may have potential as anticancer drugs. For example, it was reported that compounds with O-methylation on the A ring of the styrylbenzene structure displayed significant anticancer effectsCitation14.
Cyano is highly polar, small in size, has electron-withdrawing properties and is able to react with key amino acid residues of active sites on a variety of proteinsCitation15. In a previous experiment, compound 3 (), which had a cyano group on the ethylene bond, was found to have better selective toxic effect against HeLa cells with fewer toxic effects on non-cancerous L-02 cells when compared with the commercially available drug taxolCitation2.
On the basis of the previous study, we retained the trimethoxy groups on the A benzene ring, introduced a cyano group into the vinyl bridge to maintain its Z isomer, and either changed the substituents on the B benzene ring or replaced the B benzene ring with other heterocyclic structures to design various styrylbenzene derivatives. We also replaced the 3,4,5-trimethoxy groups with p-methyl groups to further study the effect of trimethoxy groups on anticancer activity and selective toxic effects ().
Materials and methods
Chemistry
1H NMR and 13C NMR spectra were obtained on an AV-300 (Bruker, Switzerland), and using TMS as internal standard. The chemical shifts are given in ppm referenced to the respective solvent peak, and coupling constants (J) are reported in hertz (Hz). High-resolution mass spectra were measured on a MALDI-TOF/TOF mass spectrometer (Bruker Dartonik, Germany). IR spectra were recorded (in KBr) on a FTIR1730 instrument. All reagents were obtained commercially and used were of analytical grade. Reaction processes were tracked by TLC on silica gel plates. Melting points of target compounds were determined in the open glass capillaries and were uncorrected.
General procedure for synthesis of compounds 6a-h, 7a-c, 13a-d, and 14a-b
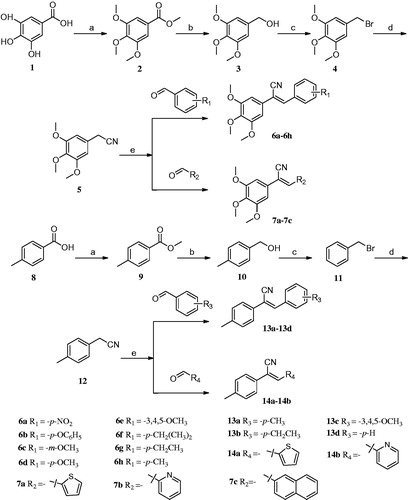
3,4,5-Trihydroxybenzoic acid (1) and 4-methylbenzoic acid (8) were used as starting materials to synthesise 5-(bromomethyl)-1,2,3-trimethoxybenzene (4) and (bromomethyl) benzene (11), respectively. Next, a mixture of 10 mmol of compound 4 or 11, 1.485 g (15 mmol) of TMSCN (trimethylsilyl cyanide), 4.725 g (15 mmol) of TBAF (tetrabutyl-ammonium fluoride), and 30 ml of acetonitrile was refluxed for 4–6 h with stirring. The reflux process was monitored by TLC. The reaction mixture was cooled to room temperature (RT), and then was added to 200 ml of ice water with severe stirring. The mixture was filtered, washed with 50% methanol, and purified by recrystallisation from 95% ethanol, yielding a final product of compound 5 or 12Citation16–20.
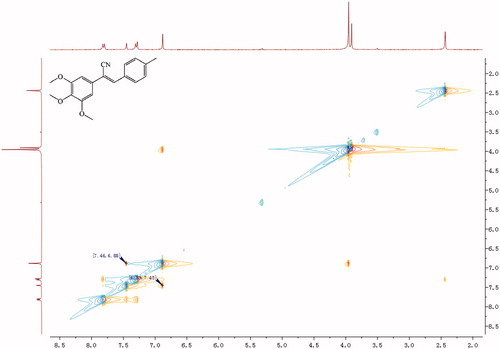
A mixture of 1 mmol compounds 5 or 12, 1 mmol properly substituted benzaldehyde or aromatic heterocyclic aldehyde, and 10 ml of methanol was heated to 60 °C with stirring. After 30 min, sodium methoxide (0.027 g, 0.5 mmol) was added to the mixture, and the mixture was kept at 60 °C for 4–6 h. The reaction was monitored by TLC. Once complete, the reaction mixture was cooled to RT, and the precipitate was separated by filtration and recrystallised from methanol to give the target final compound (Scheme 1). Because of a cyano-steric effect, the final structure was fixed as the Z isomer, confirmed by the nuclear Overhauser effect (NOE) (). The detailed information on the structural and physiochemical characteristics of the final target compounds can be found in the Supplementary material.
Figure 2. NOE (nuclear overhauser effect) result of compound 6h.
Scheme 1. Reagents and conditions: (a) (CH3)2SO4, (CH3)2CO, K2CO3, reflux; (b) LiAlH4, THF, 0 °C-rt, 4–6 h; (c) CH2Cl2, PBr3, 0 °C-rt, 3–6 h; (d) CH3CN, TMSCN, TBAF, reflux, 4–6 h; (e) CH3OH, CH3ONa, aromatic aldehydes, 4–6 h.
Biological evaluation
Materials
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The propidium iodide (PI) and Annexin V-FITC apoptosis detection kit were purchased from Invitrogen (Eugene, OR, USA).
Cell lines and cell culture
All human cell lines were used in this study. MGC-803 late-stage differentiation gastric cancer cell, A549 lung cancer cell, HepG2 hepatoma cell, AGS gastric cancer cell, BEL-7402 liver cancer cell, HCT-116 colorectal cancer cell, HeLa cervical cancer cell, SGC-7901 early differentiation gastric cancer cell, L-02 normal liver cell, MCF-7 breast cancer cell, and MCF-10A normal breast cell were initially purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). RPMI-1640 media, Dulbecco’s modified Eagle’s medium (DMEM), and foetal bovine serum (FBS) were provided from Gibco Company (Grand Island, NY, USA). The cells were maintained in DMEM or RPMI-1640, supplemented with 10% FBS, 100 IU/mL penicillin and 100 mg/mL streptomycin (Grand Island, NY, USA), and at 37 °C in a humidified atmosphere containing 5% CO2.
Cell growth inhibition assay
Cells were plated in 96-well plates at a density of 30–40% to ensure exponential growth throughout the experimental period, and then allowed to adhere for 1 dayCitation21. Next, the cells were treated with four serial concentrations (1, 10, 50, and 100 µM) of each compound. Taxol, CA-4, and CA-4P (phosphorylated form of CA-4) were used as positive controls. After 48 h incubation, MTT solution was added to each well at a final concentration of 2 mg/mL, and incubated for an additional 4 h. Next, the MTT solution was removed and 150 µL of dimethyl sulphoxide (DMSO)/well was added (Sigma-Aldrich, St Louis, MO, USA). The plates were shaken vigorously at RT to ensure complete solubilisation, and absorption at 492 nm was determined. At least three independent experiments were conducted, and the results were summarised in .
Table 1. In vitro anti-proliferative activity of the synthesised compounds, CA-4, CA-4P, and taxol against eleven cell linesTable Footnotea (IC50 (µMTable Footnoteb))
Analysis for cell cycle by flow cytometry
BEL-7402 cells were plated 6-well plates with 5.0 × 105 cells/well for overnight (O/N) and were synchronised at the G0 phase by serum starvation (0.1% FBS in RPMI-1640) for 24 h. Then the cells were incubated with 0.01, 0.05, 0.1 µM of 6h and 0.1 μM of taxol, respectively. Vehicle group was treated with 0.1% DMSO. Following 12 h of incubation, the cells were detached with 0.05% trypsin/5 mM EDTA and centrifuged at 1000 rpm for 10 min, and fixed in 70% ethanol at −20 °C for O/N. Then, the cells were subsequently analysed the cell cycle distribution by flow cytometry using a FACSCalibur flow cytometer with Cell Quest software (Becton-Dickinson, Franklin Lakes, NJ, USA), plotting at least 30,000 events per sampleCitation22. The percentage of cells in the G0/G1, S, and G2/M phases of the cell cycle were determined using the ModFit LT version 4.0 software package (Verity Software, Topsham, ME, USA).
Analysis of apoptosis by flow cytometry
Apoptosis was detected using an apoptosis detection kit. BEL-7402 cells were plated in 6-well plates (5.0 × 105 cells/well) and incubated at 37 °C overnight. Exponentially growing cells were then incubated with 6h at either 0.01 or 0.1 µM, or taxol at 0.1 µM. Vehicle group was treated with 0.1% DMSO. Following 12 h of incubation, cells were analysed for apoptosis as previously describedCitation23, using a FACSCalibur flow cytometer with Cell Quest software (Becton-Dickinson, Franklin Lakes, NJ, USA).
Cell migration assay
Quantitative cell migration was performed using 24-well Boyden chambers (Corning, NY, USA) as describedCitation7. Briefly, transwells with 8-µm pore size filters were inserted into 24-well plates. RPMI-1640 (500 µL) containing 10% FBS was added to the lower chamber, and 100 µL of a serum-free cell suspension (1 × 105 cells) was placed in the upper chamber. The BEL-7402 cells were pre-treated with indicated concentrations of 6h, taxol, or DMSO. The plates were incubated at 37 °C with 5% CO2 for 24 h, and the cells on the upside were removed using cotton swaps. After several rinses, the cells adhering in the lower layer of insert were fixed with methanol and stained with 1× crystal violet solution. The number of migrated cells in nine randomly selected fields from triplicate chambers was counted in each experiment under a phase-contrast microscope at 200× magnification.
Colony formation assay
A previously described colony formation assay was usedCitation7 with slight modification. 1 × 104 BEL-7402 cells/well were seeded into 6-well plates and then stimulated with the indicated concentration of 6h, taxol, or DMSO. The plates were removed from incubation when colonies were large enough to count (>50 cells). Colonies were then fixed and stained with 1 × Giemsa solution, rinsed, allowed to dry, and counted. All the experiments were repeated independently at least three times. The LD50 value for 6h was determined using SigmaPlot II.
Western blotting
BEL-7402 cells were harvested in logarithmic growth phase and the adherent and floating cells were collected after 12 h of drug treatment. The collected cells were resuspended in cold RIPA buffer (100 mM NaCl, 100 mM Tris (pH7.4), 10% Triton X-100, 1 mM PMSF, pH7.5) with 0.1 µM leupetin protease inhibitor as well as 1 µg/mL pepstatin and cracked on ice for 30 min. Then according to the reference to did immunoblotting assayCitation22.
Molecular modelling
The molecular docking study was performed using Discovery Studio 2017 Client. In this study, the structure of tubulin (PDB code: 1SA0) downloaded from protein database was chosen for docking. The 3D structures of compounds were provided using Chem 3D, the pdbqt format files of ligand, and the autodock format of protein were provided using AutoDockTools version 1.5.6.
Results and discussion
Chemistry
As shown in Scheme 1, through series of reactions, such as esterification, reduction, bromination, substitution, and Knoevenagel condensation, two series of derivatives were synthesisedCitation16–20. In brief, 3,4,5-trihydroxybenzoic acid (1) or p-methylbenzoic acid (8) as the starting material, dimethyl sulphate in the presence of acetone as solvent, potassium carbonate as a catalyst, compound 2 or 9 was obtained. Then, the compound was reduced respectively under ice-cooling with lithium aluminium hydride as a reducing agent, anhydrous tetrahydrofuran as solvent to give compound 3 or 10. The resulting compound 3 or 10 was dissolved in anhydrous CH2Cl2 and replaced by PBr3 under ice bath to give compound 4 or 11. Compound 5 or 12 was prepared by dissolving compound 4 or 11 in acetonitrile, with TMSCN and TBAF as catalysts. The series of target compounds were prepared by substituting aromatic aldehydes with compound 5 or 12 in the presence of methanol as solvent and sodium methoxide as catalyst. Before biological evaluation, the compounds were characterised via IR, 1H NMR, and 13C NMR spectrometry as well as high-resolution mass spectrometry, and the Z isomer was confirmed by NOE.
Biological evaluation
In vitro cytotoxicity assay and structure-activity relationship (SAR) studies
According to standard protocolCitation2, preliminary in vitro anti-proliferative activities of the synthesised compounds were evaluated by MTT assay, using nine human cancer cell lines (MGC-803, A549, HepG2, AGS, BEL-7402, HCT116, HeLa, SGC-7901, and MCF-7) and two normal human cell lines (L-02 and MCF-10A), and compared with those of CA-4, CA-4P, and taxol.
Results of the screening are summarised in . Most compounds showed good anti-proliferative activities against cancer cells including MGC-803, AGS, BEL-7402, HCT-116, and MCF-7. Three alkyl-substituted compounds (6f, 6g, and 6h), p-methoxy-substituted 6d, thiophene-2-yl derivative 7a, and naphthalene-1-yl derivative 7c, all with 3,4,5-trimethoxy groups on the A benzene ring, had lower IC50 values (<0.01–7.21 µM) in MGC-803. The order of anticancer activity was p-CH3 ≈ p-CH2CH3 > thiophene-2-yl > naphthalene-1-yl > p-OCH3 > p-(CH2)2CH3. Compounds 6g and 6h exhibited particularly strong inhibitory activities, with IC50 values less than 0.01 µM, making them more active than all control agents tested. Similarly, 6d, 6f, 6g, 6h, 7a, and 7c exhibited better anticancer activity (IC50 = 0.05, 1.33, 4.38, 0.51, 8.01, 3.51 µM, respectively) in AGS cells. In this assay, 6d exhibited activity similar to the positive control drug taxol (IC50 = 0.02 µM), and stronger activity than CA-4 (IC50 = 3.42 µM) and CA-4P (IC50 = 4.73 µM). Compounds 6f, 6g, 6h, and 7c also showed stronger activity than CA-4 and CA-4P against AGS cells. The order of anticancer activity was p-OCH3 > p-CH3 > p-(CH2)2CH3 > naphthalene-1-yl > p-CH2CH3 > thiophene-2-yl. Analysis using human hepatocellular carcinoma cells (BEL-7402) revealed that compound 6h had the best anticancer activity (IC50 = 0.15 µM), much stronger than CA-4 (IC50 = 2.02 µM) and CA-4P (IC50 = 5.45 µM). The high anticancer activity of 6h was followed by compounds 6g, 6f, 6d, 7a, and 7c with IC50 values of 0.65, 0.72, 0.75, 1.82, and 2.53 µM, respectively. The order of anticancer activity was p-CH3 > p-CH2CH3 > p-(CH2)2CH3 > p-OCH3 > thiophene-2-yl > naphthalene-1-yl. Compounds 6f, 6g, and 6h exhibited better anticancer activities (IC50 = 0.17, 0.34, and 0.61 µM, respectively) than CA-4P (IC50 = 0.59 µM) on HCT116 cells. Compounds 6f, 6h, and 7c expressed strong anticancer activity against MCF-7 (human breast adenocarcinoma) cells. Remarkably, compound 6h was 6-fold more active than taxol against MGC-803 cells and exhibited similar activity to taxol against A549 and BEL-7402 cells. Compound 6h was superior to CA-4 and CA-4P against almost all cancer cell lines tested, including MGC-803, A549, HepG-2, AGS, and BEL-7402 cells. However, 17 of the synthesised compounds displayed poor anticancer activity against HeLa and SGC-7901 cancer cell lines. In addition, among the compounds having 3,4,5-trimethoxy groups on the A benzene ring, p-NO2, p-OBn, m-OCH3, and pyridine-2-yl substituted compounds (6a, 6b, 6c, and 7b) expressed extremely poor in vitro anticancer activities against all nine cancer cell lines, and 3,4,5-trimethoxy substituted compound 6e, which has a heavy steric effect, also showed weak anticancer activity against all cancer cell lines tested. Synthesised compounds having p-methyl groups on the A benzene ring (13a–d and 14a–b) also demonstrated poor anticancer activities against all cell lines tested.
Using these preliminary results, we identified the following SARs: (1) the anticancer activity of a structure with a 3,4,5-trimethoxy group on the A benzene ring was greater than that of a structure with a p-methyl group on the A benzene ring; and (2) in structures containing 3,4,5-trimethoxy groups on the A benzene ring, introducing a B benzene ring with small size para-electron-donating groups, such as p-CH3, p-OCH3, and p-CH2CH3, or exchanging B benzene with an electron-rich aromatic heterocyclic structure, such as thiophene, significantly improves anticancer activity. Based on these observations, we expect that in structures containing 3,4,5-trimethoxy groups on the A benzene ring, electron density and steric hindrance effects on the B benzene ring strongly influence the anticancer activity of the (Z)-styrylbenzene derivatives bearing a cyano group synthesised.
Selective inhibition of cancer cell growth
Lack of selective cytotoxicity is the main factor restricting the tolerated dose of most conventional chemotherapeutic agentsCitation2. To address this, we compared the toxicity of all synthesised compounds with CA-4, CA-4P, and taxol on normal human cell lines L-02 and MCF-10A. The calculated selectivity index (SI) of each compound is depicted in . Compounds 6g and 6h exhibited 10,000-fold higher selectivity for MGC-803 cells than for normal L-02 cells, which is a much higher selectivity than displayed by taxol. In addition, many compounds expressed significantly higher selectivity for cancer cells than for L-02 cells, including 6d for AGS, 6f and 6g for BEL-7402 and HCT116, 6h for A549, AGS, BEL-7402, and HCT116 cells. However, none of the compounds showed strong selectivity for MCF-7 (breast cancer) cells over MCF-10A (normal breast) cells. The control compounds CA-4 and CA-4P displayed poor selectivity for cancer cells over normal cells although they induced strong cytotoxicity in cancer cells. On the basis of these results, we speculated that 6h was the best candidate for further mechanism analysis. The strong toxicity of 6h for MGC-803 cells (IC50 < 0.01 µM) is likely to introduce experimental error even with small changes in concentration; therefore, BEL-7402 cells, for which 6h demonstrated moderate toxicity and selectivity, were selected for further study of 6h.
Table 2. Selectivity index values of compounds relative to the effects on normal cell L-02 or MCF-10A cells.
Cell cycle regulation by compound 6h and Western blot analysis
The cell-cycle is a series of events that result in cell division, duplication, and proliferation. Numerous cytotoxic compounds exert their anti-proliferative effect by inducing cell-cycle arrest, apoptosis, or bothCitation2. These mechanisms are considered effective anticancer strategiesCitation21. To study the mechanism by which compound 6h reduced the viability of BEL-7402 cells, we used flow cytometry to analyse cell-cycle distribution. As shown in , 6h increased the percentage of G2/M cell population in a concentration-dependent manner from 23.09% to 59.99% after 12 h incubation, and the trend was similar in the S cell population (4.44–19.35%). After treatment with 0.1 µM 6h, the cell distribution at G0/G1, G2/M, and S phases was very similar to the 0.1 µM taxol treatment group. This finding suggests that 6h induces cell-cycle arrest at the S and G2/M phases.
Figure 3. Compound 6h decreases the expression of cyclin A and cyclin D1 and increases the expression of cyclin B1, thus reducing the cell cycle progression of BEL-7402 cells. (A) Cell numbers in the phases of the cell cycle were analyzed on 12 h after stimulation. Values were obtained from three separate experiments and represent the mean ± SD. *p < 0.05. **p < 0.01. ***p < 0.001 compared to respective control group. (B) Immunoblots were probed using antibodies directed against cyclin A, B1, and D1, and β-actin. Whole-cell protein lysates were extracted from BEL-7402 cells cultured after 12 h. Results are representative of three separate experiments.
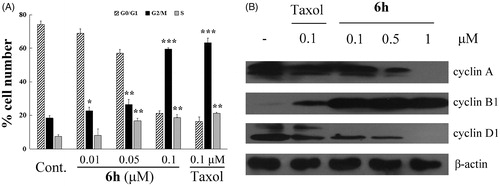
Since entry into the S phase in the cell-cycle requires the accumulation of cell-cycle activation-related cyclinsCitation7, we assessed the expression levels of cyclins, such as cyclin A and cyclin D1 to determine whether the decreased cell proliferation by 6h involved these proteins. The 6h treatment group had lower expression of cyclin A and D1 compared with the vehicle group, this reduced expression was dose-dependent, and similar to that of the taxol treatment group at 0.1 µM (). Cyclin B1 plays an important role in the transition from interphase to mitotic phase and governs cell cycle progression by enhancing cell-cycle distribution in the G2/M fractionCitation24, and it has been reported that expression of cyclin B1 is increased by exposure to anticancer agentsCitation25. In our experiment, 6h treatment induced increased expression of cyclin B1 compared with treatment with vehicle or 0.1 µM of taxol. These results suggest that the down-regulation of cyclins A and D1 and up-regulation of cyclin B1 are related to the decrease in cell proliferation induced by 6h treatment on BEL-7402 cells.
Apoptotic effects by compound 6h
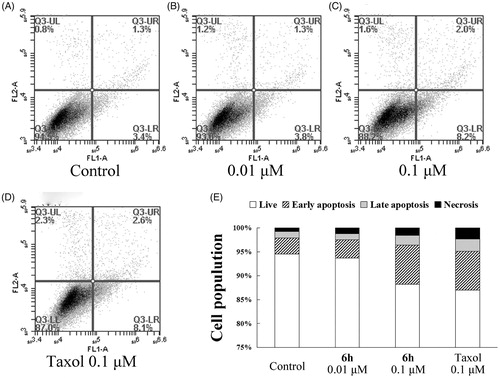
To further investigate whether 6h induces apoptosis, BEL-7402 cells were treated with vehicle, 6h (0.01 or 0.1 µM), or taxol (0.1 µM) for 12 h, then stained with Annexin V-FITC and PI. As shown in , the percentage of total apoptotic cells (early + late apoptotic cells) increased in a dose-dependent manner with 6h treatment, but the effect was weak. Treatment with 0.1 µM 6h resulted in a similar number of total apoptotic cells to the taxol treatment group (10.2% and 10.7%, respectively), both of which were higher than the vehicle treatment group (4.7%). These results indicate that apoptosis plays only a slightly important role in the inhibition of BEL-7402 cell proliferation by 6h.
Figure 4. Apoptosis induction in BEL-7402 cells after treatment for 12 h with (A) 0.1% DMSO (vehicle); (B) 0.01 μM 6h; (C) 0.1 μM 6h; (D) 0.1 μM taxol. (E) The results of the cell apoptosis.
Cell migration and colony formation inhibition by compound 6h
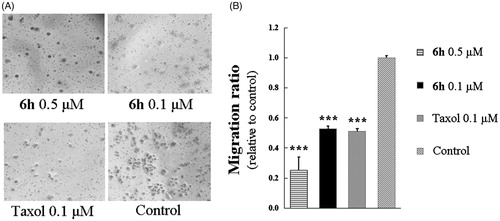
Aggressive tumours have a strong ability to proliferate and migrate, and in many cases, cell mobility affects cell proliferationCitation2. We used a transwell migration assay to investigate the effect of compound 6h on the migration of BEL-7402 cells. As shown in , treatment with 0.1 or 0.5 µM of 6h resulted in migration ratios that were markedly decreased compared with the control group. A similar finding was observed for taxol at 0.1 µM.
Figure 5. Transwell assay of compound 6h showing inhibition of BEL-7402 cell migration. (A) The images of stained BEL-7402 cells adhering in the lower layer of insert of transwell with phase-contrast microscopy (200× magnification). (B) The relative migration ratio of BEL-7402 cells. Data expressed as mean ± SD (n ≥ 3). ***p < 0.001 compared to control group.
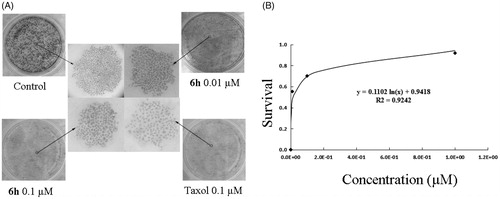
The LD50 of 6h was determined using a colony formation assay with BEL-7402 cells over a relatively long exposure time (> 10 days) to 6h. As shown in , 6h had a dose-dependent inhibitory effect on colony formation, with an LD50 value of 0.056 ± 0.0047 µM, further confirming the anticancer activity of the compound. Compared with the dense and large cell colonies of control group, the 6h treatment groups showed sparse and small cell colonies which was observed in a dose-dependent manner. Trypan blue staining was used to further investigate the cytotoxicity of 6h. Treatment of cells with 6h at concentrations ranging from 0.001 µM to 10 µM resulted in percentages of dead cells that were negligible (data not shown). These results indicate that 6h has a strong anti-proliferative effect on BEL-7402 cells in the absence of evident cytotoxicity.
Figure 6. Colony formation ability was inhibited by 6h treatment. (A) The images of stained colonies under phase-contrast microscopy. (B) The curve was fitted as the inhibition ratio of colony formation against the concentration. Values are represented as mean ± SD from three independent colony formation experiments.
Molecular docking
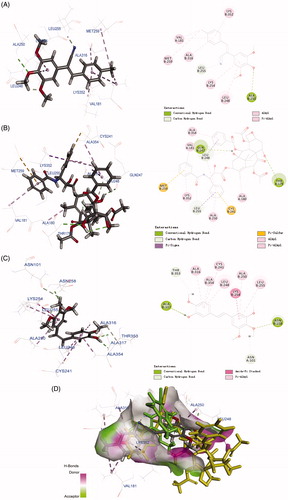
To investigate the possible binding modes of the compound with tubulin, 6h, a well-behaved and selectively toxic compound, was used in molecular docking studies. The structure of tubulin used in the docking study was obtained from the Protein Data Bank (PDB code: 1SA0, https://www.rcsb.org/structure/1SA0)Citation26. All docked conformations are ranked based on docking scores. Taxol and CA-4 were used as positive controls. As shown in , 6h, taxol, and CA-4 interacted with tubulin residues differently. Compound 6h formed abundant chemical bonds with the amino acid residues on the β chain of tubulin and showed strong affinity, whereas the number of chemical bonds formed with the amino acid residues on the α chain was sparse and showed poor affinity. The cyano group tended to bind to the β subunit but not the α subunit. In the case of the cis isomer of CA-4, the entire structure made multiple contacts with the β subunit. The 2D diagram effectively displays these structural features, highlighting several binding regions on the β subunit, including Cys241, Leu248, Ala316, and Ala354. Taxol bound between the α and β subunits of tubulin, the three benzene rings docked into the β subunit side while the rest of the compound docked into the α subunit. The 2D image shows several binding sites on the α subunit, including Val181, but most of the binding contacts are on the β subunit. The superposition of the three compounds in the biding pocket clearly shows the binding posture of 6h and taxol are similar, whereas the biding location of CA-4 is completely different from them (). On the basis of the molecular modelling results, we suggest that 6h selectively inhibits cancer cell proliferation without toxicity to normal cell proliferation by targeting both the α and β subunits of tubulin.
Figure 7. Molecular modelling of compounds in complex with tubulin (PDB: 1SA0). Shown is the proposed binding mode and interaction between tubulin and selected compounds. (A) 6h, (B) taxol, and (C) CA-4. The compounds and important amino acids in the binding pockets are shown in stick model. (D) Superposition of 6h, taxol, and CA-4 in the pocket of tubulin in surface representation. The compounds are shown in stick model with carbon atoms in grey (6h), yellow-green (taxol), or green (CA-4).
Conclusions
A series of (Z)-styrylbenzene derivatives, containing a cyano group, were designed, synthesised, and evaluated for anti-proliferative properties against nine human cancer cell lines and two non-cancerous human cell lines. Most derivatives exhibited significant anticancer activity against five of the cancer cell lines, including MGC-803 and BEL-7402 cells. SAR analysis indicated that in structures containing 3,4,5-trimethoxy groups on the A benzene ring, the electron density and steric effect of the B benzene ring strongly affect the anticancer activity of the derivatives. Most compounds, especially 6h, exhibited notably high potency against a set of cancer cell lines. Compound 6h possessed the highest anticancer activity against MGC-803 cells, being 6-fold more potent than taxol and CA-4. The IC50 value of 6h in L-02 cells was more than 10,000-fold higher than in MGC-803 cells and more than 666-fold higher than in BEL-7402 cells, indicating that 6h exhibits selective toxic effects. In addition, 6h arrested BEL-7402 cells in the G2/M phase of the cell cycle and slightly induced apoptosis. Mechanistic studies suggested that the blockade of G2/M phase was associated with up-regulation of cyclin B1 and down-regulation of cyclins A and D1. Molecular modelling results suggested the selectivity of 6h to inhibit only cancer cell proliferation was achieved by targeting both the α and β subunits of tubulin. Compound 6h inhibited the migration of BEL-7402 cells and the formation of cell colonies. In summary, these newly developed compounds showed marked biological activity in vitro and have potential for further development as a novel class of selective anticancer agents.
Supplemental_material.doc
Download MS Word (323.9 KB)Acknowledgements
We thank Sarah Bubeck, Ph.D., from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Mahapatra DK, Bharti SK, Asati V. Anti-cancer chalcones: structural and molecular target perspectives. Eur J Med Chem 2015;98:69–114.
- Ma J, Li JJ, Tian YS. Synthesis and bioactivity evaluation of 2,3-diaryl acrylonitrile derivatives as potential anticancer agents. Bioorg Med Chem Lett 2017;27:81–5.
- Huang XC, Jin L, Wang M. Design, synthesis and in vitro evaluation of novel dehydroabietic acid derivatives containing a dipeptide moiety as potential anticancer agents. Eur J Med Chem 2015;89:370–85.
- Al-Obaid AM, Abdel-Hamide SG, El-Kashef HA, et al. Substituted quinazolines, part 3. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs. Eur J Med Chem 2009;44:2379–91.
- Cragg GM, Newman DJ. Nature: a vital source of leads for anticancer drug development. Phytochem Rev 2009;8:313–31.
- Zheng SL, Zhong Q, Mottamal M, et al. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-A4 as anticancer agents. J Med Chem 2014;57:3369–81.
- Madadi NR, Penthala NR, Howk K, et al. Synthesis and biological evaluation of novel 4,5-disubstituted 2H-1,2,3-triazoles as cis-constrained analogues of combretastatin A-4. Eur J Med Chem 2015;103:123–32.
- Qi J, Dong H, Huang J, et al. Synthesis and biological evaluation of N-substituted 3-oxo-1,2,3,4-tetrahydro-quinoxaline-6-carboxylic acid derivatives as tubulin polymerization inhibitors. Eur J Med Chem 2018;143:8–20.
- Ohsumi K, Nakagawa R, Fukuda Y, et al. Novel combretastatin analogues effective against murine solid tumors: design and structure-activity relationships. J Med Chem 1998;41:3022–32.
- Demchuk DV, Samet AV, Chernysheva NB, et al. Synthesis and antiproliferative activity of conformationally restricted 1,2,3-triazole analogues of combretastatins in the sea urchin embryo model and against human cancer cell lines. Bioorg Med Chem 2014;22:738–55.
- Banimustafa M, Kheirollahi A, Safavi M, et al. Synthesis and biological evaluation of 3-(trimethoxyphenyl)-2(3H)-thiazole thiones as combretastatin analogs. Eur J Med Chem 2013;70:692–702.
- Madadi NR, Penthala NR, Song L, et al. Preparation of 4,5 disubstituted-2H-1,2,3-triazoles from (Z)-2,3-diaryl substituted acrylonitriles. Tetrahedron Lett 2014;55:4207–11.
- Carr M, Greene LM, Knox AJS, et al. Lead identification of conformationally restricted β-lactam type combretastatin analogues: synthesis, antiproliferative activity and tubulin targeting effects. Eur J Med Chem 2010;45:5752–66.
- Samanta S, Lim TL, Lam YL. Synthesis and in vitro evaluation of West Nile Virus protease inhibitors based on the 2-{6-[2-(5-phenyl-4H-{1,2,4] triazol-3-ylsulfanyl)acetylamino]benzothiazol-2-ylsulfanyl}acetamide scaffold. ChemMedChem 2013;8:994–1001.
- Li YT, Wang JH, Pan CW, et al. Syntheses and biological evaluation of 1,2,3-triazole and 1,3,4-oxadiazole derivatives of imatinib. Bioorg Med Chem Lett 2016;26:1419–27.
- Srivastava V, Darokar MP, Fatima A, et al. Synthesis of diverse analogues of oenostacin and their antibacterial activities. Bioorg Med Chem 2007;15:518–25.
- Chakraborty S, Das UK, Ben-David Y, et al. Manganese catalyzed α-olefination of nitriles by primary alcohols. J Am Chem Soc 2017;139:11710–3.
- Petch D, Anderson RJ, Cunningham A, et al. Design and synthesis of EGFR dimerization inhibitors and evaluation of their potential in the treatment of psoriasis. Bioorg Med Chem 2012;20:5901–14.
- Lieber S, Scheer F, Meissner W, et al. (Z)-2-(2-bromophenyl)-3-{[4-(1-methyl-piperazine)amino]phenyl}-acrylonitrile (DG172): an orally bioavailable PPARβ/δ-selective ligand with inverse agonistic properties. J Med Chem 2012;55:2858–68.
- Tanpure RP, Strecker TE, Chaplin DJ, et al. Regio- and stereospecific synthesis of mono-beta-d-glucuronic acid derivatives of combretastatin A-1. J Nat Prod 2010;73:1093–101.
- Liu CF, Shen QK, Li JJ, et al. Synthesis and biological evaluation of novel 7-hydroxy-4-phenylchromen-2-one-linked to triazole moieties as potent cytotoxic agents. J Enzyme Inhib Med Chem 2017;32:1111–9.
- Tian YS, Kim HJ, Kim HM. Rho-associated kinase (ROCK) inhibition reverses low cell activity on hydrophobic surfaces. Biochem Biophys Res Commun 2009;386:499–503.
- Zhang HJ, Zhang GR, Piao HR, et al. Synthesis and characterisation of celastrol derivatives as potential anticancer agents. J Enzyme Inhib Med Chem 2017;33:190–8.
- Kamal A, Ashraf M, Basha ST, et al. Design, synthesis and antiproliferative activity of new conjugates of E7010 and resveratrol as tubulin polymerization inhibitors. Org Biomol Chem 2016;14:1382–94.
- Clarke PR, Allan LA. Cell-cycle control in the face of damage-a matter of life or death. Trends Cell Biol 2009;19:89–98.
- Liu Y-N, Wang J-J, Ji Y-T, et al. Design, synthesis, and biological evaluation of 1-methyl-1,4-dihydroindeno[1,2-c]pyrazole analogues as potential anticancer agents targeting tubulin colchicine binding sites. J Med Chem 2016;59:5341–55.