
Abstract
DNA topoisomerase IB (TOP1) is a validated target for discovery and development of antitumor agents. Four TOP1 poisons are clinically used for tumor treatment now. In spite of their effectiveness in solid tumors, these camptothecin (CPT) poisons suffer from many shortcomings. Therefore, many investigations have focused on the discoveries of non-CPT poisons and catalytic inhibitors. Herein, we systematically study the antitumor activity of CYB-L10, a novel indolizinoquinolinedione TOP1 catalytic inhibitor discovered in our laboratory. The results indicated that CYB-L10 mainly acts on TOP1 in cancer cells and is not a substrate of the P-glycoprotein. In addition, CYB-L10 can induce apoptosis of HCT116 cells, shows high cytotoxicity against 60 human clinical cancer cell lines (NCI60) with the mean-graph midpoint for growth inhibition of all cancer cell lines of 0.050 µM concentration and obvious antitumor efficiency in vivo in the HCT116 xenograft model.
Introduction
DNA topoisomerase IB (TOP1) is an essential enzyme that controls the DNA topology structure in many cellular metabolic processes, including replication, transcription, and recombinationCitation1–4. TOP1 functions through a nucleophilic tyrosine residue (Tyr 732), which cleaves one phosphodiester backbone of DNA double strand, and covalently attaches to the 3′-end of the nicked DNA to form a transient enzyme-DNA covalent complex (TOP1cc)Citation2,Citation5. Inhibition of TOP1 catalytic activity or trapping of TOP1cc can result in DNA damage, which triggers apoptotic mechanisms and other cell death processesCitation6–8.
TOP1 inhibitors are grouped into two types as TOP1 “poisons” and “catalytic inhibitors” based on their molecular mechanism of action. TOP1 poisons are able to trap TOP1cc to prevent further religation and thus leading to irreversible DNA strand breaksCitation7,Citation9,Citation10. Camptothecin (CPT) and many of its derivatives are the well-known TOP1 poisons. At present, four CPT derivatives have been approved for clinical treatment of tumor, including topotecan and irinotecan approved by FDACitation7,Citation10, belotecan (in South Korea) and 10-hydroxy camptothecin (in China)Citation7,Citation11,Citation12. Unlike poisons, TOP1 catalytic inhibitors act at the upstream stage of the catalytic DNA cleavage reaction of enzymes, and prevent the formation of TOP1ccCitation13–16.
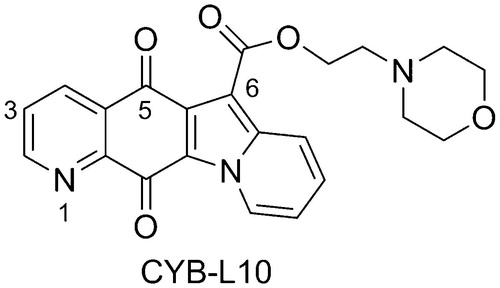
In spite of their effectiveness in solid tumors, CPT poisons suffer from many shortcomings, including bone marrow dose-limiting toxicity, severe gastrointestinal toxicityCitation17, poor solubility, chemically instability under physiological pH, and drug efflux-mediated resistanceCitation10. Therefore, many investigations have focused on the discoveries of non-CPT poisons and the catalytic inhibitorsCitation13,Citation14,Citation18–21. In our previous study, the indolizinoquinolinedione derivatives have been discovered as a new class of TOP1 catalytic inhibitorsCitation13,Citation15,Citation22, which can inhibit TOP1 catalytic cleavage reaction, and prevent the formation of TOP1ccCitation13. Further structural modification led to the discovery of several TOP1 catalytic inhibitors, among which CYB-L10 () showed good cytotoxicity and higher TOP1 inhibition than CPT without TOP1-mediated unwinding effectCitation15. Therefore, it is worthy to systematically study its antitumor activity. Herein, we report the activity of CYB-L10 in vitro against 60 clinical cancer cell lines and in vivo in HCT116 xenograft mice model.
Figure 1. Chemical structure of CYB-L10.
Methods and materials
General experiments
The human wild-type cancer cell lines HCT116 and DU-145, the resistant cell lines HCT116-siTop1 and RC0.1 were a kind gift from Dr. Y. Pommier (Laboratory of Molecular Pharmacology, Center for Cancer Research, NCI, NIH). The human wild-type cancer cell lines MCF-7, and HepG2, and the resistant cell lines MCF-7/ADR and HepG2/ADR were a kind gift from Dr. X. Z. Bu (School of Pharmaceutical Sciences, Sun Yat-sen University). CYB-L10 (molecular weight: 405.41) was synthesized according to our reported method (compound’s code: 26)Citation15. The structure was determined by NMR and MS spectra. The purity of CYB-L10 was determined to be more than 95% through HPLC.
Cell culture and MTT assay
The cells were cultured in RPMI-1640 or DMEM medium at 37 °C in a humidified atmosphere with 5% CO2. All cells to be tested in the following assays had a passage number of 3–6.
For the drug treatment experiments, the cancer cells were treated with CYB-L10 (predissolved in DMSO) at a five-dose assay concentration of 0.01, 0.1, 1, 10, and 100 µM. After incubation for 72 h at 37 °C, the MTT solution (50 µL, 1 mg/mL) in PBS (PBS without MTT as the blank) was fed to each well of the culture plate (containing 100 µL medium). After 4 h incubation, the formazan crystal formed in the well was dissolved with 100 µL of DMSO for optical density reading at 570 nm. The GI50 value was calculated by nonlinear regression analysis (GraphPad Prism).
NCI60 assay
The NCI60 (National Cancer Institute 60) tumor cell drug discovery panel was developed as a tool to assess the anticancer activity of compounds against a range of cell lines derived from nine cancer cell types, including hematological malignancies, lung, central nervous system, melanoma, colorectal, renal, breast, ovarian, and prostateCitation18,Citation23,Citation24. CYB-L10 was tested using the protocols by the NCI, which has been described previously. Briefly, CYB-L10 was tested at a five-dose assay concentration of 0.01, 0.1, 1, 10, and 100 µM for a period of 48 h. The data consist of concentration values (GI50) for each cell line at which the concentrations of compound that resulted in 50% cell growth inhibition. The overall antiproliferative potential is quantified as a mean-graph midpoint (MGM).
Flow cytometry
HCT116 cells (3.0 × 105 cells/mL) were grown in culture medium on 6-well plates and incubated in the presence or absence of CYB-L10 (1, 3, and 9 µM) for 24 h. And then, the cells were harvested and washed with cold PBS buffer, resuspended in 1 × binding buffer, and then stained with 5 µL Annexin V-FITC and 5 µL propidium iodide (KeyGEN BioTECH, Nanjing, China) for 15 min in dark. The stained cells were analyzed by using flow cytometry (BD, FACSCalibur, Franklin Lakes, NJ, USA) within 1 h. The experiments were repeated independently for three times.
Pharmacokinetic study in rat
Male Sprague-Dawley rats (weighing 220–250 g, n = 2) were treated with CYB-L10 predissolved in 10% DMSO and 10% Kolliphor® HS15 (a nonionic solubilizer) by intravenous injection (i.v., 1 mg/kg) and intragastric administration (i.g., 5 mg/kg), respectively. Blood samples (200 µL) were collected into heparinized tubes via the jugular vein at the following times: 0.083, 0.25, 0.5, 1, 2, 5, 7, and 24 h after dosing. Plasma samples (100 µL) were obtained after centrifugation for 10 min at 3000 rpm and stored at −20 °C until used for analysis. The plasma was detected through LC-MS/MS.
In vivo antitumor activity
Athymic nude mice bearing the nu/nu gene were obtained from Laboratory Animal Center of Sun Yat-sen University and maintained in pathogen-free conditions to establish the model of xenografts of HCT116. All animals were used under the Policy on the Care and Use of Laboratory Animal of Sun Yat-sen University. Male nude mice 4–5 weeks old weighing 12–15 g were used. HCT116 tumor preinduced in the mice by subcutaneously injecting of HCT116 cells (100 µL, 1 × 107 cells) was implanted. When the implanted tumors had reached a volume of about 80 mm3, the mice were randomly divided into three groups (n = 5) and administered by i.p. injection at a frequency of once every 3 days. The testing groups were treated with CYB-L10 at 20 mg/kg and 6.7 mg/kg dose, respectively. The control group was treated with an equivalent volume of saline. Tumor volumes (V) were monitored by caliper measurement of the length and width, and calculated using the formula: V = (larger diameter) × (smaller diameter)2/2, and growth curves were plotted using average tumor volume within each experimental group at the set time points. At the end of treatment, the animals were sacrificed, and the tumors were removed and weighed. The tumor weight inhibition (TWI) was calculated according to the formula: TWI = (1 − Mean tumor weight of the experimental group/Mean tumor weight of the control group) × 100%. Statistical comparisons were conducted using a one-way analysis of variance, followed by Tukey’s test.
Results and discussion
Cytotoxicity for NCI60 cell lines
CYB-L10 () was potent both on TOP1 inhibition and cytotoxicityCitation15, but exhibit very weak inhibition against TOP2 at 25 μM (Figure S1). It was selected and submitted to National Cancer Institute (NCI, USA) for a developmental therapeutics assay against the 60 clinical tumor cell lines representing nine tissue types (NCI60)Citation23–25. The tumor cells were incubated with CYB-L10 for 48 h and stained with sulphorhodamine B dye. Cell growth inhibition (GI50) was calculated relative to the control without compound. As shown in , CYB-L10 exhibited high cytotoxicity with a mean graph midpoint (MGM) for growth inhibition of all human cancer cell lines of 0.050 µM, and its GI50 values below the tested minimum concentration (10 nM) for 15 tumor cell lines.
Table 1. Cytotoxicity of CYB-L10 against NCI60 cell lines.
Cytotoxicity of CYB-L10 in drug-resistant cell lines
The cytotoxicity of CYB-L10 in drug-resistant cell lines was evaluated by using the MTT assay and summarized in . HCT116-siTop1 subline was developed by transfection of colon cancer parental cells HCT116 with short hairpin RNA vectors expressing siRNA for TOP1Citation26. Comparing to the parental cell line HCT116, HCT116-siTop1 subline showed 8.3-fold resistant to CPT, of which TOP1 is the only known cellular targetCitation10,Citation27, and about 7-fold resistant to CYB-L10, implying that TOP1 may be the major cellular target of CYB-L10.
Table 2. Cytotoxicity of the CYB-L10 in drug-resistant human cancer cell lines.
The prostate cancer cell RC0.1 has an R364H mutation in the TOP1 relating to the wild-type parental cell DU-145Citation28. The TOP1 with R364H mutation is catalytically active, but lead to RC0.1 cells resistant to CPT because the R364 residue is close to the catalytic tyrosine and can stabilize the open form of TOP1ccCitation29,Citation30. The RC0.1 cells were highly resistant to TOP1 poison CPT (396.3-fold) and less resistant to CYB-L10 (40.6-fold), implying that the binding site of CYB-L10 on TOP1 is different from that of CPT.
P-glycoprotein (Pgp) mediated drug efflux is generally responsible for classical multiple drug resistanceCitation31. The chemotherapeutic agent doxorubicin (DOX) is a substrate of Pgp, and has been found highly resistant for breast cancer MCF-7/ADR (77.8-fold) and hepatocellular HepG2/ADR sublines (47.6-fold), both with overexpressed PgpCitation32. However, CYB-L10 appeared not to be a substrate of the Pgp ().
Apoptosis analysis of CYB-L10
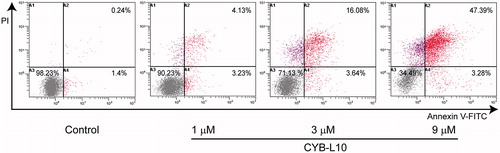
To estimate the effect of CYB-L10 on apoptosis, flow cytometry analysis using double staining with annexin V-FITC/PI was carried out for HCT116 cells. As shown in , after being treated with CYB-L10 (1, 3, and 9 μM) for 24 h, compared to the untreated control group, the apoptotic cells in the treated group showed an increase in a dose-dependent manner. CYB-L10 induced the major population of HCT116 cells into the late apoptotic stage 47.39% at 9 μM concentration.
Figure 2. The flow cytometry histograms analysis. HCT116 cells were treated with CYB-L10 at 1, 3, and 9 μM, respectively for 24 h (n = 3).
Pharmacokinetic study of CYB-L10 in vivo
The in vivo pharmacokinetic (PK) study of CYB-L10 was conducted in SD rats. The SD rats were divided into two groups (n = 2) and treated by i.v. injection at 1 mg/kg dose and i.g. at 5 mg/kg dose, respectively. The plasma samples were collected up to 24 h postdose and measured for the concentration of CYB-L10. The PK parameters were calculated and summarized in . By i.v. administration, the AUC0→∞ was 241 ± 40 h·ng/mL, and T1/2 was 18.4 ± 5.12 h. After oral administration, the Tmax, Cmax and AUC0→∞ were 0.46 ± 0.10 h, 24.7 ± 6.59 ng/mL and 76.0 ± 16.5 h·ng/mL, respectively. The bioavailability (F) was 6.3%.
Table 3. PK parameter of CYB-L10.
Antitumor activity in vivo of CYB-L10
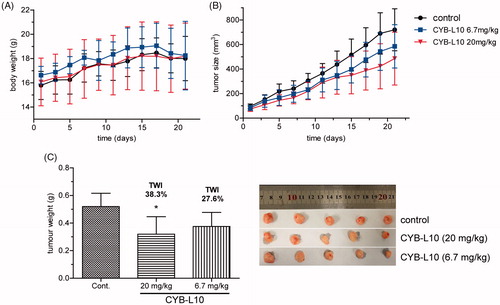
A human colon cancer HCT116 xenograft nude mice model was established to evaluate the antitumor efficiency of CYB-L10 in vivo. Mice were randomly divided into three groups and administered i.p. injection at a frequency of once every 3 days. Two groups were treated with CYB-L10 at 20 mg/kg and 6.7 mg/kg dose, respectively. The negative control group was treated with an equivalent volume of saline. As shown in , administration of CYB-L10 obviously reduced the tumor volume of the nude mice in a dose-dependent manner comparing to the negative control group. Finally, the tumor weight inhibitions (TWI) of CYB-L10 administration groups were 38.3% (20 mg/kg, p < 0.05) and 27.6% (6.7 mg/kg), respectively (). Meanwhile, the weight of the body of the mice in the CYB-L10 administration groups had no obvious loss comparing to the negative control group ().
Figure 3. The antitumor efficiency of CYB-L10 in vivo. The effects of CYB-L10 on body weight (A), tumor size (B), and tumor weight (C) at the dose of 6.7 mg/kg and 20 mg/kg in the HCT116 xenograft model. Statistically significant difference in mean tumor weight compared with the control, *p < 0.05.
Conclusion
Indolizinoquinolinedione derivative CYB-L10 serves as a novel TOP1 catalytic inhibitor with high cytotoxicity and higher TOP1 inhibition than that of CPT. Further study indicated that CYB-L10 exhibits high cytotoxicity against 60 clinical cancer cell lines from NCI (NCI60) with MGM value of 0.050 µM and induce apoptosis of HCT116 cells in the late apoptotic stage, implying CYB-L10 is a good anticancer candidate. The drug-resistant cell assays indicated that CYB-L10 may mainly act to TOP1 in tumor cells and is not a substrate of the Pgp, a drug efflux protein involving in multiple drug resistance. CYB-L10 was also evaluated on PK and antitumor efficiency in vivo, and found to have long T1/2 value and obvious antitumor efficiency in HCT116 xenograft nude mice model with TWI of 38.3% at 20 mg/kg dose.
Supplemental_material.pdf
Download PDF (223.6 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 2001;70:369–413.
- Stewart L, Redinbo MR, Qiu X, et al. A model for the mechanism of human topoisomerase I. Science 1998;279:1534–41.
- Berger JM, Gamblin SJ, Harrison SC, et al. Structure and mechanism of DNA topoisomerase II. Nature 1996;379:225–32.
- Pommier Y, Sun Y, Huang SN, et al. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 2016;17:703–21.
- Szczesny B, Brunyanszki A, Ahmad A, et al. Time-dependent and organ-specific changes in mitochondrial function, mitochondrial DNA integrity, oxidative stress and mononuclear cell infiltration in a mouse model of burn injury. PLoS One 2015;10:e0143730.
- Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev 2009;109:2894–902.
- Pommier Y, Leo E, Zhang H, et al. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 2010;17:421–33.
- Ashour ME, Atteya R, El-Khamisy SF. Topoisomerase-mediated chromosomal break repair: an emerging player in many games. Nat Rev Cancer 2015;15:137–51.
- Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol 2013;8:82–95.
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006;6:789–802.
- Zhang R, Li Y, Cai Q, et al. Preclinical pharmacology of the natural product anticancer agent 10-hydroxycamptothecin, an inhibitor of topoisomerase I. Cancer Chemoth Pharm 1998;41:257–67.
- Shao J, Huang F, Fu J. Clinical study on treatment of non-small cell lung cancer with 10-hydroxycamptothecin. Chin J Clin Oncol 2003;30:26–8.
- Wu N, Wu XW, Agama K, et al. A novel DNA topoisomerase I inhibitor with different mechanism from camptothecin induces G2/M phase cell cycle arrest to K562 cells. Biochemistry 2010;49:10131–6.
- Ganguly A, Das B, Roy A, et al. Betulinic acid, a catalytic inhibitor of topoisomerase I, inhibits reactive oxygen species-mediated apoptotic topoisomerase I-DNA cleavable complex formation in prostate cancer cells but does not affect the process of cell death. Cancer Res 2007;67:11848–58.
- Yu LM, Zhang XR, Li XB, et al. Synthesis and biological evaluation of 6-substituted indolizinoquinolinediones as catalytic DNA topoisomerase I inhibitors. Eur J Med Chem 2015;101:525–33.
- Qin XJ, Yu Q, Yan H, et al. Meroterpenoids with antitumor activities from guava (Psidium guajava). J Agric Food Chem 2017;65:4993–9.
- Lian Q, Xu J, Yan S, et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res 2017;27:784–800.
- Syrovets T, Büchele B, Gedig E, et al. Acetyl-boswellic acids are novel catalytic inhibitors of human topoisomerases I and IIa. Mol Pharmacol 2000;58:71–81.
- Castelli S, Vieira S, D, Annessa I, Katkar P, et al. A derivative of the natural compound kakuol affects DNA relaxationof topoisomerase IB inhibiting the cleavage reaction. Arch Biochem Biophys 2013;530:7–12.
- De Camargo MS, Da Sliva MM, Correa RS, et al. Inhibition of human DNA topoisomerase IB by nonmutagenic ruthenium(II)-based compounds with antitumoral activity. Metallomics 2016;8:179–92.
- Takarada JE, Guedes APM, Correa RS, et al. Ru/Fe bimetallic complexes: synthesis, characterization, cytotoxicity and study of their interactions with DNA/HSA and human topoisomerase IB. Arch Biochem Biophys 2017;636:28–41.
- Cheng Y, An LK, Wu N, et al. Synthesis, cytotoxic activities and structure-activity relationships of topoisomerase I inhibitors: indolizinoquinoline-5,12-dione derivatives. Bioorg Med Chem 2008;16:4617–25.
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813–23.
- Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol Cancer Ther 2010;9:1451–60.
- Reinhold WC, Sunshine M, Varma S, et al. Using CellMiner 1.6 for systems pharmacology and genomic analysis of the NCI-60. Clin Cancer Res 2015;21:3841–52.
- Miao ZH, Player A, Shankavaram U, et al. Nonclassic functions of human topoisomerase I: genome-wide and pharmacologic analyses. Cancer Res 2007;67:8752–61.
- Hsiang YH, Hertzberg R, Hecht S, et al. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 1985;260:14873–8.
- Urasaki Y, Laco GS, Pourquier P, et al. Characterization of a novel topoisomerase I mutation from a camptothecin resistant human prostate cancer cell line. Cancer Res 2001;61:1964–9.
- Staker BL, Hjerrild K, Feese MD, et al. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA 2002;99:15387–92.
- Staker BL, Feese MD, Cushman M, et al. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 2005;48:2336–45.
- Wu CP, Hsieh CH, Wu YS. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm 2011;8:1996–2011.
- Zhu J, Wang R, Lou L, et al. Jatrophane diterpenoids as modulators of P-glycoprotein-dependent multidrug resistance (MDR): advances of structure-activity relationships and discovery of promising MDR reversal agents. J Med Chem 2016;59:6353–69.