
Abstract
Phosphoinositide-dependent protein kinase-1 (PDK1) is an important protein in mediating the PI3K-AKT pathway and is thus identified as a promising target. The catalytic activity of PDK1 is tightly regulated by allosteric modulators, which bind to the PDK1 Interacting Fragment (PIF) pocket of the kinase domain that is topographically distinct from the orthosteric, ATP binding site. Allosteric modulators by attaching to the less conserved PIF-pocket have remarkable advantages such as higher selectivity, less side effect, and lower toxicity. Targeting allosteric PIF-pocket of PDK1 has become the focus of recent attention. In this review, we summarise the current advances in the structure-based discovery of PDK1 allosteric modulators. We will first present the three-dimensional structure of PDK1 and illustrate the allosteric regulatory mechanism of PDK1 through the modulation of the PIF-pocket. Then, the recent advances of PDK1 allosteric modulators targeting the PIF-pocket will be recapitulated detailly according to the structural similarity of allosteric modulators.
1. Introduction
Phosphoinositide-dependent protein kinase-1 (PDK1) is a major regulator of the AGC family of kinases that phosphorylate and activate at least 23 related AGC protein kinasesCitation1, such as p70 S6 kinase (S6K)Citation2,Citation3, p90 ribosomal S6 kinase (RSK)Citation4,Citation5, serum and glucocorticoid-induced protein kinase (SGK)Citation6,Citation7, PKC isoformsCitation8,Citation9, protein kinase C-associated kinase 2 (PRK2)Citation10, and PKB/AKTCitation11–13. They can initiate tumorigenesis through the constitutive activation of kinases induced by oncogenic mutationsCitation14–16. Moreover, PDK1 plays a key role in the PI3K-AKT pathway, which is one of the most common deregulations in human cancersCitation17–20. Thus, selective modulators of PDK1 may have utility as anti-cancer agentsCitation21–24.
PDK1 possesses three ligand binding sites: the substrate binding site, the catalytic ATP binding site, and the PDK1 Interacting Fragment (PIF) binding siteCitation25. The PIF-pocket, which is a hydrophobic surface pocket, has two functions: the recruitment of the downstream substrate kinases harbouring the hydrophobic motif (HM) and the stimulation of the intrinsic activity of PDK1Citation26. Initially, a lot of efforts was devoted to the development of inhibitors targeting the ATP binding site which is relatively conserved in more than 500 protein kinases encoded by the human genomeCitation27–35. However, ATP-competitive inhibitors generally suffer from a low selectivity for PDK1, affecting other protein kinases unsatisfactorily and subsequently leading to potential side effectsCitation36. As an alternative, there is increasingly attractive in the discovery of non-ATP-competitive inhibitors of PDK1 in drug developmentCitation37,Citation38.
A possible strategy to develop non-ATP-competitive inhibitors is the design of selective allosteric modulators of PDK1 through binding to their allosteric sites, which are spatially and topographically distinct from its orthosteric, ATP binding siteCitation39,Citation40. Allostery is a very efficient mechanism which regulates the function of biological macromolecules through the binding of an effector to an allosteric site distinct from the orthosteric, active siteCitation41–47. Compared to orthosteric ligands that bind to highly conserved orthosteric site, allosteric modulators, by targeting less conserved allosteric sites, offer several remarkable advantages such as greater selectivity, fewer side effects, and lower toxicityCitation46,Citation48–50. Therefore, allostery has been a crucial strategy for the development of non-ATP-competitive kinase inhibitorsCitation51,Citation52.
The PIF-pocket, which is distant from the ATP binding site, can act as an allosteric site of PDK1Citation53,Citation54. As a result, molecules directed to the PIF-pocket have much higher selectivity for PDK1 against its homologous kinasesCitation55. In contrast to complete inhibition of enzyme activity by ATP-competitive inhibitors, targeting allosteric sites may endow modulators with different functions such as inhibition or activation of protein kinases. For example, several allosteric modulators bound to the PIF-pocket can act as inhibitors or activators in the regulation of PDK1 functionCitation56. In addition, allosteric modulators do not compete with the ATP-competitive inhibitors. Indeed, they can work together in an individual proteinCitation57. Although the development of allosteric modulators still faces a few challenges such as low binding affinities and poor solubility, it represents a novel strategy for drug discovery due to their remarkable advantagesCitation58–61. In recent years, the discovery of PDK1 allosteric modulators has experienced an upsurge with a significant increase in their number.
In this review, we summarise the current advances in the development of PDK1 allosteric modulators. We will first present the crystal structure of PDK1 and the binding mechanisms of PDK1 allosteric modulators briefly. Then, the recent discovery of PDK1 allosteric modulators targeting the PIF-pocket will be elucidated detailly, which may help the design of PDK1 allosteric modulators with an improved potency and selectivity for the treatment of human diseases.
2. The three-dimensional structure of PDK1
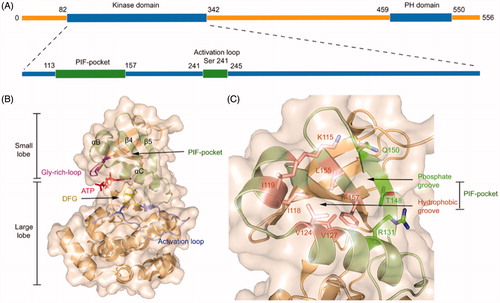
PDK1 is one of the most ancient and conserved protein kinases, which belongs to the AGC protein kinases. Unlike other AGC kinases, the catalytic domain of PDK1 does not possess a HM that is related to autophosphorylationCitation62. The full-length of PDK1 protein contains 556 amino acid residues that comprise an N-terminal serine-threonine kinase domain and a C-terminal pleckstrin homology (PH) domain ()Citation63,Citation64. Within the kinase domain, the polypeptide chain can be further subdivided into two lobes where the C-terminal lobe (C-lobe) is much larger than the N-terminal lobe (N-lobe) ()Citation65. The activation loop (T-loop) is located between the large C-lobe and the helix αC of the N-lobe and positioned after a conserved DFG motif. Notably, phosphorylation of residue Ser241 on the T-loop is strictly required for PDK1 kinase activityCitation66,Citation67. Between the two lobes is the ATP binding site whose roof is formed by the Gly-rich-loopCitation68. In the small N-lobe, the αB and αC helixes and the adjacent β4 and β5 strands form a ∼5 Å deep pocket, named the PIF-pocket, which enables to recognise the phosphorylated HM of substrate kinasesCitation69. The PIF-pocket consists of a separate hydrophobic groove and a positively charged phosphate-binding groove (). Previous studies have shown that the hydrophobic groove rather than the phosphate groove is the determinant for the phosphorylation of substrate kinases by PDK1, and the phosphate groove only serves a key factor in maximising phosphorylation of PDK1 substratesCitation70. In the PIF-pocket, residue Leu155 on the β-sheet 5 is located at the centre and other residues such as Lys115, Ile118, and Ile119 on the αB helixes and Val124 and Val127 on the αC helixes wrap the PIF-pocket. When individual residues Lys115, Ile119, and Glu150 were mutated into Ala, the affinity of each PDK1 mutant to its substrates was reduced. Previous studies have confirmed that residue Leu155 is essential for the ability of PDK1 to interact with the HM of substrates through the PIF-pocket. Interestingly, when Leu155 was mutated by Ala, it increased the activity of PDK1, which may be instructive for the design of allosteric activators of PDK1Citation71.
Figure 1. (A) The general structure of the peptide chain of PDK1. (B) Structural features of the catalytic core of PDK1. The PIF-pocket is shown in pale green, the activation loop in slate, the Gly-rich-loop in magenta, the DFG in yellow, and ATP in red. (C) Structural features of the PIF-pocket of PDK1. The residues that form the phosphate groove are shown in green, while the other residues that form the hydrophobic groove are shown in salmon.
3. Binding mechanisms of PDK1 allosteric modulators
It is well-established that the activity of protein kinases is usually regulated by the phosphorylation of T-loop residues, which triggers conformational changes in their catalytic domain and subsequently facilitates the entry of substrates and ATP to the kinase domainCitation72–74. The optimal activation of AGC kinases family requires both phosphorylation of residues located at the T-loop and HMCitation75,Citation76. In the active state, PDK1 is identified as the primary kinase to phosphorylate T-loop of other AGC kinasesCitation77,Citation78. Except PKB, almost all of the PDK1 substrates must dock their own C-terminal HMs into the PIF-pocket of PDK1 for phosphorylation and activationCitation79–81. In the meanwhile, the resulting docking can enhance the catalytic activity of PDK1 through an allosteric communication from the allosteric PIF-pocket to the orthosteric ATP binding siteCitation82–84.
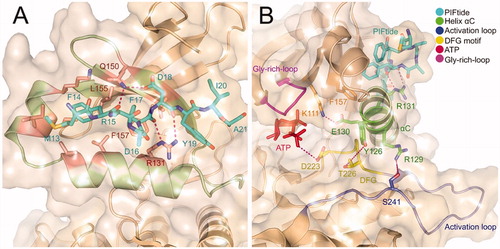
The above-phosphorylated residues that bind to the PIF-pocket can sometimes be replaced by the acidic residues of short peptides such as PIFtideCitation85. PIFtide is a 24-amino acid polypeptide derived from the PDK1 substrate PRK2, which can stimulate PDK1 activity toward a short peptide substrate but disrupt recruitment and phosphorylation of the full-length substrates S6K and SGKCitation86–88. PIFtide is the most effective HM peptide toward PDK1 with a Kd of 4 3 ∼ 90 nM and a 7-fold enhanced activation for PDK1. Sadowsky et al. previously elucidated the orientation of PIFtide in the PIF-pocket of PDK1 and explored the subtle relationship between PIFtide localisation and kinase activation through disulphide trapping approachCitation56. Then, Rettenmaier et al. determined the first high-resolution crystal structure of the PDK1-PIFtide complexCitation57 (). This complex illustrates the interaction of PIFtide with the PIF-pocket and the mechanism by which conformational changes at the PIF-pocket are transmitted to the ATP binding site. There are two obvious interactions in the complex. One is the hydrogen bonding interaction that is formed by the conserved negatively charged residue Asp18 of PIFtide with the residue Gln150 of PDK1 and the other is the salt bridge interaction that is formed by the residue Asp16 of PIFtide with the Arg131 of PDK1. In addition, the side chain aromatic rings of the conserved residues Phe14, Phe17, and Tyr19 of PIFtide occupy three hydrophobic sub-pockets, respectively. Residues Met13, Ile20, and Ala21 of PIFtide protrude the PIF-pocket. The docking of PIFtide to the PIF-pocket not only causes the above conformational changes in the pocket, but these changes are in turn transmitted to the ATP binding site through the critical component of the helix αC (). The interaction of PIFtide with the residue Arg131 causes a slight movement of the helix αC, which further leads to partial changes of residues Arg129, Tyr126, Glu130. The hydrogen bonding interactions between these residues with the residue Ser241 on the T-loop, Thr226 on the DFG motif, and Lys111 adjacent to the Gly-rich-loop conduct the allosteric signal to the ATP binding site. To reveal the relative energetic contribution of each residue from PIFide, alanine scanning mutagenesis method was applied to identify the binding energy hotspots. Five hotspot residues of PIFtide were then identified, including Met13, Phe14, Phe17, Asp18, and Tyr19.
Figure 2. (A) Ribbon representation of the X-ray structure of PDK1-PIFtide complex (PDB ID: 4RRV). The residues that form the PIF-pocket are shown in salmon, while the other residues from PIFtide are shown in cyan. (B) Ribbon representation of the connection of the helix αC with the activation loop, DFG, and Gly-rich-loop. The helix αC and related residues are shown in green, DFG and related residues are shown in yellow, activation loop and related residues are shown in slate, and Gly-rich-loop is shown in magenta.
Since phosphorylation can be mimicked by the docking of PIFtide, it is reasonable that non-peptide small molecules may also mimic the interactions required to trigger corresponding conformational changes. Engel et al. confirmed that small molecules can provide all the necessary requirements for conformational changes, resulting in the enhanced activation of PDK1Citation85. Stroba et al. obtained the first co-crystal of the allosteric small molecule bound to the PIF-pocket of PDK1Citation89. By virtue of fluorescence-based assay and deuterium exchange experiments, Hindie et al. observed the local changes at the PIF-pocket and the conformational changes at the T-loop of the ATP binding site, which are induced by the binding of allosteric modulators at the PIF-pocketCitation90. Further experiments indicated that both the hydrophobic interactions formed between the aromatic rings of allosteric modulators and the PIF-pocket and the ionic interactions formed between the carboxyl group of allosteric modulators and residues Thr148 or Arg131 from the PIF-pocket played critical roles in the binding of allosteric modulators.
PIF-pocket can recruit the downstream substrate kinases harbouring the HM and stimulate the intrinsic activity of PDK1. Therefore, when an allosteric modulator binds to the PIF-pocket, it changes the intrinsic activity of PDK1 by competition with the HM peptide of natural substrate kinases. The previous studies showed that blocking of PIF-pocket was the primary effect by these binding molecules, although such molecules may be allosteric activators of PDK1 in vitro essayCitation55,Citation91. Furthermore, the activation of most downstream kinases of PDK1 requires docking of substrate kinases to the PIF-pocket expect for AKTCitation92. As a result, regardless of whether an allosteric modulator activates or inhibits PDK1 in vitro, a large probability behaves as an inhibitor for the entire signalling pathway in vivo. Here, we only focus on the allosteric modulators targeted to the PIF-pocket with their effects on PDK1 itself in vitro, regardless of their effects on the signalling pathway.
4. Allosteric activators of PDK1
In recent years, a major stride has been made in the discovery of PDK1 allosteric activators. Based on the identified allosteric activators, we classify these molecules into six classes according to their similarity of chemical structures, including diary carboxylic acid derivatives (), benzoazepin-2-one derivatives (), disulphide derivatives (), diaryl sulphonamide derivatives (), benzimidazole derivatives (), and oxypyridine derivatives (). We focus on the detailed PDK1-modulator interactions at the PIF-pocket for the best potent molecule from each class.
Figure 3. Structure and biochemical characterisation of diary carboxylic acid derivatives. AC50: compound concentration required for 50% of maximum activation of PDK1; Amax: maximum activation of PDK1 compared to DMSO control (=100%); T308tide: a synthetic substrate peptide in the radioactivity-based kinase assay that does not interact with the PIF-pocket but could be phosphorylated by PDK1.
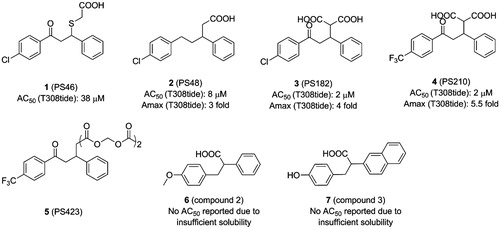
Figure 4. Structure and biochemical characterisation of benzoazepin-2-one derivatives. EC50: compound concentration is required to result in 50% decrease of HTRF signal in a HTRF competition assay, representing the ability of compounds to displace PIFtide peptide; Amax: maximum activation of PDK1 compared to DMSO control (=100%).
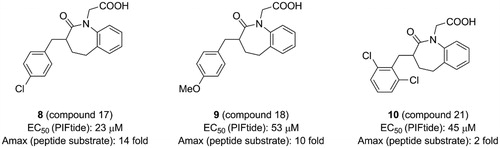
Figure 5. Structure and biochemical characterisation of disulphide derivatives.
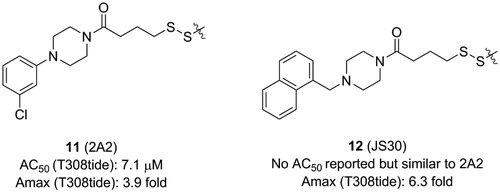
Figure 6. Structure and biochemical characterisation of diaryl sulphonamide derivatives. Kd: the ability of compounds to displace PIFtide in a FP competitive binding assay.
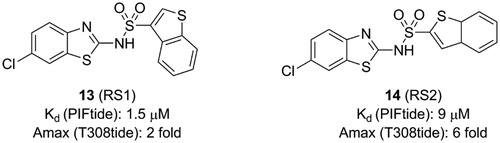
Figure 7. Structure and biochemical characterisation of benzimidazole derivatives. EC50: compound concentration is required to result in 50% displacement of the fluorophore-labeled PIFtide from the PIF-pocket in a FP competitive binding assay.
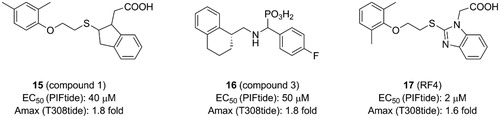
Figure 8. Structure and biochemical characterisation of oxypyridine derivatives. EC50: concentration that resulted in 50% displacement of the PIFtide from the PIF-pocket in an AlphaScreen interaction-displacement assay.
4.1. Diary carboxylic acid derivatives
Based on the pharmacophore model defined by the C-terminal HM of PKA that possesses key residues Phe347 and Phe350, Engel et al. in silico screened a chemical library consisting of 60,000 low-molecular-weight molecules and then tested potential molecules in vitroCitation85,Citation93. Finally, PS46 (1, ) was identified for its high activation level with a low AC50 (AC50=concentrations that gives 50% of maximum activation). Site-directed mutagenesis at the centre of PIF-pocket eradicated the binding of 1 to PDK1, strongly supporting the notion that PIF-pocket is a bona fide target of this molecule. Similarly, 1 disrupted the interaction between PDK1 and PIFtide in a surface plasmon resonance (SPR) assay. Consistent with above phenomena, the phosphorylation of substrates S6K and SGK by PDK1, which are required to dock into the PIF-pocket of PDK1, was blocked by 1. Most importantly, when residue Arg131 was mutated by Met or Ala to lose its positive charge, the catalytic activity of PDK1 mutant could not be increased by 1. But when Arg131 was mutated by Lys to retain its positive charge, the catalytic activity of PDK1 mutant was not affected. Moreover, when the carboxyl group of 1 was converted to an ester group, the catalytic activity of PDK1 showed no change. Further computational docking revealed that the salt bridge interaction between the carboxylic acid group of 1 and the residue Arg131 of PIF-pocket is essential for the allosteric activation of PDK1. Finally, the selectivity of this molecule was proven to be excellent by testing on other AGC kinases. Taken together, 1 is the first reported small molecule which can enhance the catalytic activity of PDK1 by targeting the allosteric PIF-pocket.
1 contains a chiral centre which requires to evaluate the contribution of each enantiomer to the total activity. The sulphide group also has potential oxidisation and is prone to arise retro-Michael reactions. By replacing the chiral centre with a double bond and retaining the three hybridised C-atoms in the chain to connect the two benzene rings, Stroba et al. synthesised PS48 (2, ) that showed a 4-fold lower AC50 than 1Citation89. Importantly, the crystal structure of 2 bound to PDK1 was determined. The comparison between the cocrystal structure of PDK1-2 complex with the structure of PDK1 alone reveals details of the allosteric mechanism by 2 (). The PDK1-2 complex shows that the carboxylic acid group of 2 interacts with residues Arg131 and Thr148 of PDK1 through salt bridge and hydrogen bonding interactions and with residue Gln150 via water-mediated hydrogen bonding interaction. Residue Leu155 separates the hydrophobic groove into two sub-pockets that are occupied by the two benzene rings of 2. Especially, the right benzene ring forms edge-to-face CH-π interactions with residue Phe157 and the chlorine atom of the benzene ring on the left forms a halogen bond with residue Lys115. In addition, the residues Arg131, Glin150, and Phe157 rotate about 90° and the rotation of residue Lys115 reaches 180°. The resulting movement of these residues enlarges the depth of the PIF-pocket and stabilises the helix αC. The stabilisation of the helix αC limits the conformational shift of the T-loop, Gly-rich-loop, and DFG motif, thereby increasing the rigidity of the ATP binding site. Furthermore, Hindie et al. verified the molecular mechanism of allosteric changes induced by 2 through fluorescence-based assay and deuterium exchange experimentsCitation90. Overall, 2 binding caused local changes in the PIF-pocket, which allosterically triggered conformational changes at the T-loop and the ATP binding site. Through analysis of structure-activity relationships (SAR) of 2, the minimal structural requirements for an allosteric modulator toward PDK1 were proposed: two aromatic groups linked by an aliphatic chain, a side chain with a free carboxyl group, and a V-shaped overall conformation of the aryl rings.
Figure 9. (A) Comparison of the X-ray structure of unbound PDK1 (pink; PDB ID: 3HRC) with the X-ray structure of PDK1-2 complex (pale yellow; PDB ID: 3HRF). (B) Comparison of the X-ray structure of PDK1-2 complex with the X-ray structure of PDK1-3 complex (slate; PDB ID: 4AW0). (C) Comparison of the X-ray structure of PDK1-3 with the X-ray structure of PDK1-4 complex (salmon; PDB ID: 4AW1).
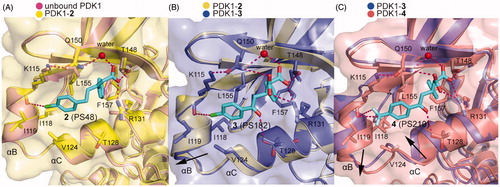
To further improve potency of 2, Wilhelm et al. optimised this activator by retaining the ketone group of 1 and introducing the shorter carboxyl side chain from 2Citation55, because both the keto group and the carboxyl group play a key role in the binding affinity of allosteric activators by offering additional ionic interactions. Thus, the two carboxyl groups were introduced in the optimisation, yielding PS182 (3, ) with an AC50 value of 2.5 μM and a 4-fold enhanced activation for PDK1. Although there was an improvement of affinity through a dicarboxyl moiety, optimisation of the ring substituents may achieve a better result. Obviously, this conjecture is true for the identification of PS210 (4, ) where the chlorine moiety in the benzene ring of 3 was replaced by a trifluoromethyl group. It showed that 4, with an AC50 value of 2 μM, displayed a 5.5-fold enhanced activation for PDK1. Notably, both 3 and 4 increased the thermal stability of PDK1 whereas 2 had almost no effect measured by differential scanning fluorimetry (DSF) assays.
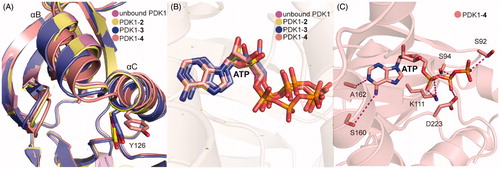
Both 3 and 4 were co-crystallized by soaking each molecule with PDK1 crystal. As shown in , the overall binding mode of PDK1-3 complex was very similar to that of PDK1-2 complex. Two hydrophobic sub-pockets are also occupied by the benzene rings of 3, and the CH-π interactions between the right benzene ring with residue Phe157 are remained. However, the second carboxyl group of 3 forms an additional salt bridge with residue Arg131 of the PIF-pocket, and directly interacts with residue Gln150. It is also noteworthy for the interaction between the residue Thr128 of PDK1 and the keto group of 3. Deep in the pocket, 3 causes a downward movement of the helix αB, which can drive the change of the Gly-rich-loop of the ATP binding site. This conformational change leads to reshape the phosphate groups of ATP. As shown in , aside from the above interactions, the trifluoromethyl substituent of 4 forms additional hydrophobic interactions with the residues of helix αB such as Val124, Ile118 and Ile119. These interactions cause the helix αB to move further by 1.1 Å in a direction perpendicular to the first motion. Comparing the helix αB and the helix αC in the crystal structures of PDK1-2, PDK1-3, PDK1-4, and unbound PDK1, it can be found that the helix αB gradually moves down with the increase of PDK1 activation factor, but the displacement of the helix αC seems to be irregular (). The allosteric signal is transmitted to the ATP binding site through the Gly-rich loop. Further comparison of ATP structure in the four structures shows that the β, γ-phosphates of ATP in the PDK1-4 complex rotated largely (). The resulting movement increases the interaction of ATP with the residues Asp223, Lys111 and Ser94 (). Overall, the affinity of allosteric activators is proportional to the number of contacts that are formed by activators with the PIF-pocket. Additionally, the substituents on the left benzene ring have a further optimised space to exert more influence on the helix αB.
Figure 10. (A) Comparison of the helix αB and helix αC of unbound PDK1 (magenta), PDK1-2 complex (pale yellow), PDK1-3 complex (slate), PDK1-4 complex (salmon). (B) Comparison of the ATP of unbound PDK1 (magenta), PDK1-2 complex (pale yellow), PDK1-3 complex (slate), PDK1-4 complex (salmon). (C) Cartoon/surface representations of the interaction between ATP with the ATP binding site of PDK1-4 complex.
The diary carboxylic acid derivatives have poor permeability due to the negative charge of the carboxyl group. To improve the permeability and explore the pharmacologic effects of these modulators more efficiently, carboxyl groups were replaced by ester groups, yielding PS423 (5, ). This molecule could be cleaved to produce the homologous diary carboxylic acid derivatives by cellular esterases and was identified as a prodrug of 4 that can readily permeate cell membrane. Further experiments revealed that 5 did not affect the activity of PDK1 in vitro, which once again confirmed the necessity of carboxylic acid groups for the activation of PDK1.
The fragment screening method is an orthogonal complement of high throughput screening for searching ligands for biomacromolecules. Since fragments are limited to low molecular weights, the efficient sampling of chemical space with ∼10 K fragments or even less can be accomplished. Moreover, the screened fragments are usually hydrophilic and can be easily chemically modified, which is more suitable as a starting point for drug development. Stockman et al. developed an NMR fragment screening approach for identifying small molecules binding to both the ATP and the PIF-pocket of PDK1Citation94. Then, by means of saturation transfer difference (STD) NMR experiments, the selected molecules can be distinguished between binding at the ATP binding site and at the allosteric PIF-pocket. Through screening, a fragment library of 10,000 diverse molecules, compounds 2 (6, ) and 3 (7, ) bound to the allosteric PIF-pocket were obtained. The two molecules were inactive at 313 μM in the Kinase-Glo assay, but they did not show any inhibition of PDK1. However, they showed 33 ± 13% and 30 ± 12% activation using the Calliper assay (a kinase activity assay through measuring the incorporation of a phosphate into a fluorescent tagged substrate peptide), respectively, compared to the control reaction at the 313 μM. However, it is difficult to determine AC50 values of both molecules owing to their poor solubility. Interestingly, the chemical structures of two molecules resemble diary carboxylic acid derivatives. All of them share one negatively charged carboxyl group and two hydrophobic benzene rings. The difference is that the length of the linker connecting the two aromatic rings is different. The flexibility of the longer carbon chain may explain the increased activity of 1 compared to both 6 and 7. However, more specific allosteric details require co-crystallization of both 6 and 7 with PDK1.
4.2. Benzoazepin-2-one derivatives
Wei et al. analysed the above allosteric activators of PDK1 acquired by virtual screening and NMR-based fragment screeningCitation95. All structures of these molecules contain a negatively charged carboxylate and two aromatic hydrophobic groups. The carboxylate mimics the phosphate group of the substrate kinases and the effect of the two benzene rings are very similar to the phenylalanine residues of the HM. Based on this evidence, a suite of benzoazepin-2-one derivatives targeting the PIF-pocket of PDK1 were designed de novo through virtual docking. After in silico evaluation and further SAR exploration by using 1 as the positive control with an EC50 value of ∼133 μM, three excellent molecules, compound 17 (8, ) with an EC50 value of ∼23 μM, compound 18 (9, ) with an EC50 value of ∼53 μM, and compound 21 (10, ) with an EC50 value of ∼45 μM, were obtained.
Benzoazepin-2-one derivatives were designed using diary carboxylic acid derivatives as the template, so there are many similarities of chemical structures between them such as two benzene rings and one carboxylic acid. The difference is that the original hydrophobic chain in the diphenylpropionic acid derivatives is replaced by the benzoazepin-2-one scaffold. This substitution is beneficial for binding. First, the rigid benzodiazepine replaces the flexible hydrophobic chain to aid in the orientation of the benzene ring and the carboxylic acid. Then, the extra ketone group was designed, supposing that it may form an additional interaction with the residue Thr128 of PIF-pocket such as 4. These could explain the higher affinity of 8, 9 and 10 compared to 1. Unexpectedly, 9 and 10 have the similar affinity, but show different levels of PDK1 activation. However, the co-crystal structures of these small molecules with PDK1 are still unavailable and the detailed mechanisms are unclear. Further research of the activation mechanism by benzoazepin-2-one derivatives is required to ascertain.
4.3. Disulphide derivatives
Sadowsky et al. developed a disulphide trapping method that allows small molecules to interrogate allosteric sites of protein kinases and subsequently assesses the effects of binding on protein structure and functionCitation56,Citation96. Initially, they prepared six PDK1 Cys mutants in which residues at six positions around the PIF-pocket were mutated by a cysteine (K115C, I119C, V124C, R131C, T148C, and Q150C). A small library consisting of 480 disulphide fragments was screened and each mutant trapped a series of distinct disulphide fragments which showed a good selectivity.
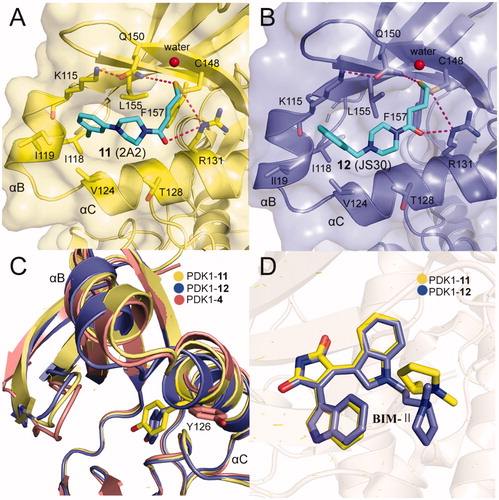
The best fragment is 2A2 (11, ) with an EC50 value of ∼7.1 μM, which is special to the mutant PDK1T148C and enhanced the activation of PDK1T148C by about 3.9-fold. Optimisation of 11 resulted in JS30 (12, ), which achieved a 6.3-fold enhanced activation of PDK1T148C. The activation of PDK1 by 12 was slightly higher than the previously best molecule 4 which activated PDK1 by 5.5-fold. However, the activated ability of the three molecules toward PDK1 is different, so comparison of their cocrystal complexes is useful in the exploration of the mechanism of allosteric activation ( and ). The binding modes of the two disulphide molecules 11 and 12 at the PIF-pocket are similar to that of 4. The remarkable difference is that the sulphur atom of 11 and 12 replaces the carboxylic acid group of 4 to form interactions with residues Arg131, Cys148, and Gln150. Especially, the disulphide bond formed by 11 and 12 with the mutated residue Cys148 is stronger than the corresponding hydrogen bond in the PDK1-4 complex. This may be why 11 and 12 have a reasonable affinity for the PIF-pocket. In addition, since there were no benzene rings at the right end of 11 and 12, the conformation of the critical residue Phe157 at the PIF-pocket does not undergo changes. Comparison of the conformational changes of the PIF-pocket shows that the helix αB has moved downward and the pyrrole ring of ATP-competitive ligand bis-mercaptomaleimide-II (BIM-II) has rotated about 90°in the 12-bound PDK1 compared to the 11-bound PDK1 ( and ). This is consistent with the diary carboxylic acid derivatives by which the downward movement of the helix αB is positively correlated with the protein activity.
Figure 11. (A) Cartoon/surface representations of the interaction between 12 with the PIF-pocket of PDK1 (pale yellow; PDB ID: 3ORZ). (B) Cartoon/surface representations of the interaction between 12 with the PIF-pocket of PDK1 (slate; PDB ID: 3OTU). (C) Comparison of the helix αB and helix αC of PDK1-11 complex, PDK1-12 complex and PDK1-4 complex (salmon). (D) Comparison of the BIM-IIof PDK1-11 complex and PDK1-12 complex.
4.4. Diaryl sulphonamide derivatives
Different from the covalent allosteric activators acquired through the disulphide trapping method, Rettenmaier et al. identified the noncovalent allosteric activators targeting the PIF-pocket of PDK1 by the site-directed chemical screenCitation57,Citation97. Fluorescence polarisation (FP) competitive binding assay was used to identify molecules that can disrupt the interaction between PIF-pocket and PIFtide. Initially, 154,000 molecules were screened at a single dose through the FP assay. Then, 1280 molecules in dose-response mode were further selected for testing using the FP and SPR assays. The top 10 hits were repurchased for kinase activity assay, and a diaryl sulphonamide derivative with the most potent activity was identified. To further optimise this parent molecule, a series of iterative synthesis was conducted. Testing by FP, SPR, and kinase activity assays, the regioisomers RS1 (13, ) and RS2 (14, ) with a Kd of ∼1.5 and ∼9 μM were obtained, respectively. Moreover, 13 and 14 stimulated PDK1 activity toward a short peptide substrate by 2-fold and 6-fold, respectively.
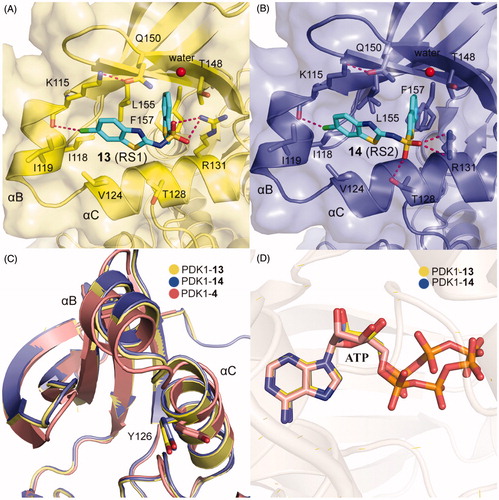
Compared with diary carboxylic acid derivatives, 13 and 14 also had excellently biochemical characterisation, but the chemical structures of 13 and 14 were different from that of diary carboxylic acid derivatives. First of all, the benzene rings at both ends are replaced by the benzo heterocycles. With reference to previous benzoazepin-2-one derivatives, this is expected to increase the rigidity of the entire molecule and thereby aids in the orientation of the functional groups. Then, the substitution of a sulphonamide group with a carboxylic acid group greatly improves the cell permeability of this molecule. In the experiment, 13 can diffuse freely into the cells, which is very helpful for the drug-forming properties of the molecule. The two molecules have performed well on the affinity of the PIF-pocket. However, after obtaining the co-crystal structures of these two molecules with PDK1, it is found that the two molecules still have a lot of space for improvement ( and ). The sulphur atom on the thiophene at the right end was originally expected to possess an equivalent property to 12, which could interact with residue Thr148. Therefore, if the sulphur atom on the five-membered ring can be transferred to the six-membered ring, the affinity may be optimised. In addition, in the modification of 3 to 4, the chlorine atom at the left end can be attempted to be replaced by the trifluoromethyl group. When comparing the binding modes of 13 and 14, 14 has an extra hydrogen bonding interaction with the residue Thr128 at the helix αC, which may play a role in the more activation of PDK1 by 14 than 13. Finally, the conformational changes of the helix B at the allosteric PIF-pocket and the ATP molecule induced by both 13 and 14 ( and ) are similar to those by 11 and 12 ( and ).
Figure 12. (A) Cartoon/surface representations of the interaction between 13 with the PIF-pocket of PDK1 (pale yellow; PDB ID: 4RQK). (B) Cartoon/surface representations of the interaction between 14 with the PIF-pocket of PDK1 (slate; PDB ID: 4RQV). (C) Comparison of the helix αB and helix αC of PDK1-13 complex, PDK1-14 complex and PDK1-4 complex (salmon). (D) Comparison of the ATP of PDK1-13 complex, PDK1-14 complex, and PDK1-4 complex.
4.5. Benzimidazole derivatives
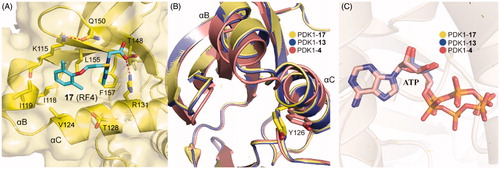
Rettenmaier et al. had tried to use a structure-based virtual screening which preformed against both crystal structures and comparative models to identify ligands bound to the PIF-pocket of PDK1Citation98. Based on the crystal structure of PDK1-2 complex, a group of six structural models of the PIF-pocket were created. A chemical library of 6,300 property-matched molecules which are commercially available was generated. These molecules were docked against all six structural models for virtual screening. Then the selected molecules were tested by FP competitive binding assay to identify whether the hits were bound to the PIF-pocket of PDK1. Compound 1 (15, ) with an EC50 value of ∼40 μM and compound 3 (16, ) with an EC50 value of ∼50 μM were identified, respectively. Furthermore, in order to optimise these two molecules, 518 commercially available analogues were extracted from the ZINC database by means of analogue-by-catalogue searching. Finally, 15 analogues were selected based on the scoring equal to or even better the two parent molecules. Among these molecules, RF4 (17, ) is the most potent compound with an EC50 value of ∼2 μM.
The requirements for virtual screening are based on existing models, including the formation of two hydrogen bonds with residues Arg131 and Thr148 and occupying the hydrophobic sub-pockets. It is noteworthy that the 2,6-dimethyl substituted benzene ring of 17 packed tightly into the hydrophobic sub-pocket lined by residues Ile119 and Leu155 of PDK1 (), indicating that the 2,4-dimethyl group of 15 was not perfect spatially. This is beneficial for the optimisation of the left benzene ring of 4 or 12. In addition, 13 and 17 have approximate activation effectiveness, which are quite different from 4. This phenomenon is consistent with the conformational changes of the helix αB, helix αC and ATP binding site ( and ).
Figure 13. (A) Cartoon/surface representations of the interaction between 17 with the PIF-pocket of PDK1 (pale yellow; PDB ID: 4XX9). (B) Comparison of the helix αB and helix αC of PDK1-17 complex, PDK1-13 complex (slate) and PDK1-4 complex (salmon). (C) Comparison of the ATP of PDK1-17 complex, PDK1-13 complex, and PDK1-4 complex.
4.6. Oxypyridine derivatives
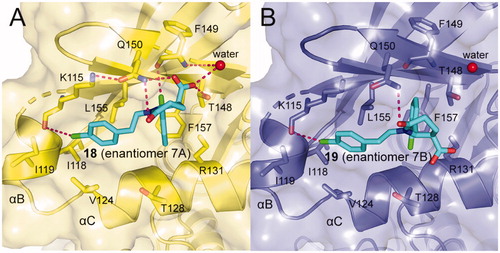
ANCHOR.QUERY is a method that efficiently allows to obtain active molecules from the known molecule by combination of computation and synthesisCitation99. The molecules acquired through this approach can be synthesised in one step by multicomponent reaction (MCR) chemistry. Based on above advantages, Kroon et al. applied ANCHOR.QUERY approach for the new scaffolds that bind to the allosteric PIF-pocket of PDK1Citation100. The starting point of their investigation was based on the cocrystal structure of PDK1-2 complex. Through virtual screening and following SAR research, the racemic compound 7 was synthesised. The ability of the racemic to disrupt the PDK1-PIFtide interaction was different, with IC50 values of 7.0 μΜ for enantiomer 7 A (18, ) and 15 μM for enantiomer 7B (19, ), respectively. In both molecules, the 2-chlorophenyl substituent that acts as an anchor is located in the deep PIF-pocket, but shows a different orientation ( and ). In the structure of 18-bound PDK1, the 2-chlorophenyl substituent forms a short contact with the residue Phe149, while in the structure of 19-bound PDK1 it turns about 180°, with no contacts with Phe149. In addition, the extra interactions formed by the carboxylic acid group and the amide group of 18 can explain the higher affinity of 18 than 19.
Figure 14. (A) Cartoon/surface representations of the interaction between 18 with the PIF-pocket of PDK1 (pale yellow; PDB ID: 5ACK). (B) Cartoon/surface representations of the interaction between 19 with the PIF-pocket of PDK1 (slate; PDB ID: 5ACK).
5. Allosteric inhibitors of PDK1
Although past research has focussed on the development of allosteric activators, a few allosteric inhibitors have also been discovered. We try to explain the inhibition mechanism to better understand the activation mechanism.
5.1. Disulphide derivatives
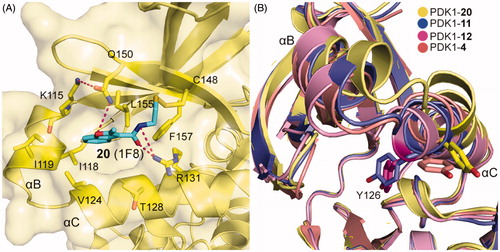
By means of the disulphide trapping method, Sadowsky et al. also unexpectedly discovered an allosteric inhibitor of PDK1, 1F8 (20, ), which inhibited the activation of PDK1T148C by 68% and with an EC50 value of ∼7.2 μM. Like other disulphide derivatives such as 11 and 12, the affinity of 20 toward the PIF-pocket also primarily relies on the disulphide bond formed with the mutated residue Cys148 (). The difference is that the hydrogen bonding interactions of 20 with residues Arg131 and Gln150 are established by the amide group and the oxygen atom on the furan ring, respectively. The allosteric inhibition mechanism of 20 can be inferred by the displacements of helices αB and αC (). From 12, 4, 11 to 20, the activity of PDK1 bound to these allosteric small molecules were 630%, 550%, 390%, and 32%. In this process, the position of the helix αB rises in turn. Unfortunately, the obvious rule was not observed between the position of the key helix αC and kinase activity. But the residue Tyr126 on the helix αC gradually moves away from the ATP binding site as the PDK1 activity decreases. It is generally speculated that the allosteric inhibition of PDK1 is caused by the helices αB and αC being away from the active site.
Figure 15. Structure and biochemical characterisation of the allosteric inhibitors directed to the PIF-pocket of PDK1. EC50 (T308tide): compound concentration is required for 50% of maximum effect. EC50 (PDKtide): compound concentration is required to result in 50% displacement of the PDKtide from the PIF-pocket or ATP site in an AlphaScreen interaction-displacement assay.
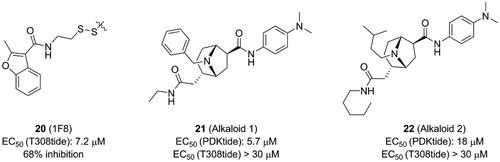
Figure 16. (A) Cartoon/surface representations of the interaction between 20 with the PIF-pocket of PDK1 (pale yellow; PDB ID: 3ORX). (B) Comparison of the helix αB and helix αC of PDK1-20 complex (pale yellow), PDK1-11 complex (slate), PDK1-12 complex (pink) and PDK1-4 complex (salmon).
5.2. Alkaloid derivatives
Bisubstrate ligand approach, characterised by design of bisubstrate analogues binding to two ligand-binding sites, is promising for developing efficient modulators of protein kinases with multiple binding sitesCitation101,Citation102. The three ligand-binding sites possessed by PDK1 were known according to previous studies. Based on the above evidence, Bobkova et al. developed a sensitive ultrahigh throughput enzymatic assay to identify modulators of PDK1Citation103. They designed a biotinylated fusion peptide that could bind to both ATP binding site and PIF-pocket. This biotinylated fusion peptide was used as the substrate for phosphorylation by PDK1, which allowed them to identify ATP-competitive inhibitors and allosteric modulators of PIF-pocket. After screening over a million of molecules library, the specificity of molecules binding to PIF-pocket was evaluated by thermal shift binding assay and AKT-tide enzymatic assay. Then, alkaloid 1 (21, ) with an EC50 value of ∼5.7 μM and alkaloid 2 (22, ) with an EC50 value of ∼18 μM were identified, respectively. They exhibited remarkable selectivity against other kinases, but the affinities were very low and showed unmeasurable effect by the AKT-tide enzymatic assay.
To explore the possible interaction of alkaloid derivatives with PDK1, 21 was docked into the PIF-pocket of PDK1. The binding mode indicated that the interactions were dominated by hydrophobic interactions between the 21 and the PIF-pocket of PDK1. The dimethylphenylamine group of 21 occupied a hydrophobic pocket outlined by residues Val127 and Phe157. Additionally, the benzene ring occupied an adjacent hydrophobic site formed by residues Ile118, Ile119, Val124, Val127, and Leu155. However, 21 does not contain a carboxyl moiety, thus it is incapable to form the ionic interaction with residue Arg131 from the PIF-pocket and to further induce conformational changes resembled other allosteric activators of PDK1. The different interactions between alkaloid derivatives with hydrophobic groove may be responsible for the inhibition of PDK1 by such small molecules. Obviously, although 21 and 20 are both allosteric inhibitors of PDK1, their inhibitory mechanisms at the PIF-pocket may be different. Further studies of the allosteric inhibition mechanism of PDK1 by 21 require co-crystallization of this molecule with PDK1.
6. Conclusions
The PIF-pocket was initially found to recruit and phosphorylate downstream kinases, and further studies showed that the HM peptides of downstream kinases bound to the PIF-pocket allosterically stimulated the intrinsic activity of PDK1. Therefore, allosteric modulators can be designed to mimic the HM peptide of downstream kinases by attached to the PIF-pocket. To date, seven classes of PDK1 allosteric modulators have been reported based on the similarity of chemical structure, including diary carboxylic acid derivatives, benzoazepin-2-one derivatives, disulphide derivatives, diaryl sulphonamide derivatives, benzimidazole derivatives, oxypyridine derivatives, and alkaloid derivatives. The vast majority of modulators are allosteric activators that activate PDK1 catalytic activity when they are bound to the PIF-pocket, whereas the allosteric inhibitors can also be observed such as 20 that inhibited PDK1 activity. The binding of allosteric activators to the PIF-pocket can stabilise the closure between the PIF-pocket and the leaf, which further stabilised the DFG motif, the activation loop, and the ATP binding site, leading to the stabilisation of catalytic PDK1 conformation. However, allosteric inhibitors binding to the PIF-pocket caused the realignment of helices αB and αC in the inactive conformation, leading to the inhibition of PDK1 activity. Among the reported PDK1 allosteric modulators, 4, 13, and 17 showed good binding affinities to the PDK1. Comparison of the three compounds reveals that their interactions with the PIF-pocket are similar. They have two aromatic rings at a distance from each other that occupy the hydrophobic part of the PIF-pocket and are engaged in hydrophobic interactions. There is a fatty chain linking the two aromatic rings that provides a flexible effect. The carbon on the fatty chain can be substituted with an acyl group that can form hydrogen bonds with the residues from the PIF-pocket. Therefore, it is feasible that the future design of PDK1 allosteric modulators may retain the carboxylic acid moiety, the two aromatic rings, and the fatty chain.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Mora A, Komander D, Van Aalten DMF, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 2004;15:161–70.
- Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 1999;253:100–9.
- Bayascas JR, Leslie NR, Parsons R, et al. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/-) mice. Curr Biol 2005;15:1839–46.
- Frodin M. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J 2000;19:2924–34.
- Jensen CJ, Buch MB, Krag TO, et al. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J Biol Chem 1999;274:27168–76.
- Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J 1999;339:319–28.
- Park J, Leong MLL, Buse P, et al. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J 1999;18:3024–33.
- Balendran A, Biondi RM, Cheung PCF, et al. A 3-phosphoinositide-dependent protein kinase-1 (PDK1) docking site is required for the phosphorylation of protein kinase Cζ (PKCζ) and PKC- related kinase 2 by PDK1. J Biol Chem 2000;275:20806–13.
- Frey U, Morris RGM, Good JAL, et al. Protein kinase C Isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998;281:2042–5.
- Flynn P, Mellor H, Casamassima A, Parker PJP. Rho GTPase control of protein kinase C-related protein kinase activation by 3-phosphoinositide-dependent protein kinase. J Biol Chem 2000;275:11064–70.
- Bauer AF, Sonzogni S, Meyer L, et al. Regulation of protein kinase C-related Protein Kinase 2 (PRK2) by an intermolecular PRK2-PRK2 interaction mediated by its N-terminal domain. J Biol Chem 2012;287:20590–602.
- Manning B, Cantley L. AKT/PKB signaling: navigating downstream. Cell 2007;129:1261–74.
- Stephens L, Anderson K, Stokoe D, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 1998;279:710–4.
- Flynn P, Wong M, Zavar M, et al. Inhibition of PDK-1 activity causes a reduction in cell proliferation and survival. Curr Biol 2000;10:1439–42.
- Manuscript A, Magnitude S. Analysis of 3-phosphoinositide-dependent kinase-1 signaling and function in ES cells. Exp Cell Res 2008;31:1713–23.
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010;11:9–22.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005;4:988–1004.
- Manuscript A, Magnitude S. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008;31:1713–23.
- Ni D, Liu D, Zhang J, Lu S. Computational insights into the interactions between calmodulin and the c/nSH2 domain of p85α regulatory subunit of PI3Kα: implication for PI3Kα activation by calmodulin. Int J Mol Sci 2018;19:151.
- Schmitt DL, Sundaram A, Jeon M, et al. Spatial alterations of de novo purine biosynthetic enzymes by Akt-independent PDK1 signaling pathways. PLoS One 2018;13:1–15.
- Li W, Qian L, Lin J, et al. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget 2017;8:65143–51.
- Peng F, Wang JH, Fan WJ, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018;37:1062–74.
- Wang F, Shan S, Huo Y, et al. MiR-155-5p inhibits PDK1 and promotes autophagy via the mTOR pathway in cervical cancer. Int J Biochem Cell Biol 2018;99:91–9.
- Maurer M, Su T, Saal LH, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res 2009;69:6299–307.
- Biondi RM. Phosphoinositide-dependent protein kinase 1, a sensor of protein conformation. Trends Biochem Sci 2004;29:136–42.
- Lopez-Garcia LA, Schulze JO, Fröhner W, et al. Allosteric regulation of protein kinase PKCζ by the N-terminal C1 domain and small compounds to the PIF-pocket. Chem Biol 2011;18:1463–73.
- Gassel M, Breitenlechner CB, Rüger P, et al. Mutants of protein kinase A that mimic the ATP-binding site of protein kinase B (AKT). J Mol Biol 2003;329:1021–34.
- Kim KH, Wissner A, Floyd MB, et al. Benzo[c][2,7]naphthyridines as inhibitors of PDK-1. Bioorg Med Chem Lett 2009;19:5225–8.
- Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. Chem Med Chem 2008;3:1810–38.
- Angiolini M, Banfi P, Casale E, et al. Structure-based optimization of potent PDK1 inhibitors. Bioorganic Med Chem Lett 2010;20:4095–9.
- Erlanson DA, Arndt JW, Cancilla MT, et al. Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg Med Chem Lett 2011;21:3078–83.
- Lee ACH, Ramanujulu PM, Poulsen A, et al. Thieno[3,2-d]pyrimidin-4(3H)-one derivatives as PDK1 inhibitors discovered by fragment-based screening. Bioorganic Med Chem Lett 2012;22:4023–7.
- Wucherer-Plietker M, Merkul E, Müller TJJ, et al. Discovery of novel 7-azaindoles as PDK1 inhibitors. Bioorg Med Chem Lett 2016;26:3073–80.
- Chen T, Sorna V, Choi S, et al. Fragment-based design, synthesis, biological evaluation, and SAR of 1H-benzo[d]imidazol-2-yl)-1H-indazol derivatives as potent PDK1 inhibitors. Bioorganic Med Chem Lett 2017;27:5473–80.
- O’Brien NJ, Brzozowski M, Wilson DJD, et al. Synthesis and biological evaluation of substituted 3-anilino-quinolin-2(1H)-ones as PDK1 inhibitors. Bioorganic Med Chem 2014;22:3781–90.
- Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discov Today 2007;12:622–33.
- Kirkland LO, McInnes C. Non-ATP competitive protein kinase inhibitors as anti-tumor therapeutics. Biochem Pharmacol 2009;77:1561–71.
- Kiselyov A, Balakin KV, Tkachenko SE, Savchuk NP. Recent progress in development of non-ATP competitive small-molecule inhibitors of protein kinases. Mini Rev Med Chem 2006;6:711–7.
- Yang J, Cron P, Thompson V, et al. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell 2002;9:1227–40.
- Nagashima K, Shumway SD, Sathyanarayanan S, et al. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric kinase inhibitor. J Biol Chem 2011;286:6433–48.
- Lu S, Zhang J. Designed covalent allosteric modulators: an emerging paradigm in drug discovery. Drug Discov Today 2017;22:447–53.
- Lu S, Huang W, Zhang J. Recent computational advances in the identification of allosteric sites in proteins. Drug Discov Today 2014;19:1595–600.
- Lu S, Li S, Zhang J. Harnessing allostery: a novel approach to drug discovery. Med Res Rev 2014;34:1242–85.
- Lu S, Ji M, Ni D, Zhang J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov Today 2018;23:359–65.
- Huang M, Song K, Liu X, et al. AlloFinder: a strategy for allosteric modulator discovery and allosterome analyses. Nucleic Acids Res 2018;46:W451–8.
- Lu S, Jang H, Muratcioglu S, et al. Ras conformational ensembles, allostery, and signaling. Chem Rev 2016;116:6607–65.
- Lu S, Jang H, Gu S, et al. Drugging Ras GTPase: a comprehensive mechanistic and signaling structural view. Chem Soc Rev 2016;45:4929–52.
- Nussinov R, Tsai CJ. Allostery in disease and in drug discovery. Cell 2013;153:293–305.
- Fang Z, Grütter C, Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem Biol 2013;8:58–70.
- Lu S, Zhang J. Small molecule allosteric modulators of G-protein-coupled receptors: drug–target interactions. J Med Chem 2018; DOI: 10.1021/acs.jmedchem.7b01844.
- Garuti L, Roberti M, Bottegoni G. Non-ATP competitive protein kinase inhibitors. Curr Med Chem 2010;17:2804–21.
- Changeux JP, Christopoulos A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell 2017;19:4–21.
- Schulze JO, Saladino G, Busschots K, et al. Bidirectional allosteric communication between the ATP-binding site and the regulatory PIF pocket in PDK1 protein kinase. Cell Chem Biol 2016;23:1193–205.
- Fr W, Lopez-garcia LA, Neimanis S, et al. 4-Benzimidazolyl-3-phenylbutanoic acids as novel pif-pocket-targeting allosteric inhibitors of protein kinase PKC ζ. J Med Chem 2011;54:6714–23.
- Wilhelm A, Lopez-Garcia LA, Busschots K, et al. 2-(3-oxo-1,3-diphenylpropyl)malonic acids as potent allosteric ligands of the PIF pocket of phosphoinositide-dependent kinase-1: development and prodrug concept. J Med Chem 2012;55:9817–30.
- Sadowsky JD, Burlingame MA, Wolan DW, et al. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc Natl Acad Sci USA 2011;108:6056–61.
- Rettenmaier TJ, Sadowsky JD, Thomsen ND, et al. A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1. Proc Natl Acad Sci 2014;111:18590–5.
- Wenthur CJ, Gentry PR, Mathews TP, Lindsley CW. Drugs for allosteric sites on receptors cody. Annu Rev Pharmacol Toxicol 2014;54:165–84.
- Ni D, Song K, Zhang J, Lu S. Molecular dynamics simulations and dynamic network analysis reveal the allosteric unbinding of monobody to H-Ras triggered by R135K mutation. Int J Mol Sci 2017;18:2249.
- Wylie AA, Schoepfer J, Jahnke W, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017;543:733–7.
- Urwyler S. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol Rev 2011;63:59–126.
- Gagliardi PA, di Blasio L, Primo L. PDK1: a signaling hub for cell migration and tumor invasion. Biochim Biophys Acta Rev Cancer 2015;1856:178–88.
- Di Blasio L, Gagliardi PA, Puliafito A, Primo L. Serine/threonine kinase 3-phosphoinositide-dependent protein kinase-1 (PDK1) as a key regulator of cell migration and cancer dissemination. Cancers (Basel) 2017;9:1–17.
- Y MD, Casamayor A, Currie RA, et al. Characterisation of a plant 3-phosphoinositide-dependent protein kinase-1 homologue which contains a pleckstrin homology domain. FEBS Lett 1999;451:220–6.
- Alessi DR, Deak M, Casamayor A, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 1997;7:776–89.
- Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell 2000;103:185–8.
- Casamayor A, Morrice NA, Alessi DR. Phosphorylation of ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem J 1999;292:287–92.
- Gagliardi PA, Puliafito A, Primo L. PDK1: at the crossroad of cancer signaling pathways. Semin Cancer Biol 2018;48:27–35.
- Biondi RM, Komander D, Thomas CC, et al. High resolution crystal structure of the human PDK1 catalytic domain de® nes the regulatory phosphopeptide docking site. EMBO J 2002;21:4219–28.
- Deak M, Collins BJ, Murray-Tait V, et al. In vivo role of the phosphate groove of PDK1 defined by knockin mutation. J Cell Sci 2005;118:5023–34.
- Collins BJ, Deak M, Arthur JSC, et al. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 2003;22:4202–11.
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell 2002;109:275–82.
- Pawson T, Scott JD. Protein phosphorylation in signaling-50 years and counting. Trends Biochem Sci 2005;30:286–90.
- Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem Rev 2001;101:2209–42.
- Raimondi C, Calleja V, Ferro R, et al. A small molecule inhibitor of PDK1/PLCγ 31 interaction blocks breast and melanoma cancer cell invasion. Sci Rep 2016;6:1–14.
- Biondi RM, Cheung PCF, Casamayor A, et al. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J 2000;19:979–88.
- Biondi RM, Nebreda AR. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 2003;372:1–13.
- Alessi DR. Discovery of PDKI, one of the missing links in insulin signal transduction. Biochem Soc 2001;3:1–14.
- Tu YK, Hong YY, Chen YC. PDK1: the major transducer of pI 3-kinase actions. Life Sci J 2009;6:23–7.
- Arencibia JM, Pastor-Flores D, Bauer AF, et al. AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim Biophys Acta - Proteins Proteomics 2013;1834:1302–21.
- Stokoe D, Stephens LR, Copeland T, et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 1997;277:567–70.
- Morten F, Antal TL, Dummler BA, et al. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J 2002;21:5396–407.
- Park J, Li Y, Kim SH, et al. Characterization of fragmented 3-phosphoinsitide-dependent protein kinase-1 (PDK1) by phosphosite-specific antibodies. Life Sci 2013;93:700–6.
- Milburn CC, Deak M, Kelly SM, et al. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J 2003;375:531–8.
- Engel M, Hindie V, Lopez-Garcia LA, et al. Allosteric activation of the protein kinase PDK1 with low molecular weight compounds. EMBO J 2006;25:5469–80.
- Balendran A, Casamayor A, Deak M, et al. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr Biol 1999;9:393–404.
- Biondi RM, Kieloch A, Currie RA, et al. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J 2001;20:4380–90.
- Yang J, Cron P, Good VM, et al. letters crystal structure of an activated Akt/Protein Kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol 2002;9:940–4.
- Stroba A, Schaeffer F, Hindie V, et al. 3,5-Diphenylpent-2-enoic acids as allosteric activators of the protein kinase PDK1: structure-activity relationships and thermodynamic characterization of binding as paradigms for PIF-binding pocket-targeting compounds. J Med Chem 2009;52:4683–93.
- Hindie V, Stroba A, Zhang H, et al. Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat Chem Biol 2009;5:758–64.
- Busschots K, Lopez-Garcia LA, Lammi C, et al. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chem Biol 2012;19:1152–63.
- Medina JR. Selective 3-phosphoinositide-dependent kinase 1 (PDK1) inhibitors: dissecting the function and pharmacology of PDK1. J Med Chem 2013;56:2726–37.
- Haesendonckx S, Tudisca V, Voordeckers K, et al. The activation loop of PKA catalytic isoforms is differentially phosphorylated by Pkh protein kinases in Saccharomyces cerevisiae. Biochem J 2012;448:307–20.
- Stockman BJ, Kothe M, Kohls D, et al. Identification of allosteric PIF-pocket ligands for PDK1 using NMR-based fragment screening and1H-15N TROSY experiments. Chem Biol Drug Des 2009;73:179–88.
- Wei L, Gao X, Warne R, et al. Design and synthesis of benzoazepin-2-one analogs as allosteric binders targeting the PIF pocket of PDK1. Bioorg Med Chem Lett 2010;20:3897–902.
- Erlanson DA, Wells JA, Braisted AC. Tethering: fragment-based drug discovery. Annu Rev Biophys Biomol Struct 2004;33:199–223.
- Shoichet BK. Virtual screening of chemical libraries. Nature 2004;432:862–5.
- Castranova V, Asgharian B, Sayre P, et al. Small-molecule allosteric modulators of the protein kinase PDK1 from structure-based docking. J Med Chem 2015;58:1922–2013.
- Koes D, Khoury K, Huang Y, et al. Enabling large-scale design, synthesis and validation of small molecule protein-protein antagonists. PLoS One 2012;7:e32839.
- Kingdom U, Kingdom U, London F, et al. Discovery of a potent allosteric kinase modulator by combining computational and synthetic methods. Angew Chem Int 2015;21:129–39.
- Xu Z, Nagashima K, Sun D, et al. Development of high-throughput TR-FRET and AlphaScreen assays for identification of potent inhibitors of PDK1. J Biomol Screen 2009;14:1257–62.
- Macarron R. Critical review of the role of HTS in drug discovery. Drug Discov Today 2006;11:277–9.
- Bobkova EV, Weber MJ, Xu Z, et al. Discovery of PDK1 Kinase Inhibitors with a Novel Mechanism of Action by Ultrahigh Throughput Screening. J Biol Chem 2010;285:18838–46.