
Abstract
A series of sixteen benzenesulfonamide derivatives has been synthesised and tested as inhibitors of Vibrio cholerae carbonic anhydrase (CA) enzymes, belonging to α-CA, β-CA, and γ-CA classes (VchCAα, VchCAβ, and VchCAγ). The determined Ki values were compared to those of selected human CA isoforms (hCA I and hCA II). Structure-affinity relationship analysis highlighted that all tested compounds proved to be active inhibitors of VchCAα at nanomolar concentration. The VchCAβ activity was lower to respect inhibitory efficacy toward VchCAα, whereas, these benzenesulfonamide derivatives failed to inhibit VchCAγ. Interestingly, compound 7e combined the best activity toward VchCAα and VchCAβ. In order to obtain a model for binding mode of our inhibitors toward bacterial CAs, we carried out docking simulations by using the available crystal structures of VchCAβ.
GRAPHICAL ABSTRACT
1. Introduction
The gram-negative bacterium Vibrio cholerae is responsible for the secretory diarrheal disease cholera that infects millions of people worldwide and might cause high mortality if left untreated. While the rehydration therapy is an effective treatment approach as well as the use of antibiotics in severe infections, there is a pressing need for agents that prophylactically could prevent the injurious effects of a cholera infection.
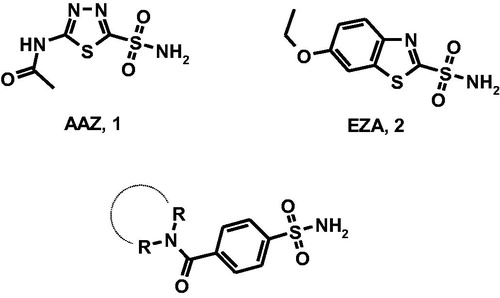
There is evidence that protein ToxT is one of the transcription factors activating the V. cholerae virulence gene expression; therefore, ToxT is a target for the development of cholera therapeuticsCitation1,Citation2. ToxT activity is (a) negatively controlled by the unsaturated fatty acids (UFAs) of the bile and (b) enhanced by the presence of bicarbonate in the upper small intestine where V. cholerae preferentially colonises. Therefore, bicarbonate is likely an important chemical signal during the cholera diseaseCitation3,Citation4. Generally, bacteria increase cytosolic bicarbonate levels through bicarbonate transporter proteins and carbonic anhydrases (CAs, EC 4.2.1.)Citation2. It has been demonstrated that V. cholerae can increase the cytosolic bicarbonate levels through the hydration of CO2 by the action of CAs since V. cholerae lacks bicarbonate transporter proteins in its genomeCitation3. Thus, the CAs represent an attractive molecular target for the development of innovative anti-infective agentsCitation3. The metalloenzyme CAs are grouped into seven genetically distinct families, named α-, β-, γ-, δ-, ζ-, η-, and ɵ-CAs, with different structures and active site architecture as well as metal ions in the catalytic region (zinc, iron, cobalt, and cadmium)Citation5. Vibrio cholerae encodes carbonic anhydrase belonging to α-CA, β-CA, and γ-CA classes (VchCAα, VchCAβ, and VchCAγ)Citation6, that could represent potential targets for the development of anti-infectives targeting V. cholerae colonisationCitation7–9. The α- and β-CAs are Zn(II) metalloenzymes, whereas γ-CAs use Fe(II) as metal ion; however they are also active with Zn(II) or Co(II). The metal ion is coordinated by three His residues in the α- and γ-classes, and one His and two Cys residues in the β-class. Several classes of CA inhibitors (CAIs)Citation10,Citation11 have been identified; among them, sulfonamide derivatives have been shown to anchor the metal-coordinated water molecule/hydroxide ion within catalytic site thus inducing CA inhibition. The prototype sulfonamide inhibitor acetazolamide (, AAZ, 1) is clinically used as diuretic and anticonvulsant agent in humans; additionally, AAZ displays inhibitory effects toward bacterial isozymes. Moreover, as confirmation of the role of bicarbonate in virulence and colonisation processes, the carbonic anhydrase inhibitor ethoxzolamide (EZA, 2) proved to decrease the virulence gene expression and to reduce the growth of pathogenCitation7.
Figure 1. Chemical structures of well-known CAIs acetazolamide (AAZ, 1) and ethoxzolamide (EZA, 2) and designed 4-(cycloalkyl)-1-carbonylbenzenesulfonamides.
The principal drawback using CAIs as antiinfectives agents is the lack of selectivity towards the pathogenic versus human isoformsCitation7. For this reason, many research groups are continually involved in the synthesis of new CAIs or in the modification/optimisation of the existing inhibitors, which are commonly tested on CAs from mammalian and pathogenic organismsCitation10,Citation11. Most of these inhibitors represent interesting leads towards the optimisation of new antibiotic agents showing excellent inhibitory efficiency and selectivity for the target CAs over the human (h) off-target isoform hCA ICitation10,Citation11. In the course of our efforts to identify novel selective CAIs, we have synthesised and tested a series of quinolone/isoquinoline-arylsulfonamides showing high affinity toward human CAsCitation12–22. Specifically, we have reported the discovery of a set of heteroaryl-N-carbonylbenzenesulfonamides as a class of potent inhibitors of druggable CA isoforms (hCA II, hCA VII, hCA IX, and hCA XIV)Citation15,Citation17,Citation18,Citation20–22.
Recently, we have studied a new small library of CAIs containing azepine/piperidine/piperazine nucleus linked to benzenesulfonamide fragment and disclosed several compounds that demonstrated inhibitory effects in low nanomolar range toward hCAsCitation23. This series of compounds has been rationally designed by using the cycloalkylamine nucleus as the core for binding contact in the middle area of catalytic site (). The visual inspection of their binding mode in co-crystal adducts with hCA II and hCA VII confirmed that the sulfonamide portion is crucial for CA recognition processCitation23. By considering the high inhibitory effects of this series of potent sulfonamide compounds, we decided to further study their CA inhibitory profile thus exploiting their capability to act as potential anti-infective agents. Therefore, the primary purpose of our work is to provide new information about structure-affinity relationship relative to the inhibition of the three classes from V. cholerae, VchCAα, VchCAβ, and VchCAγ . Particularly, our idea was to study in depth the role of hydrophobic interactions within VchCA cavity.
2. Materials and methods
2.1. Chemistry
All reagents were used without further purification and bought from common commercial suppliers. Microwave-assisted reactions were carried out in a Focussed Microwave TM Synthesis System, Model Discover (CEM Technology Ltd Buckingham, UK). Melting points were determined on a Buchi B-545 apparatus (BUCHI Labortechnik AG Flawil, Switzerland) and are uncorrected. By combustion analysis (C, H, N) carried out on a Carlo Erba Model 1106-Elemental Analyser we determined the purity of synthesised compounds; the results confirmed a ≥ 95% purity. Merck Silica Gel 60 F254 plates were used for analytical TLC (Merck KGaA, Darmstadt, Germany). For detection, iodine vapour and UV light (254 nm) were used. Flash Chromatography (FC) was carried out on a Biotage SP1 EXP (Biotage AB Uppsala, Sweden). 1H NMR spectra were measured in dimethylsulfoxide-d6 (DMSO-d6) and CDCl3 with a Varian Gemini 300 spectrometer (Varian Inc. Palo Alto, California, USA); chemical shifts are expressed in δ (ppm) and coupling constants (J) in hertz. All exchangeable protons were confirmed by addition of D2O. Rf values were determined on TLC plates using a mixture of DCM/MeOH (96/4) as eluent. All compounds have been re-synthesised following the previously reported procedure, that is briefly described belowCitation23.
2.1.1. Synthetic procedures for benzenesulfonamides 5a–e, 6a, 7a–f, 8a–d
Pathway i: To a solution of 4-(aminosulfonyl)benzoic acid (4) (6 mmol) in THF (15 ml) the carbonylimidazole (6 mmol) (CDI) at 0 °C was added. The obtained mixture was stirred at room temperature for 3 h and then the appropriate amine derivative (15 mmol) in DMF (5 ml) was added dropwise. The reaction mixture was stirred at room temperature for 2 h. The solvent was removed in vacuo; by adding of aqueous solution of NaHCO3 (5 ml) we obtained compounds 5a and 5d as crude products, which were purified through crystallization by a mixture of Et2O and EtOH (1:1)
Pathway ii: A mixture of 4-(aminosulfonyl)benzoic acid (4) (2 mmol) and N,N,N,N-tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate (HBTU) (2 mmol) in DMF(2 ml) was stirred at room temperature for 1 h. Then, a solution of the appropriate amine derivative (2 mmol) in TEA (2 mmol) was added dropwise. The reaction mixture was left overnight, then quenched with water (10 ml) and extracted with EtOAc (3 × 10 ml). The organic phase was dried with Na2SO4 and the solvent was removed in vacuo. The residue was purified by flash chromatography (DCM/MeOH 96:4), crystallized by treatment with a mixture of Et2O and EtOH (1:1) to give the desired final compounds 5b, 5c, 6a, 7a–f, and 8a–d as white powders; the chemical characterisation of all re-synthesised compounds was in good agreement with literature (see Supporting Material).
2.2. Preparation of the bacterial CAs
The three bacterial enzymes were prepared accordingly to the procedure reported by our groupsCitation24. Briefly, the GeneArt Company (Invitrogen), specialised in gene synthesis, designed the genes encoding for the bacterial α, β, and γ- CAs. The BL21 DE3 competent cells (Agilent) were transformed with the expression vector pET15-b containing the gene encoding for one of the three CA-classes. Subsequently, bacterial cells were induced with 1 mM IPTG and, after 30 min, treated with 0.1 M ZnCl2. After 4 h, cells were harvested and disrupted by sonication at 4 °C. After centrifugation at 12,000×g for 45 min, the supernatant was incubated with His Select HF nickel affinity gel resin (Sigma) equilibrated in lysis buffer for 30 min. The protein was eluted with the wash buffer containing 200 mM imidazole. Collected fractions were dialysed against 50 mM Tris/HCl, pH 8. At this stage of purification, the protein was at least 95% pure.
2.3. Carbonic anhydrase inhibition assay
An Applied Photophysics stopped-flow instrument (Leatherhead, Surrey (UK)) has been used for assaying the CA catalysed CO2 hydration activityCitation25. Phenol red (at a concentration of 0.2 mM) has been used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM TRIS (pH 8.3) as buffer, and 20 mM NaClO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters (by Lineweaver–Burk plots) and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The un-catalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10–100 mM) were prepared in distilled-deionized water, and dilutions up to 0.01 mM were done after that with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature before assay, to allow for the formation of the E–I complex or the eventual active site-mediated hydrolysis of the inhibitor. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlierCitation26–28, and represent the mean from at least three different determinations. The human CA isoforms were recombinant ones obtained in-house. All salts/small molecules were of the highest purity available, from Sigma-Aldrich (Milan, Italy).
2.4. Modelled structure of the opening of the active site of β–carbonic anhydrase from V. cholerae
The structure used to perform docking studies was built on different steps. First of all, the crystal structure of closed β-CA from V. cholerae was retrieved from the RCSB Protein Data Bank (PDB code 5CXK)Citation29. The water molecules were discarded, then hydrogen atoms were added to the protein by using Discovery Studio 2.5.5 softwareCitation30. In the second step, a superimposition of residues Cys42, Asp44, Arg46, His98, Cys101, Zn301 of β-CA from V. cholerae on corresponding residues Cys47, Asp49, Arg51, His103, Cys106, Zn228 of chain A of X-ray complex of AAZ with β-CA from the unicellular green alga Coccomyxa (Co-CA, PDB code 3UCJ)Citation31 was performed using PyMOL software (https://pymol.org).
The above-mentioned superimposition was useful to modify the orientation of the side-chains of the two amino acids Asp44 and Arg46 of β-CA from V. cholerae using as template the conformation of Asp49 and Arg51 of β-CA from Coccomyxa. The modified structure of β-CA from V. cholerae was minimised using the conjugate gradient algorithm of NAMD. Minimisation was performed for 500 steps keeping all atoms fixed except for the backbones of residue 43–47 and the side-chains of Asp44 and Arg46: in detail, to maintain the correct orientation of carboxylate moiety of Asp44 and guanidinium group of Arg46 also these groups were kept fixed.
The atom types were assigned using force field CHARMM v22 and the atomic charges according to Gasteiger–Marsili method by VegaCitation32. Furthermore, in the obtained structure, assuming a similar binding mode of the AAZ in both of the β-CAs, we inserted in the catalytic site of β-CA from V. cholerae the X-ray conformation of the acetazolamide retrieved from β-CA from Coccomyxa. The side-chains of the residues around the inhibitor AAZ have been minimised by LigandScoutCitation33.
2.5. Docking studies
The complex obtained was used to perform docking studies by GOLD softwareCitation34 and the AAZ was discarded. The ligand structures were constructed by Vega 3.1.1 and the energy was minimised by using the Conjugate Gradient method (1000 steps). The minimised ligands were docked in their corresponding proteins using Gold Suite 5.0.1. The region of interest, which was used by the Gold programme, was defined as containing the residues within 10 Å of the original position of the acetazolamide in the model structure.
A scaffold constraint (penalty = 5.0) was used to restrict the solutions in which the sulfonamide moiety was able to coordinate the metal within the catalytic binding site. ChemPLP was chosen as the fitness function. The standard default settings were used in all the calculations and the ligands were submitted to 100 genetic algorithm runs. The “allow early termination” command was deactivated. Results differing by less than 0.75 Å in the ligand − all atom RMSD were clustered together. The conformations with the highest fitness values were chosen for both analysis and representation.
All obtained complexes were minimised keeping the Zn ion fixed and using the conjugated gradients algorithm by NAMDCitation35. The results were displayed using the PyMOL software (https://pymol.org).
3. Results and discussion
3.1. Chemistry
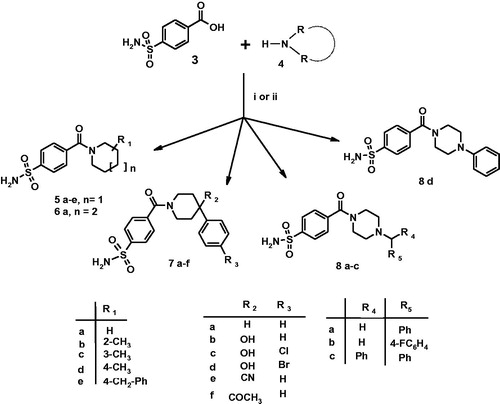
As displayed in Scheme 1, the studied 4-(cycloalkyl)-1-carbonylbenzenesulfonamides 5a–e, 6a, 7a–f, 8a–d have been re-synthesised following a synthetic procedure previously reported by usCitation23; experimental details can be found in “Materials and Methods” section as well as in Supporting Material.
Scheme 1. Reagents and conditions: (i) carbonyldiimidazole (DCI), THF, r.t., 3 h, then RR’NH, DMF r.t., 2 h; (ii) RR’NH, HBTU, DMF, TEA, r.t., overnight.
3.2. Carbonic anhydrase inhibition
The inhibitory effects of this series of sixteen 4-(cycloalkyl-1-carbonyl)benzenesulfonamide derivatives 5a–e, 6a, 7a–f, 8a–c, and 8d were evaluated against VchCAα, VchCAβ, and VchCAγ by using the stopped-flow carbon dioxide hydrase assay. The obtained results are summarised in and compared with the well-known inhibitor AAZ (1). Moreover, the Ki values measured against bacterial CAs were compared with an affinity towards selected human CA isoforms hCA I and hCA IICitation23.
Table 1. Inhibition of hCA I, hCA II, VchCAα, VchCAβ, and VchCAγ for compounds 5a–e, 6a, 7a–f, 8a–c and 8d and acetazolamide (1, AAZ) by a stopped flow CO2 hydrase assay.
As reported in , all studied compounds proved to be inhibitors of the VchCAα and displayed Kis falling in a wide range of 43.3 and 843.2 nM. In terms of structure-affinity relationship considerations, the data collected in suggested that the unsubstituted 4-(piperidine-1-carbonyl)benzenesulfonamide (5a), the three methyl-substituted analogues 5b, 5c, 5d as well as cyclohomologue compound 6a were less active than corresponding 4-aryl-substituted compounds 7a, 7 b, 7c, 7d, 7e, and 7f. Compound 7a (Ki value of 43.3 nM) demonstrated the best activity having the phenyl ring linked to 4-position of piperidine nucleus. The introduction of an additional hydroxyl group, nitrile or acetyl functionalities at C–4 position of piperidine nucleus induces a weak decrease of affinity. The presence of 4′-chlorophenyl ring and hydroxyl group at 4-position of piperidine nucleus of compound 7c afforded equi-active inhibitor of unsubstituted 4-phenyl analog 7a. Among the series of benzenesulfonamides 8a–d containing the piperazine core, the 4-(4-phenylpiperazine-1-carbonyl)benzenesulfonamide (8d) displayed the best inhibitory effect (Ki value of 47.0 nM), thus confirming that the presence of a bulky aromatic group anchored to the 4-position of cycloalkylamine nucleus generally improves the affinity against VchCAα. It is interesting to note the impact of the introduction of a methylene spacer between piperazine core and phenyl moiety: it was found a ten-fold reduction of activity of the compound 4-(4-benzylpiperazine-1-carbonyl)benzenesulfonamide (8a) (Ki = 470 nM) when compared to parent compound 8d. This evidence might be due to a different binding within the catalytic cavity. Moreover, the introduction of a fluorine atom in the para position of the phenyl ring of benzylpiperazine-sulfonamide 8b (Ki value of 95.7 nM) improves the affinity toward VchCAα. On the contrary, the presence of a benzhydryl moiety (compound 8c, Ki = 275.5 nM) seems well tolerated to respect the benzyl-fragment (compound 8a).
Concerning the activity on VchCAβ, several compounds proved to be inhibitors at low micromolar concentration showing inhibition data ranging from 806.4 nM to 6612 nM. Overall, the presence of a 4-aryl moiety resulted advantageous for inhibitory effects toward VchCAβ. The best affinity was found for 4-(4-cyano-4-phenyl-piperidine-1-carbonyl)benzenesulfonamide compound 7e (Ki = 806.4 nM). The presence of other small substituents, such as acetyl for compound 7f or hydroxyl group for compounds 7 b, 7c, and 7d did not significantly influence the inhibitory effects toward VchCAβ. Moreover, the replacement of piperidine nucleus with piperazine one resulted in divergent effects: (a) it was found a completely loss of inhibitory activity for unsubstituted compound 8a, whereas compounds 8b, 8c, and 8d shared similar potency as moderate inhibitors of VchCAβ class.
On the contrary, all studied compounds were totally ineffective inhibitors against VchCAγ enzyme. Despite these compounds demonstrated interesting selectivity against VchCAα to respect β- and γ-classes, they proved to be active inhibitors of ubiquitously expressed hCAI and hCA II at low nanomolar concentration, thus impairing their employment as therapeutics in humans.
3.3. Modeling
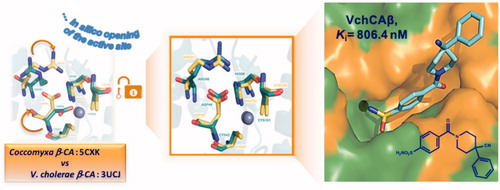
In order to gain more information about the binding interaction of the most active compounds into the enzymatic catalytic site, we carried out a theoretical structural study for understanding and revealing the binding mode of inhibitors toward bacterial CA classes. The crystal structure of apoenzyme VchCAβ has been recently reported by Ferraroni et al. (PDB code 5CXK)Citation29, whereas, the determined structures of VchCAα and VchCAγ from V. cholerae are not currently available. The crystal structures of β-CA are reported for other speciesCitation29,Citation36–39; these structural data furnished information about the homology of their 3-D folds as well as the shape of catalytic site, that displays the zinc coordinated by two cysteines, one-histidine and one-aspartic amino acid residue (the so-called “closed active site”). Particularly, for VchCAβs the catalytic zinc ion is coordinated by Cys42, Asp44, His98, and Cys101. (See schematic representation in Supporting Material). The catalytic site of VchCAβs assumes the so-called “closed active site” conformation as an inactive state. The pH-tuned movement of the Asp 44 residue from metal ion converts the “closed active site” to the “open active site”. Since the available 3 D structure the β-CA from V. cholerae was solved only in the “closed active site” configurationCitation29, the rational design of novel inhibitors through a classical structure-based approach is currently hampered. To overcome the lack of this crucial structural information, in the first step of our theoretical study, we performed a simulation for the opening of the active site of β-CA from V. cholerae. In more details, we superimposed the 3 D X-ray structure of “closed active site” of β-CA from V. cholerae (PDB code 5CXK)Citation29 to “open active site” of available β-CA from Coccomyxa in complex with inhibitor AAZ (1) (PDB code 3UCJ)Citation31. Based on the conformation of side chain of crucial residues Asp49 and Arg51 of β-CA from Coccomyxa, we successively modified the orientation of the corresponding side-chain of Asp44 and Arg46, that are considered responsible for proton shuttling and “closed state” of β-CA. By assuming that inhibitor 1 displays a similar binding mode in these two β-CAs, we extracted AAZ from β-CA of Coccomyxa and then docked this inhibitor in the built 3 D structure of β-CA from V. cholerae; in the last step, we minimised all system. As a result, we obtained the hypothetical “open active site” of β-CA from V. cholerae as the “first in silico model”, useful for mapping the inhibitor binding interactions; our idea was to translate these data for understanding the inhibitory effects of the above mentioned sixteen 4-(cycloalkyl-1-carbonyl)benzensulfonamide derivatives (5a–e, 6a, 7a–f, 8a–d) toward VchCAβ.
(A) displays AAZ (1) bound directly to the “hypothetic open” active site of β-CA from V. cholerae. As expected, the sulfonamide moiety anchors the catalytic zinc ion; additionally, the nitrogen of the N-acetamido group forms a hydrogen bond to the oxygen atom of Gly102, the thiadiazole ring establishes π/π interaction with Tyr83 which is reinforced by further hydrogen bond contacts between heterocyclic nitrogen atoms and hydroxyphenyl substituent. This network of interactions might explain the affinity for VchCAβ at submicromolar concentration (Ki value of 451 nM, ), thus supporting the reliability of our modelled VchCAβ catalytic cavity. Then we studied the docking poses into our “modelled” VchCAβ catalytic cavity for all 4-(cycloalkyl-1-carbonyl)benzensulfonamide derivatives (5a–e, 6a, 7a–f, 8a–d) bearing the sulfonamide as a minimal structural requirement to anchor the zinc ion in the deep catalytic site of all CAs.
Figure 2. Docking poses into our “modelled” VchCAβ catalytic cavity for acetazolamide (A), sulfonamide derivatives 5a (B) and 7e (C). The interactions between the VchCAβ and inhibitors AAZ, 5a and 7e were examined using PyMOL and LIGPLUSCitation40 software; residues involved in hydrophobic interactions and hydrogen bonds (yellow dashed lines) and Zn ion coordination are shown.
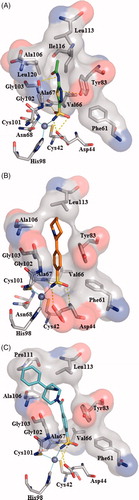
In , we described the best pose of prototype unsubstituted compound 5a showing the ability to create a network of interactions with the deep area of the catalytic site near to zinc ion. As expected, the sulfonamide group is engaged in polar contacts with the CA cavity, whereas the remaining portion of molecule occupies the middle area establishing few hydrophobic interactions with residues Tyr83, Leu113, and Ala106. Therefore, we hypothesised that these interactions could be not sufficient to inhibit VchCAβ ( Ki >10,000 nM ). This consideration might be applicable to justify the low efficacy demonstrated by sulfonamide derivatives 5 b–d and 6a structurally related to parent compound 5a. Interestingly, the best active inhibitor 7e (Ki = 806.4 nM, see ) demonstrated the ability to form additional interactions with a cluster of hydrophobic residues Ala106, Pro111, and Leu 113 when compared with unsubstituted analog 5a, thus suggesting that the 4-phenyl substituent of compound 7e is a crucial fragment to make more favourable hydrophobic contacts in the top of the cavity. This result indicates that the ligands having a good affinity towards VchCAβ are expected to establish additional hydrogen bonding interactions or hydrophobic contacts in the middle or top region of the enzymatic cavity.
4. Conclusions
In conclusions, a small series of benzenesulfonamides has been screened as inhibitors of V. cholerae carbonic anhydrases. Compound 7e demonstrated interesting affinity against VchCAα and VchCAβ with Ki values of 89.9 and 806.4 nM, respectively. The predicted binding mode of compound 7e in the modelled catalytic site of VchCAβ suggests that the introduction of an extra aromatic ring might improve the contacts in the top area of the catalytic cavity, thus furnishing suggestions for the rational design of new compounds targeting V. cholerae CAs.
Supplemental Material
Download MS Word (244.6 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Peterson KM, Gellings PS. Multiple intraintestinal signals coordinate the regulation of Vibrio cholerae virulence determinants. Pathog Dis 2018;76.
- Muanprasat C, Chatsudthipong V. Cholera: pathophysiology and emerging therapeutic targets. Future Med Chem 2013;5:781–98.
- Thomson JJ, Withey JH. Bicarbonate increases binding affinity of Vibrio cholerae ToxT to virulence gene promoters. J Bacteriol 2014;196:3872–80.
- Cobaxin M, Martinez H, Ayala G, et al. Cholera toxin expression by El Tor Vibrio cholerae in shallow culture growth conditions. Microb Pathog 2014;66:5–13.
- Lomelino CL, Andring JT, McKenna R. Crystallography and its impact on carbonic anhydrase research. Int J Med Chem 2018;2018:9419521.
- Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704.
- Modak JK, Liu YC, Machuca MA, et al. Structural basis for the inhibition of Helicobacter pylori alpha-carbonic anhydrase by sulfonamides. PLoS One 2015;10:e0127149.
- Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704.
- Capasso C, Supuran CT. Inhibition of bacterial carbonic anhydrases as a novel approach to escape drug resistance. Curr Top Med Chem 2017;17:1237–48.
- Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.
- De Luca L, Mancuso F, Ferro S, et al. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur J Med Chem 2018;143:276–82.
- Bruno E, Buemi MR, Di Fiore A, et al. Probing molecular interactions between Human Carbonic Anhydrases (hCAs) and a novel class of benzenesulfonamides. J Med Chem 2017;60:4316–26.
- Bruno E, Buemi MR, De Luca L, et al. In vivo evaluation of selective carbonic anhydrase inhibitors as potential anticonvulsant agents. ChemMedChem 2016;11:1812–8.
- Buemi MR, De Luca L, Ferro S, et al. Carbonic anhydrase inhibitors: design, synthesis and structural characterization of new heteroaryl-N-carbonylbenzenesulfonamides targeting druggable human carbonic anhydrase isoforms. Eur J Med Chem 2015;102:223–32.
- De Luca L, Ferro S, Damiano FM, et al. Structure-based screening for the discovery of new carbonic anhydrase VII inhibitors. Eur J Med Chem 2014;71:105–11.
- Gitto R, Damiano FM, Mader P, et al. Synthesis, structure-activity relationship studies, and X-ray crystallographic analysis of arylsulfonamides as potent carbonic anhydrase inhibitors. J Med Chem 2012;55:3891–9.
- Gitto R, Damiano FM, De Luca L, et al. Synthesis and biological profile of new 1,2,3,4-tetrahydroisoquinolines as selective carbonic anhydrase inhibitors. Bioorg Med Chem 2011;19:7003–7.
- Mader P, Brynda J, Gitto R, et al. Structural basis for the interaction between carbonic anhydrase and 1,2,3,4-tetrahydroisoquinolin-2-ylsulfonamides. J Med Chem 2011;54:2522–6.
- Gitto R, Agnello S, Ferro S, et al. Identification of potent and selective human carbonic anhydrase VII (hCA VII) inhibitors. ChemMedChem 2010;5:823.
- Gitto R, Agnello S, Ferro S, et al. Identification of 3,4-Dihydroisoquinoline-2(1H)-sulfonamides as potent carbonic anhydrase inhibitors: synthesis, biological evaluation, and enzyme-ligand X-ray studies. J Med Chem 2010;53:2401–8.
- Gitto R, Ferro S, Agnello S, et al. Synthesis and evaluation of pharmacological profile of 1-aryl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-sulfonamides. Bioorg Med Chem 2009;17:3659–64.
- Buemi MR, Di Fiore A, De Luca L, et al. Exploring structural properties of potent human carbonic anhydrase inhibitors bearing a 4-(cycloalkylamino-1-carbonyl)benzenesulfonamide moiety. Eur J Med Chem 2019;163:443–52.
- De Luca V, Del Prete S, Supuran CT, et al. Protonography, a new technique for the analysis of carbonic anhydrase activity. J Enzyme Inhib Med Chem 2015;30:277–82.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Del Prete S, Vullo D, De Luca V, et al. Anion inhibition profiles of alpha-, beta- and gamma-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem 2016;24:3413–7.
- Del Prete S, Vullo D, De Luca V, et al. Anion inhibition profiles of the complete domain of the eta-carbonic anhydrase from Plasmodium falciparum. Bioorg Med Chem 2016;24:4410–4.
- De Luca V, Vullo D, Del Prete S, et al. Cloning, characterization and anion inhibition studies of a gamma-carbonic anhydrase from the Antarctic bacterium Colwellia psychrerythraea. Bioorg Med Chem 2016;24:835–40.
- Ferraroni M, Del Prete S, Vullo D, et al. Crystal structure and kinetic studies of a tetrameric type II β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr D Biol Crystallogr 2015;71:2449–56.
- Discovery Studio 2.5.5 Accelrys. Biovia, San Diego, CA; 2009. [cited 2019 Jun 13]. Available from: http://www.accelrys.com.
- Huang S, Hainzl T, Grundstrom C, et al. Structural studies of beta-carbonic anhydrase from the green alga Coccomyxa: inhibitor complexes with anions and acetazolamide. PLoS One 2011;6:e28458.
- Pedretti A, Villa L, Vistoli G. VEGA - An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput-Aided Mol Des 2004;18:167–73.
- Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model 2005;45:160–9.
- Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802.
- Cronk JD, O'Neill JW, Cronk MR, et al. Cloning, crystallization and preliminary characterization of a beta-carbonic anhydrase from Escherichia coli. Acta Crystallogr D Biol Crystallogr 2000;56:1176.
- Rowlett RS, Tu C, Lee J, et al. Allosteric site variants of Haemophilus influenzae beta-carbonic anhydrase. Biochemistry 2009;48:6146–56.
- Covarrubias AS, Bergfors T, Jones TA, et al. Structural mechanics of the pH-dependent activity of beta-carbonic anhydrase from Mycobacterium tuberculosis. J Biol Chem 2006;281:4993–9.
- Nishimori I, Minakuchi T, Vullo D, et al. Inhibition studies of the beta-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium with sulfonamides and sulfamates. Bioorg Med Chem 2011;19:5023–30.
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011;51:2778–86.