
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Twenty novel talmapimod analogues were designed, synthesised and evaluated for the in vivo anti-inflammatory activities. Among them, compound 6n, the most potent one, was selected for exploring the mechanisms underlying its anti-inflammatory efficacy. In RAW264.7 cells, it effectively suppressed lipopolysaccharides-induced (LPS-induced) expressions of iNOS and COX-2. As illustrated by the western blot analysis, 6n downregulated both the NF-κB signalling and p38 MAPK phosphorylation. Further enzymatic assay identified 6n as a potent inhibitor against both p38α MAPK (IC50=1.95 µM) and COX-2 (IC50=0.036 µM). By virtue of the concomitant inhibition of p38α MAPK, its upstream effector, and COX-2, along with its capability to downregulate NF-κB and MAPK-signalling pathways, 6n, a polypharmacological anti-inflammatory agent, deserves further development as a novel anti-inflammatory drug.
1. Introduction
So far, inflammatory diseases, especially the chronic inflammatory disorders, have continuously to be a major global health concern due to the lack of effective and well-tolerated drugsCitation1–3. Currently, the majority of anti-inflammatory therapies have been focussed on two distinct strategies, the first directly interferes with the biological function of the pro-inflammatory mediators by interacting with them or their targets, and the second blocks the production of pro-inflammatory mediatorsCitation4. However, given the complex mechanism underlying some inflammatory diseases, which are involved with multiple signalling pathways, the legendary magic bullet, a drug with high potency and selectivity towards a specific biological target, is insufficient for curing themCitation4–7. Two approaches are capable of achieving multi-dimensional regulation of disease-related signalling pathways, including the drug combination and polypharmacology featuring simultaneous modulation of multiple targets with a single drug moleculeCitation8,Citation9. Between them, the latter benefits from the potential to obviate the drug–drug interactions and minimise the combined off-target effectsCitation10–13.
Talmapimod 1, as a highly selective p38α mitogen-activated protein kinase (p38α MAPK) inhibitor developed by Scios. Inc. from compound 2Citation14, has been advanced to Phase-II clinical trials for the treatment of rheumatoid arthritis, multiple myeloma and bone marrow diseasesCitation15. From an internal programme to prepare butylphthalide derivatives, an undesired compound 6a was obtained via the previously designed synthetic route. Owing to its structural similarity to 1 and 2, we are intrigued by the potential of 6a and its derivatives as anti-inflammatory agents (). Hence, on the basis of 6a, a series of talmapimod analogues were designed and synthesised as shown in . With the attempt to validate their anti-inflammatory efficacy, these talmapimod analogues were first evaluated in vivo. The most potent compound 6n was further tested for the inhibitory activity against nitric oxide (NO) production in RAW264.7 cells, and its anti-inflammatory mechanism was investigated by western blot. Additionally, according to the results of mechanism study, compounds with promising anti-inflammatory activity in vivo were selected for evaluating the p38α MAPK and cyclooxygenases (COXs) inhibitory activities. Finally, molecular docking studies were conducted to elucidate the possible binding modes with these proteins.
Figure 1. Structures and potencies of 1, 2 and talmapimod analogue 6a.
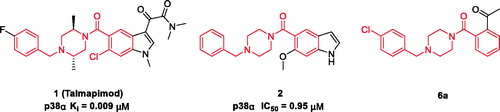
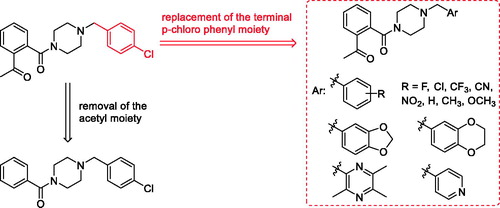
Figure 2. The design of talmapimod analogues.
2. Experimental
2.1. Chemistry
Starting materials, reagents and solvents were purchased from common commercial suppliers. If necessary, purification was carried out prior to use. Melting points were uncorrected and determined on a WRS-1B apparatus. 1H and 13 C NMR spectra were recorded on Bruker Avance 400 II (400 MHz) spectrometer using DMSO-d6 with tetramethylsilane (TMS) as internal standard. ESI-MS were obtained by Thermo Q-Exactive spectrometer.
2.1.1. General procedure for target compounds 6a-6s
3-(Iodomethyl)-3H-isobenzofuran-1-one (4). Iodine (9.0 g, 36 mmol) was added in a solution of 2-vinylbenzoic acid (2.7 g, 18 mmol) in CH3CN (30 ml). The reaction mixture was stirred at 25 °C for 1 h under N2 atmosphere and quenched with saturated Na2S2O3 solution. The mixture was extracted with EA. The EA layer phase was washed successively with water, NaHCO3, Na2S2O3, dried over Na2SO4 and concentrated to a yellow solid. The crude product was purified by recrystallization from hot ethanol, afforded the title compound as a white crystal, Yield: 43%; m.p. 86.9 – 88.4 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.92 – 7.79 (m, 2H, Ar-H), 7.74 (d, 1H J = 7.7, Ar-H), 7.69 – 7.61 (m, 1H, Ar-H), 5.66 (t, 1H, J = 4.0 Hz, CH), 3.97 (dd, 1H, J = 11.3, 3.9 Hz, CH), 3.87 (dd, 1H, J = 11.3, 4.3 Hz, CH). ESI-Mass for C9H7IO2 m/z: 274.7 [M + H]+.
1–(2-(4–(4-Chlorobenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6a) A solution of 4 (690 mg, 3.0 mmol) in DCM (10.0 ml) was added dropwise to a solution of 1–(4-chlorobenzyl)piperazine (840 mg, 4.0 mmol) and K2CO3 (700 mg, 5.0 mmol) in 20 ml DCM. The reaction mixture was stirred at 25 °C for 3 h. The mixture was washed successively with H2O, brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography utilising PE/EA (2:1) as the eluent to afforded the title compound as white solid, Yield: 35%; m.p. 97.5 – 98.4 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, 1H, J = 7.7, 1.2 Hz, Ar-H), 7.63 (dd, 1H, J = 7.5, 1.3 Hz, Ar-H), 7.55 (dd, 1H, J = 7.5, 1.3 Hz, Ar-H), 7.41 – 7.36(m, 2H, Ar-H), 7.36 – 7.31 (m, 2H, Ar-H), 7.29 (dd, 1H, J = 7.7, 1.2 Hz, Ar-H), 3.59 (t, 2H, J = 5.1 Hz, piperazine-H), 3.42 (s, 2H, CH2), 3.04 (t, 2H, J = 5.0 Hz, piperazine-H), 2.55 (s, 3H, CH3), 2.44 (t, 2H, J = 5.1 Hz, piperazine-H), 2.28 (t, 2H, J = 5.0 Hz, piperazine-H); 13 C NMR (100 MHz, DMSO-d6) δ: 198.57 (C-6, C = O, ketone), 170.28 (C-9, C = O, amide), 137.19 (C-5), 136.46 (C-15), 135.31 (C-8), 132.86 (C-2), 132.62 (C-18), 130.31 (C-3), 129.66 (C-16, 20), 128.85 (C-17, 19), 128.45(C-4), 127.24 (C-1), 62.04 (C-14, –CH2), 52.50 (C-11, –CH2, Piperazine), 52.34 (C-12, –CH2, Piperazine), 46.83 (C-13, –CH2, Piperazine), 41.62 (C-10, –CH2, Piperazine), 27.67 (C-7, –CH3). ESI-Mass for C20H21ClN2O2 m/z: 357.229 [M + H]+.
Compound 6 b-6s were prepared in a procedure similar to that described for 6a.
1–(2-(4–(4-Methoxybenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6 b) white solid; Yield: 36%; m.p. 88.4–89.6 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, J = 7.7, 1.2 Hz, 1H, Ar-H), 7.63 (d, J = 7.5, 1.3 Hz, 1H, Ar-H), 7.55 (d, J = 7.5, 1.4 Hz, 1H, Ar-H), 7.32 – 7.25 (m, 1H, Ar-H), 7.25 – 7.18 (m, 2H, Ar-H), 6.94 – 6.84 (m, 2H, Ar-H), 3.73 (s, 3H, CH3), 3.57 (d, J = 5.1 Hz, 2H, piperazine-H), 3.42 (s, 2H, CH2), 3.04 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.41 (t, J = 5.1 Hz, 2H, piperazine-H), 2.26 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.22 (C-6, C = O, ketone), 169.35 (C-9, C = O, amide), 158.78 (C-18), 137.12 (C-5), 135.80 (C-8), 132.92 (C-2), 130.54 (C-16, 20), 130.25 (C-3), 130.12 (C-15), 129.28 (C-4), 127.41 (C-1), 114.04 (C-17, 19), 61.78 (C-14, –CH2), 55.45 (C-21, –OCH3), 52.47 (C-11, –CH2, Piperazine), 52.16 (C-12, –CH2, Piperazine), 46.75 (C-13, –CH2, Piperazine), 41.54 (C-10, –CH2, Piperazine), 28.33 (C-7, –CH3). ESI-Mass for C20H24N2O3 m/z: 353.232 [M + H]+.
4-((4–(2-Acetylbenzoyl)piperazin-1-yl)methyl)benzonitrile (6c) white solid; Yield: 46%; m.p. 143.6–145.6 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, J = 7.8, 1.2 Hz, 1H, Ar-H), 7.80 (d, J = 8.2 Hz, 2H, Ar-H), 7.64 (dd, J = 7.5, 1.3 Hz, 1H, Ar-H), 7.59 – 7.49 (m, 3H, Ar-H), 7.29 (dd, J = 7.8, 1.2 Hz, 1H, Ar-H), 3.65 – 3.57 (m, 4H, piperazine-H, CH2), 3.06 (t, J = 5.0 Hz, 2H, piperazine-H), 2.56 (s, 3H, CH3), 2.46 (t, J = 5.1 Hz, 2H, piperazine-H), 2.30 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ 199.22 (C-6, C = O, ketone), 169.41 (C-9, C = O, amide), 144.61 (C-15), 137.07 (C-5), 135.70 (C-8), 132.98 (C-2), 132.65 (C-16, 20), 130.33 (C-3), 130.01 (C-17, 19), 129.32 (C-4), 127.40 (C-1), 119.36 (C-21), 110.23 (C-18), 61.60 (C-14, –CH2), 52.53 (C-11, –CH2, Piperazine), 52.29 (C-12, –CH2, Piperazine), 46.70 (C-13, –CH2, Piperazine), 41.50 (C-10, –CH2, Piperazine), 28.31 (C-7, –CH3). ESI-Mass for C20H21N3O2 m/z: 348.237 [M + H]+.
1–(2-(4–(4-(Trifluoromethyl)benzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6d) white solid; Yield: 31%; m.p. 63.1–64.5 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.99 (dd, J = 7.7, 1.3 Hz, 1H, Ar-H), 7.69 (d, J = 8.1 Hz, 2H, Ar-H), 7.64 (dd, J = 7.5, 1.3 Hz, 1H, Ar-H), 7.60 – 7.51 (m, 3H, Ar-H), 7.29 (dd, J = 7.7, 1.3 Hz, 1H, Ar-H), 3.66 – 3.57 (m, 4H, piperazine-H, CH2), 3.07 (dd, J = 5.0, 4.1 Hz, 2H, piperazine-H), 2.56 (s, 3H, CH3), 2.46 (d, J = 5.0 Hz, 2H, piperazine-H), 2.31 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.20 (C-6, C=O, ketone), 169.41 (C-9, C=O, amide), 143.51 (C-15), 137.09 (C-5), 135.71 (C-8), 132.96 (C-2), 130.31 (C-3), 129.84 (C-16, 20), 129.30 (C-4), 128.12 (JC-F = 31.6 Hz) (C-18), 127.40 (C-1), 125.53 (JC-F = 3.8 Hz) (C-17, 19), 124.79 (JC-F = 270.0 Hz) (C-21), 61.58 (C-14, –CH2), 52.55 (C-11, –CH2, Piperazine), 52.28 (C-12, –CH2, Piperazine), 46.70 (C-13, –CH2, Piperazine), 41.51 (C-10, –CH2, Piperazine), 28.29 (C-7, –CH3). ESI-Mass for C21H21F3N2O2 m/z: 391.251 [M + H]+.
1–(2-(4–(4-Nitrobenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6e) white solid; Yield: 42%; m.p. 190.8–192.4 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.29 – 8.15 (m, 2H, Ar-H), 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.69 – 7.59 (m, 3H, Ar-H), 7.55 (dd, J = 7.5, 1.3 Hz, 1H, Ar-H), 7.29 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 3.65 (s, 2H, CH2), 3.64 – 3.56 (m, 2H, piperazine-H), 3.07 (dd, J = 5.0, 4.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.48 (t, J = 5.0 Hz, 2H, piperazine-H), 2.32 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.24 (C-6, C=O, ketone), 169.42 (C-9, C=O, amide), 147.05 (C-15), 146.91 (C-18), 137.06 (C-5), 135.70 (C-8), 132.99 (C-2), 130.33 (C-3), 130.18 (C-16, 20), 129.33 (C-4), 127.40 (C-1), 123.86 (C-17, 19), 61.28 (C-14, –CH2), 52.55 (C-11, –CH2, Piperazine), 52.31 (C-12, –CH2, Piperazine), 46.70 (C-13, –CH2, Piperazine), 41.50 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3). ESI-Mass for C20H21N3O4 m/z: 368.240 [M + H]+.
1–(2-(4–(3-Nitrobenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6f) white solid; Yield: 30%; m.p. 180.1–181.8 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.18 (t, J = 1.9 Hz, 1H, Ar-H), 8.15 – 8.10 (m, 1H, Ar-H), 7.98 (dd, J = 7.7, 1.3 Hz, 1H, Ar-H), 7.79 (dd, J = 7.6, 1.2 Hz, 1H, Ar-H), 7.67 – 7.61 (m, 2H, Ar-H), 7.55 (dd, J = 7.5, 1.2 Hz, 1H, Ar-H), 7.29 (dd, J = 7.7, 1.3 Hz, 1H, Ar-H), 3.65 (s, 2H, CH2), 3.61 (d, J = 5.0 Hz, 2H, piperazine-H), 3.07 (dd, J = 5.0, 4.0 Hz, 2H, piperazine-H), 2.56 (s, 3H, CH3), 2.48 (d, J = 5.0 Hz, 2H, piperazine-H), 2.38 – 2.28 (m, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.25 (C-6, C=O, ketone), 169.40 (C-9, C=O, amide), 148.29 (C-17), 137.06 (C-5), 135.95 (C-15), 135.70 (C-8), 132.98 (C-2), 130.33 (C-3), 130.24 (C-20), 129.32 (C-4), 127.40 (C-1), 123.60 (C-16), 122.55 (C-18), 61.02 (C-14, –CH2), 52.42 (C-11, –CH2, Piperazine), 52.19 (C-12, –CH2, Piperazine), 46.70 (C-13, –CH2, Piperazine), 41.48 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3). ESI-Mass for C20H21N3O4 m/z: 368.253 [M + H]+.
1–(2-(4–(2,4-Dichlorobenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6 g) white solid; Yield: 37%; m.p. 74.9–76.4 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.99 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.66 – 7.62 (m, 1H, Ar-H), 7.60 (d, J = 2.2 Hz, 1H, Ar-H), 7.58 – 7.50 (m, 2H, Ar-H), 7.42 (dd, J = 8.3, 2.2 Hz, 1H, Ar-H), 7.29 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 3.60 (t, J = 5.0 Hz, 2H, piperazine-H), 3.58 (s, 2H, CH2), 3.10 – 3.02 (m, 2H, piperazine-H), 2.56 (s, 3H, CH3), 2.49 (d, J = 5.0 Hz, 2H, piperazine-H), 2.34 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.24 (C-6, C=O, ketone), 169.40 (C-9, C=O, amide), 137.07 (C-5), 135.73 (C-8), 135.06 (C-15), 134.63 (C-16), 132.98 (C-2), 132.72 (C-18), 132.56 (C-20), 130.32 (C-3), 129.32 (C-4), 129.16 (C-17), 127.70 (C-19), 127.41 (C-1), 58.33 (C-14, –CH2), 52.54 (C-11, –CH2, Piperazine), 52.34 (C-12, –CH2, Piperazine), 46.72 (C-13, –CH2, Piperazine), 41.52 (C-10, –CH2, Piperazine), 28.33 (C-7, –CH3). ESI-Mass for C20H20ClN2O2 m/z: 391.183 [M + H]+.
1–(2-(4–(2-Chlorobenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6 h) white solid; Yield: 44%; m.p. 133.5–134.6 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.99 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.66 – 7.62 (m, 1H, Ar-H), 7.59 – 7.48 (m, 2H, Ar-H), 7.43 (dd, J = 7.7, 1.6 Hz, 1H, Ar-H), 7.37 – 7.26 (m, 3H, Ar-H), 3.60 (d, J = 5.1 Hz, 4H, piperazine-H, CH2), 3.06 (dd, J = 5.0, 4.0 Hz, 2H, piperazine-H), 2.56 (s, 3H, CH3), 2.49 (d, J = 5.0 Hz, 2H, piperazine-H), 2.35 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.25 (C-6, C=O, ketone), 169.39 (C-9, C=O, amide), 137.09 (C-5), 135.76 (C-8), 135.73 (C-15), 133.77 (C-16), 132.98 (C-2), 131.33 (C-20), 130.32 (C-3), 129.74 (C-18), 129.31 (C-4), 129.17 (C-17), 127.52 (C-19), 127.42 (C-1), 58.96 (C-14, –CH2), 52.63 (C-11, –CH2, Piperazine), 52.40 (C-12, –CH2, Piperazine), 46.73 (C-13, –CH2, Piperazine), 41.54 (C-10, –CH2, Piperazine), 28.33 (C-7, –CH3). ESI-Mass for C20H21ClN2O2 m/z: 357.229 [M + H]+.
1–(2-(4–(3-Chlorobenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6i) white solid; Yield: 48%; m.p. 105.8–107.5 °C;1H NMR (400 MHz, DMSO-d6) δ: 7.99 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.66 – 7.62 (m, 1H, Ar-H), 7.55 (dd, J = 7.5, 1.3 Hz, 1H, Ar-H), 7.41 – 7.25 (m, 5H, Ar-H), 3.60 (t, J = 5.0 Hz, 2H, piperazine-H), 3.51 (s, 2H, CH2), 3.13 – 2.99 (m, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.45 (t, J = 5.0 Hz, 2H, piperazine-H), 2.29 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.20 (C-6, C=O, ketone), 169.38 (C-9, C=O, amide), 141.15 (C-15), 137.10 (C-5), 135.72 (C-8), 133.42 (C-17), 132.96 (C-2), 130.55 (C-19), 130.30 (C-3), 129.29 (C-4), 128.88 (C-16), 127.87 (C-18), 127.45 (C-1), 127.40 (C-20), 61.48 (C-14, –CH2), 52.47 (C-11, –CH2, Piperazine), 52.24 (C-12, –CH2, Piperazine), 46.71 (C-13, –CH2, Piperazine), 41.50 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3). ESI-Mass for C20H21ClN2O2 m/z: 357.212 [M + H]+.
1–(2-(4–(4-Fluorobenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6j) white solid; Yield: 38%; m.p. 107.6–108.8 °C; 1H NMR (400 MHz, DMSO-d6) δ: 7.99 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.40 – 7.31 (m, 2H, Ar-H), 7.28 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.20 – 7.10 (m, 2H, Ar-H), 3.59 (t, J = 5.0 Hz, 2H, piperazine-H), 3.48 (s, 2H, CH2), 3.04 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.43 (t, J = 5.0 Hz, 2H, piperazine-H), 2.27 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.23 (C-6, C=O, ketone), 169.37 (C-9, C=O, amide), 161.73 (JC-F = 242.5 Hz) (C-18), 137.09 (C-5), 135.74 (C-8), 134.51 (JC-F = 2.9 Hz) (C-15), 132.96 (C-2), 131.13 (JC-F = 8.1 Hz) (C-16, 20), 130.30 (C-3), 129.22 (C-4), 127.70 (C-1), 115.38 (JC-F = 21.0 Hz) (C-17, 19), 61.40 (C-14, –CH2), 52.49 (C-11, –CH2, Piperazine), 52.17 (C-12, –CH2, Piperazine), 46.72 (C-13, –CH2, Piperazine), 41.51 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3). ESI-Mass for C20H21FN2O2 m/z: 341.199 [M + H]+.
1–(2-(4-Benzylpiperazine-1-carbonyl)phenyl)ethan-1-one (6k) white solid; Yield: 38%; m.p. 69.7–70.9 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.36 – 7.22 (m, 6H, Ar-H), 3.59 (t, J = 5.0 Hz, 2H, piperazine-H), 3.50 (s, 2H, CH2), 3.05 (dd, J = 5.0, 4.1 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.44 (t, J = 5.0 Hz, 2H, piperazine-H), 2.29 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.21 (C-6, C=O, ketone), 169.36 (C-9, C=O, amide), 138.36 (C-15), 137.12 (C-5), 135.76 (C-8), 132.94 (C-2), 130.28 (C-3), 129.29 (C-4), 128.67 (C-16, 17, 19, 20), 127.47 (C-1), 127.41 (C-18), 62.38 (C-14, –CH2), 52.58 (C-11, –CH2, Piperazine), 52.29 (C-12, –CH2, Piperazine), 46.74 (C-13, –CH2, Piperazine), 41.53 (C-10, –CH2, Piperazine), 28.34 (C-7, –CH3). ESI-Mass for C20H22N2O2 m/z: 323.231 [M + H]+.
1–(2-(4–(4-Methylbenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6 l) white solid; Yield: 46%; m.p. 116.7–118.3 °C.1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.28 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.19 (d, J = 8.0 Hz, 2H, Ar-H), 7.13 (d, J = 8.0 Hz, 2H, Ar-H), 3.58 (t, J = 5.0 Hz, 2H, piperazine-H), 3.44 (s, 2H, CH2), 3.04 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.42 (t, J = 5.0 Hz, 2H, piperazine-H), 2.28 (s, 5H, piperazine-H, CH3). 13 C NMR (100 MHz, DMSO-d6) δ: 199.20 (C-6, C=O, ketone), 169.37 (C-9, C=O, amide), 137.13 (C-5), 136.49 (C-15), 135.75 (C-8), 135.22 (C-18), 132.94 (C-2), 130.29 (C-3), 129.29 (C-4, 17, 19), 129.24 (C-16, 20), 127.40 (C-1), 62.14 (C-14, –CH2), 52.52 (C-11, –CH2, Piperazine), 52.24 (C-12, –CH2, Piperazine), 46.73 (C-13, –CH2, Piperazine), 41.53 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3), 21.17 (C-21, –CH3). ESI-Mass for C21H24N2O2 m/z: 337.255 [M + H]+.
1–(2-(4–(2,4-Dimethoxybenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6 m) white solid; Yield: 46%; m.p. 113.9–115.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.97 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.54 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.27 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.17 (d, J = 8.3 Hz, 1H, Ar-H), 6.53 (d, J = 2.4 Hz, 1H, Ar-H), 6.49 (dd, J = 8.2, 2.4 Hz, 1H, Ar-H), 3.75 (d, J = 3.0 Hz, 6H, OCH3×2), 3.57 (t, J = 5.0 Hz, 2H, piperazine-H), 3.41 (s, 2H, CH2), 3.03 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.43 (t, J = 5.0 Hz, 2H, piperazine-H), 2.27 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.22 (C-6, C=O, ketone), 169.32 (C-9, C=O, amide), 160.06 (C-16), 158.85 (C-18), 137.14 (C-5), 135.76 (C-8), 132.94 (C-2), 131.37 (C-20), 130.27 (C-3), 129.27 (C-4), 127.41 (C-1), 117.86 (C-15), 104.88 (C-19), 98.67 (C-17), 55.83 (C-14, –CH2), 55.55 (C-22, –CH3), 55.48 (C-21, –CH3), 52.54 (C-11, –CH2, Piperazine), 52.23 (C-12, –CH2, Piperazine), 46.76 (C-13, –CH2, Piperazine), 41.56 (C-10, –CH2, Piperazine), 28.34 (C-7, –CH3). ESI-Mass for C22H26N2O4 m/z: 383.300 [M + H]+.
1–(2-(4–(2-Methoxybenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6n) white solid; Yield: 40%; m.p. 117.9–119.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.35 – 7.20 (m, 3H, Ar-H), 6.97 (dd, J = 8.3, 1.1 Hz, 1H, Ar-H), 6.92 (dd, J = 8.3, 1.2 Hz, 1H, Ar-H), 3.77 (s, 3H, OCH3), 3.59 (s, 2H, piperazine-H), 3.49 (s, 2H, CH2), 3.05 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.47 (t, J = 5.0 Hz, 2H, piperazine-H), 2.31 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.23 (C-6, C=O, ketone), 169.36 (C-9, C=O, amide), 157.79 (C-16), 137.12 (C-5), 135.76 (C-8), 132.95 (C-2), 130.31 (C-20), 130.28 (C-3), 129.28 (C-4), 128.58 (C-15), 127.42 (C-1), 125.82 (C-18), 120.57 (C-19), 111.22 (C-17), 55.76 (C-14, –CH2; C-21, –CH3), 52.69 (C-11, –CH2, Piperazine), 52.42 (C-12, –CH2, Piperazine), 46.77 (C-13, –CH2, Piperazine), 41.57 (C-10, –CH2, Piperazine), 28.33 (C-7, –CH3). ESI-Mass for C21H24N2O3 m/z: 353.251 [M + H]+.
1–(2-(4–(3-Methoxybenzyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6o) white solid; Yield: 45%; m.p. 119.3–120.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.5, 1.2 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.2 Hz, 1H, Ar-H), 7.32 – 7.19 (m, 2H, Ar-H), 6.92 – 6.85 (m, 2H, Ar-H), 6.85 – 6.78 (m, 1H, Ar-H), 3.74 (s, 3H, OCH3), 3.58 (t, J = 5.0 Hz, 2H, CH2), 3.47 (s, 2H, CH2), 3.05 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.42 (t, J = 5.0 Hz, 2H, piperazine-H), 2.28 (t, J = 5.0 Hz, 2H, piperazine-H); 13 C NMR (100 MHz, DMSO-d6) δ: 199.22 (C-6, C=O, ketone), 169.35 (C-9, C=O, amide), 158.78 (C-17), 137.12 (C-5), 135.80 (C-8), 132.92 (C-2), 130.54 (C-15), 130.25 (C-3), 130.12 (C-19), 129.28 (C-4), 127.41 (C-1), 125.37 (C-20), 114.17 (C-18), 114.04 (C-16), 61.78 (C-14, –CH2), 55.45 (C-21, –CH3), 52.47 (C-11, –CH2, Piperazine), 52.16 (C-12, –CH2, Piperazine), 46.75 (C-13, –CH2, Piperazine), 41.54 (C-10, –CH2, Piperazine), 28.33 (C-7, –CH3). ESI-MS for C21H24N2O3 m/z: 353.240 [M + H]+.
1–(2-(4-(Benzo[d][1,3]dioxol-5-ylmethyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6p) white solid; Yield: 36%; m.p. 103.4–104.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.6, 1.2 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.2 Hz, 1H, Ar-H), 7.28 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 6.92 – 6.80 (m, 2H, Ar-H), 6.75 (dd, J = 7.9, 1.3 Hz, 1H, Ar-H), 5.99 (s, 2H, OCH3), 3.58 (t, J = 5.0 Hz, 2H, piperazine-H), 3.40 (s, 2H, CH2), 3.04 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.42 (t, J = 5.0 Hz, 2H, piperazine-H), 2.26 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.21 (C-6, C=O, ketone), 169.35 (C-9, C=O, amide), 147.68 (C-19), 146.65 (C-18), 137.12 (C-5), 135.74 (C-8), 132.94 (C-2), 132.17 (C-15), 130.28 (C-3), 129.28 (C-4), 127.40 (C-1), 122.44 (C-16), 109.50 (C-17), 108.31 (C-20), 101.24 (C-21), 62.03 (C-14, –CH2), 52.42 (C-11, –CH2, Piperazine), 52.12 (C-12, –CH2, Piperazine), 46.74 (C-13, –CH2, Piperazine), 41.52 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3). ESI-Mass for C21H22N2O4 m/z: 367.239 [M + H]+.
1–(2-(4-((2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)methyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6q) white solid; Yield: 40%; m.p. 124.9–126.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.28 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 6.84 – 6.71 (m, 3H, Ar-H), 4.21 (s, 4H, OCH2×2), 3.58 (t, J = 5.0 Hz, 2H, piperazine-H), 3.37 (s, 2H, CH2), 3.04 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.41 (t, J = 5.0 Hz, 2H, piperazine-H), 2.26 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.22 (C-6, C=O, ketone), 169.35 (C-9, C=O, amide), 143.50 (C-19), 142.82 (C-18), 137.14 (C-5), 135.74 (C-8), 132.95 (C-2), 131.31 (C-15), 130.28 (C-3), 129.29 (C-4), 127.40 (C-1), 122.07 (C-16), 117.80 (C-17), 117.13 (C-20), 64.47 (C-22), 64.42 (C-21), 61.76 (C-14, –CH2), 52.50 (C-11, –CH2, Piperazine), 52.15 (C-12, –CH2, Piperazine), 46.74 (C-13, –CH2, Piperazine), 41.53 (C-10, –CH2, Piperazine), 28.33 (C-7, –CH3). ESI-Mass for C22H24N2O4 m/z: 381.248 [M + H]+.
1–(2-(4-((3,5,6-Trimethylpyrazin-2-yl)methyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6r) white solid; Yield: 47%; m.p. 119.0–120.1 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.64 (dd, J = 7.5, 1.2 Hz, 1H, Ar-H), 7.55 (dd, J = 7.5, 1.3 Hz, 1H, Ar-H), 7.29 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 3.58 (s, 2H, CH2), 3.55 (t, J = 5.0 Hz, 2H, piperazine-H), 3.01 (t, J = 5.0 Hz, 2H, piperazine-H), 2.55 (s, 3H, CH3), 2.50 (s, 3H, CH3), 2.47 (t, J = 5.0 Hz, 2H, piperazine-H), 2.40 (d, J = 4.3 Hz, 6H, CH3 × 2), 2.30 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.22 (C-6, C=O, ketone), 169.36 (C-9, C=O, amide), 149.99 (C-19), 149.88 (C-16), 147.94 (C-18), 147.61 (C-15), 137.08 (C-5), 135.73 (C-8), 132.95 (C-2), 130.28 (C-3), 129.29 (C-4), 127.41 (C-1), 61.78 (C-14, –CH2), 52.71 (C-11, –CH2, Piperazine), 52.51 (C-12, –CH2, Piperazine), 46.71 (C-13, –CH2, Piperazine), 41.53 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3), 21.54 (C-23), 21.44 (C-22), 20.93 (C-21). ESI-Mass for C21H26N4O2 m/z: 367.306 [M + H]+.
1–(2-(4-(Pyridin-4-ylmethyl)piperazine-1-carbonyl)phenyl)ethan-1-one (6 s) white solid; Yield: 44%; m.p. 129.0 – 131.2 °C. 1H NMR (400 MHz, DMSO-d6) δ: 8.55 – 8.47 (m, 2H, Py-H), 7.98 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.63 (dd, J = 7.5, 1.3 Hz, 1H, Ar-H), 7.55 (dd, J = 7.6, 1.3 Hz, 1H, Ar-H), 7.38 – 7.32 (m, 2H, Py-H), 7.29 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 3.62 (t, J = 5.0 Hz, 2H, piperazine-H), 3.54 (s, 2H, CH2), 3.07 (t, J = 5.0 Hz, 2H, piperazine-H), 2.56 (s, 3H, CH3), 2.47 (t, J = 5.0 Hz, 2H, piperazine-H), 2.30 (t, J = 5.0 Hz, 2H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 199.23 (C-6, C=O, ketone), 169.41 (C-9, C=O, amide), 150.02 (C-16), 147.59 (C-18), 137.08 (C-5), 135.72 (C-8), 132.97 (C-2), 130.32 (C-3), 129.31 (C-4), 127.41 (C-1), 124.19 (C-19), 60.90 (C-14, –CH2), 52.57 (C-11, –CH2, Piperazine), 52.35 (C-12, –CH2, Piperazine), 46.70 (C-13, –CH2, Piperazine), 41.49 (C-10, –CH2, Piperazine), 28.32 (C-7, –CH3). ESI-Mass for C19H21N3O2 m/z: 324.257 [M + H]+.
2.1.2. The procedure of synthetic for (4-benzylpiperazin-1-yl) (phenyl) methanone (8)
Benzoic acid 7 (370 mg 3 mmol) was dissolved in DCM and then oxalyl chloride (630 mg, 5 mmol) and DMF (3 drops) were added at 0 °C. After reaction at 25 °C for 2 h, the reaction mixture was concentrated in vacuo. The residue in DCM was added dropwise to a solution of 1–(4-chlorbenzyl)piperazine (840 mg, 4.0 mmol) and Et3N (610 mg, 6 mmol) at 0 °C. The reaction mixture was stirred at 25 °C for 3 h and concentrated in vacuo. To the residue was added DCM and the mixture was washed successively with H2O, brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography utilising PE/EA (2:1) as the eluent to afford the title compound as white solid; Yield: 64%; m.p. 101.0 – 101.9 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.48 – 7.42 (m, 3H, Ar-H), 7.42 – 7.36 (m, 4H, Ar-H), 7.36 – 7.31 (m, 2H, Ar-H), 3.62 (s, 2H, piperazine-H), 3.50 (s, 2H, CH2), 3.36 (s, 2H, piperazine-H), 2.38 (d, J = 26.3 Hz, 4H, piperazine-H). 13 C NMR (100 MHz, DMSO-d6) δ: 169.34 (C-7, C=O, amide), 137.27 (C-6), 136.32 (C-13), 132.04 (C-16), 131.14 (C-3), 129.95 (C-14, 18), 128.86 (C-1, 5), 128.65 (C-14, 17), 127.35 (C-2, 4), 61.31 (C-12, –CH2), 53.11 (C-9, –CH2, Piperazine), 52.61 (C-10, –CH2, Piperazine), 47.58 (C-11, –CH2, Piperazine), 41.96 (C-8, –CH2, Piperazine). ESI-Mass for C18H19ClN2O m/z: 315.196 [M + H]+.
2.2. Biological evaluation
2.2.1. Animals
Male ICR mice (20 ± 2 g) were obtained from the Experimental Animal Centre of Anhui Medical University (Hefei, China). The mice reared in a pathogen-free setting (23 ± 2 °C, 55 ± 5% humidity) with free access to water and pelleted food throughout the experimental cycle. All experimental procedures performed in accordance with guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and were approved by the Experimental Animal Ethics Committee of Anhui University of Chinese Medicine (Hefei, China).
2.2.2. DNFB-induced mouse model of ACD
After 7 days acclimatisation, the mice were randomly divided into control group, ACD model group, dexamethasone positive control group and 20 compound groups, six in each group. The control group and the model group were given the same dose of the vehicle, the positive control group was given dexamethasone 0.5 mg/kg, and the test compound groups were given compound 5 mg/kg, all of which were given by intragastric administration and were administered once a day for 6 days. Except for the control group, the 50 µL of 1% DNFB (in acetone/olive oil 4:1) was administered to the stripped epidermis, and repeated the next day. After 6 days of drug treatment, 10 µL of 1% DNFB acetone olive oil solution was applied on both sides of the right ear to stimulate inflammation, and equivalent acetone olive oil solution was applied on the left ear for comparison. After 24 h, mice were sacrificed, ears were cut off along the baseline of both ears, and round ear slices were sheared in the same part of both ears with a 6 mm hole puncher and calculate the swelling degree and inhibition rate after weighing. Calculation was carried out according to the following equations.
2.2.3. Cell culture
Murine RAW 264.7 macrophages were obtained from the American Type Culture Collection (ATCC, USA). The cells were incubated in DMEM media supplemented with 10% FBS, 100 U/mL penicillin and 100 mg/mL streptomycin at 37 °C with 5% CO2.
2.2.4. Cell viability assay
Cell viability was assessed using the MTT assay. RAW 264.7 cells were inoculated in 96-well plate at a density of 1.0 × 105 cells per well. After incubated for 24 h, the cells were treated with various concentrations of the compound. After an additional 24-h incubation, 20 µL of 0.5 mg/mL MTT reagent was added to wells and incubated for another 4 h. After 4 h, cell culture media was removed and DMOS was added into each well, and then, the optical density was measured at 570 nm. The IC50 values were determined by GraphPad Prism 6.
2.2.5. Measurement of NO production
The RAW 264.7 macrophages were inoculated at 96-well plates and were pre-treated with vehicle or 6n (0–20 µM dose range) for 2 h and then stimulated with LPS (200 ng/mL) for 24 h. NO concentration in the medium was determined using Griess reagent kit (Beyotime, China) at 540 nm with a microplate reader (MQX200, Bio-Tek, USA) and calculated the inhibition rate of NO.
2.2.6. Western blot
The cell in 96-well plates were treated as described above and then stimulated with LPS (200 ng/mL) for 24 h. The cells were harvested and lysed in an extraction lysis buffer (Beyotime Biotechnology, Shanghai, China) containing protease inhibitors. The protein concentration was determined using a BCA protein assay kit (Thermo Scientific, 23227). The whole cell lysates were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Each membrane was incubated with Tris-buffered saline (pH 7.6, containing 0.05% Tween-20 and 5% non-fat milk). The nitrocellulose membrane was incubated with the primary antibody against p-JNK1/2, JNK1/2, p-p38, p38, p-ERK1/2, ERK1/2, IκBa, NF-κB p65, COX-2, iNOS or β-Actin (Abcam, Cambridge, UK). Immunoreactive bands were detected by incubating with horseradish peroxidase-conjugated secondary antibodies, and visualised using enhanced chemiluminescence reagents (Bio-Rad, Hercules, CA).
2.2.7. p38α MAPK inhibition assay in vitro
The in vitro ability of test compounds and SB203580 (PerkinElmer, Boston, MA, USA) to inhibit p38α MAPK were measured according to the method reported by Babu J. MavunkelCitation16. In brief, after mixing the enzyme reagent with the sample, a reaction mixture containing 200 µM biotin-peptide substrate and 600 µM ATP (+100 µCi/mL γ-32 P-ATP) was added to initiate the reaction. After incubation at 30 °C for 60 min, 10 µL of 1.5% phosphoric acid solution was added to terminate the reaction. Part of the reaction solution was transferred to the well of a streptavidin-coated flash plate, washed in PBS containing 0.01% Tween and sealed. The average value of counts per minute for each group and the IC50 value was calculated.
The average fluorescence values of each well were calculated and recorded as RFU (Relative Fluorescence Unit) blank control (RFU blank), RFU 100% enzyme activity control (RFU enzyme), RFU positive drug control (RFU drug) and RFU test compound (RFU compound). The inhibition rate is calculated according to the following formula.
The IC50 value of the test compound was calculated by the concentration-inhibition reaction curve and the assays were performed in triplicate.
2.2.8. COX-1 and COX-2 inhibition assay in vitro
The COX-1/COX-2 inhibitory activity of test compounds and celecoxib were determined by COX Inhibitor Screening Kit (Fluorometric) (BioVision, Inc., Mountain View, CA, USA) according to the manufacturer's instructions. Simply, different concentrations of the test compound solution were added to the mixed solution containing COX-1/COX-2 enzyme (10 µL) and Assay Buffer (960 µL, 0.1 M Tris-HCl pH 8.0 containing 5 µM EDTA and 2 µM phenol). After the addition of the arachidonic acid solution (100 µM), the mixture was kept at 37 °C in the dark for 5 min and then added 50 µL of 1 M HCl to stop the reaction. The fluorescence value was measured with an excitation wavelength of 535 nm and an emission wavelength of 587 nm. The IC50 values were calculated as described above.
2.3. Molecular docking
The X-ray crystal structure of p38α MAPK (PDB code: 2QD9), COX-1 (PDB code: 1PGF) and COX-2 (PDB code: 1CX2) were obtained from Protein Data Bank. Before docking, the 3 D structures of 6n was generated and the energy minimisation was carried out; removing water moleculars and adding hydrogen atoms to p38α MAPK, COX-1 and COX-2 with the AutoDock ToolsCitation17. Then, the docking was performed by Autodock 4.2 programme with Lamarckian genetic algorithm to sift the best ligand enzyme interaction. The final graphical representations were rendered by PyMOLCitation18.
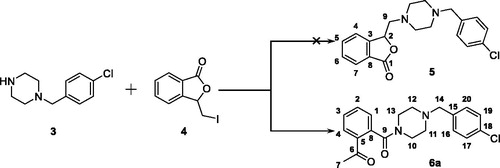
Figure 3. The generation of compound 6a, an unexpected product, from the reaction between 3 and 4.
3. Result and discussion
3.1. Chemistry
In our attempt to prepare the 3-butylphthalide derivative 5 via the nucleophilic substitution between 1–(4-chlorobenzyl)piperazine 3 and 3-(iodomethyl)isobenzofuran-1 (3H)-one 4, an unexpected main product was afforded. Despite its MS consistent with the chemical structure of 5, no characteristic signal of 2-CH appeared in the 1H NMR spectrum, while a singlet with the integral of 3 existed at 2.56 ppm. Meanwhile, two signals appeared at 170.28 and 198.57 ppm, respectively, in the 13 C NMR spectrum, indicating the existence of two carbonyl groups in the chemical structure of the product. Taken together, it was speculated that the afforded product was 6a (). The singlet at 2.56 ppm was generated from its acetyl moiety.
Figure 4. The possible mechanism for the generation of 6a.
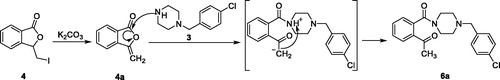
The speculated mechanism for the generation of 6a was displayed in . Intermediate 4 firstly underwent elimination to give enol ester 4a. Afterwards, the five-membered lactone was opened in the presence of 1–(4-chlorobenzyl)piperazine. Finally, the proton on the piperazine was transferred to the carbonyl alpha site to form compound 6a.
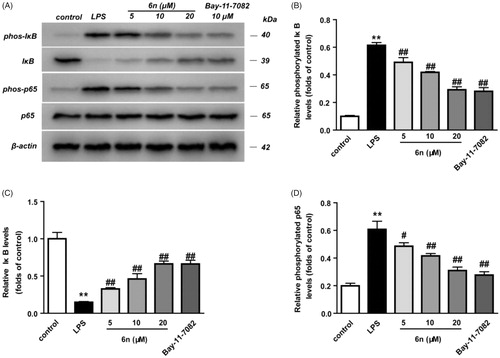
Figure 5. Compound 6n inhibited LPS-induced NF-κB activation in RAW264.7 cells. RAW264.7 cells were co-incubated with 6n (5, 10, 20 μM) and LPS (200 ng/mL) for 30 min. The levels of NF-κB p65, IκB, and their phosphorylated forms were analysed using western blot. The results were showed as means ± SD (n = 3); **p < 0.01 vs. compared with the control group; #p < 0.05, ##p < 0.01 vs. compare with LPS-stimulated group.
The synthetic route to target talmapimod analogues 6a-s and 8 was displayed in Scheme 1. 3-(Iodomethyl)isobenzofuran-1 (3H)-one 4 was prepared according to the procedure reported by Siegfried H. Reich et al.Citation19. It was treated with a series of benzyl piperazine derivatives in the presence of K2CO3 to furnish compounds 6a-s. After converting benzoic acid 7 into benzoyl chloride, it was condensed with 1–(4-chlorobenzyl)piperazine 3 to afford 8 as the product.
Scheme 1. Reagents and conditions: (a) I2, CH3CN, 25 °C; (b) K2CO3, CH3CN, 25 °C; (c) oxalyl chloride, 1–(4-chlorbenzyl)piperazine, DCM, Et3N, 25 °C.
3.2. Biological evaluation
3.2.1. Anti-inflammatory activity in vivo
To validate their anti-inflammatory efficacy, we evaluated compounds 6a-s and 8 in vivo at a p.o. dose of 5 mg/kg in a 2,4-dinitrofluorobenzenethe-induced (DNFB-induced) mouse model of allergic contact dermatitisCitation20. Dexamethasone (DEX) at a p.o. dose of 0.5 mg/kg was employed as the positive control. After the mice were sacrificed, the swelling degree and inhibition rate were calculated by weighing the same part of both ears. The results demonstrated that compounds 6f, 6j, 6n, 6p and 8 had significant inhibitory activity (p < 0.01). In particular, 6n exerted the strongest in vivo anti-inflammatory activity with the inhibition rate of 46.3%. Hence, 6n was further selected for exploring the molecular mechanisms underlying its anti-inflammatory efficacy ().
Table 1. Results of anti-inflammatory activity in vivo of compounds and DEX (Mean ± SD, n = 6).
3.2.2. Compound 6n inhibited LPS-induced inflammatory mediators in RAW264.7 cells
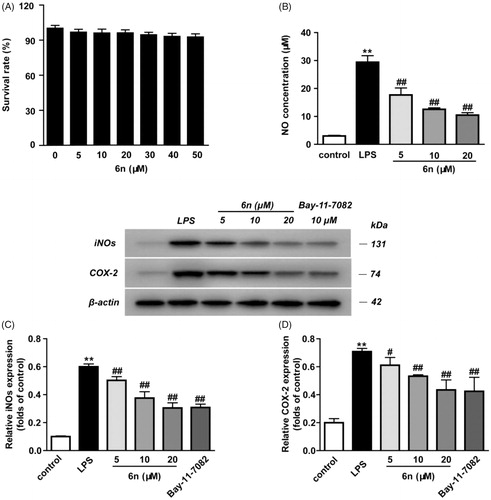
NO serves as an important inflammation mediator, and its continuous high concentration is involved with the development of inflammation-related diseasesCitation21. Besides, NO regulates inducible nitric oxide synthetase (iNOS) and cyclooxygenase 2 (COX-2)Citation22. In view of these, we examined the inhibitory effect of 6n on LPS-induced NO production and LPS-induced expressions of iNOS and COX-2 in RAW264.7 cells (). The cytotoxicity of 6n was firstly evaluated, and no significant toxicity was observed at concentrations ranging from 5 to 50 µM ( and . While the LPS (200 ng/mL) stimulation for 24 h significantly increased NO production, compound 6n reduced LPS-induced NO production in a dose-dependent manner (. Besides, 6n-treatment culminated in a dose-dependent decrease in the LPS-induced expressions of iNOS () and COX-2 (.
Figure 6. Compound 6n inhibited LPS-induced iNOS and COX-2 expressions in RAW264.7 cells. The cells were pre-treated with different concentrations of 6n and then were stimulated with LPS (200 ng/mL) for 24 h. Bay 11–7082 is the NF-κB inhibitor. Cell viability was evaluated using the MTT assay. NO production was measured using nitrite and nitrate assay. iNOS and COX-2 expression were detected by Western blot. (A) Cell viability assay; (B) Quantitative analysis of NO productions; (C) Quantitative analysis of iNOS expressions, (D) Quantitative analysis of COX-2 expressions. β-actin was used as loading control. The results were showed as means ± SD (n = 3); **p < 0.01 vs compared with the control group; #p < 0.05, ##p < 0.01 vs compared with LPS-stimulated group.
3.2.3. Compound 6n inhibited LPS-induced nuclear factor-kappa B (NF-κB) activation in RAW264.7 cells
The activation of NF-κB through proteasomal degradation and phosphorylation of inhibitory κB (IκB) led to the translocation of NF-κB p65 and its interaction with the gene promoter region in nucleus, thereby promoting the expression of iNOS and COX-2Citation3,Citation23. Thus, western blot was performed to examine the effect of 6n on LPS-induced transcriptional activity of NF-κB in RAW264.7 cells. As shown in , 2-h pre-treatment with 6n or Bay 11e7082 before LPS stimulation markedly decreased LPS-induced IκB phosphorylation () and increased cytosolic IκB (. Meanwhile, the accumulation of NF-κB p65 subunit in the nucleus was also lowered (. These results indicated that 6n exerted anti-inflammatory effects through modulation of NF-κB-signalling pathway.
3.2.4. Compound 6n inhibited LPS-induced p38 MAPK-signalling activation in RAW264.7 cells
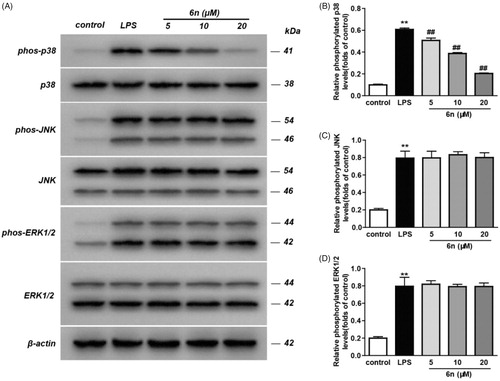
MAPKs play pivotal roles in incurring the immune-mediated inflammation by regulating the transcription and translation of a variety of crucial transcription factors, including activation protein-1 (AP-1) and NF-κBCitation24. Inflammatory stimuli trigger a signalling cascade mediated by p38 MAPK, which activates transcription and translation of genes associated with inflammatory responses such as TNF-α, IL-1β and IL-6, and further induces the expression of inflammatory mediators such as COX-2, iNOS and adhesion moleculesCitation25. Considering this, we examined the impact of 6n on the phosphorylation of the three MAPK subtypes, including p38, JNK and ERK. As a result, 6n can significantly lower p38 phosphorylation (), though there was no obvious effect on the phosphorylation of JNK () and ERK (. These results suggested that the anti-inflammatory effect of 6n was related to the inhibition of p38 phosphorylation.
Figure 7. Compound 6n inhibited LPS-induced MAPK-signalling activation in RAW264.7 cells. RAW264.7 cells were co-incubated with 6n (5, 10, 20 μM) and LPS (200 ng/mL) for 30 min. The levels of p38, JNK, ERK1/2 and their phosphorylated forms were analysed using western blot. The results were showed as means ± SD (n = 3); **p < 0.01 vs compared with the control group; #p < 0.05, ##p < 0.01 vs compare with LPS-stimulated group.
3.2.5. Anti-inflammatory activity in vitro
The down-regulation of p38 MAPK phosphorylation by 6n may be attributed to its interaction with the upstream effector. Besides, owing to the structural similarity of the target compounds to talmapimod, their inhibition against p38α MAPK was expected. This concurrent inhibition of p38α MAPK and its upstream effector would contribute to a two-spot ablation of p38α MAPK-related signalling pathway, which might be beneficial to anti-inflammatory treatment. Thereby, 6f, 6j, 6n and 8, with strong anti-inflammatory activity, were further evaluated against p38α MAPK with SB203580, a selective p38α MAPK inhibitor, as the reference. Besides, we also evaluated them against COX-2, a well-established anti-inflammatory target, given the correlation of COX-2 with MAPK signalling. The results demonstrated that 6n exhibited attractive inhibitory activity against p38α MAPK with IC50 value of 1.95 µM, along with potent inhibitory activity against COX-2 with IC50 value of 0.036 µM, which was comparable to that of Celecoxib. Besides, it inhibited COX-2 with a favourable selectivity, which was beneficial to lowering gastrointestinal intolerance. The concomitant inhibition of p38α MAPK, its upstream effector, as well as COX-2 may account for the most favourable anti-inflammatory activity of 6n. In addition, compound 6j was identified as a potent inhibitor against COX-2 with IC50 value of 0.022 µM, while compound 8 was characterised as a potent inhibitor against p38α MAPK with IC50 value of 1.05 µM ().
Table 2. P38α MAPK, COX-1 and COX-2 inhibition activity in vitro.
3.3. Molecular docking study
The molecular docking analysis of 6f, 6j, 6n and 8 was carried out to elucidate their anti-inflammatory activity in vitro. As showed in , 8 exhibited the lowest binding energy value (−8.98 kcal/mol) when they docked with p38α MAPK, and 6f bound to COX-2 active site with best binding energy value of −7.82 kcal/mol. This was consistent with the above-mentioned in vitro enzymatic experiment results.
Table 3. Docking results of 6f, 6j, 6n and 8
The prominent inhibitory activity against p38α MAPK and COX-2 contributed to the most potent in vivo anti-inflammatory activity of 6n. It bound to the p38α MAPK active site in a similar manner to the co-crystallized ligand (), with the amide carbonyl engaged in H-bond contacts with residues Met 109 and Gly 110 in the hinge region, and 2-methoxybenzyl projected into the hydrophobic pocket formed by Thr 106, Val 105, Leu 104 and Lys 53 (. Moreover, the molecular docking results also accounted for the potency of 6n against COX-2 and its COX-2 selectivity. In compared with COX-1, the active site of COX-2 featured an additional side pocket surrounded by Val 523, Leu 352 and Ser 353Citation14,Citation26–28. The 2-methoxybenzyl moiety of 6n was embedded in this pocket, with the methoxyl group forming two critical H-bonds with Arg 513 and His 90, and the piperazine nitrogen tethered to the 2-methoxybenzyl moiety formed a H-bond with Lue 352 at the mouth of the pocket. Furthermore, the acetyl on benzene ring was located at the other end of the active site and was involved with H-bond contact with Arg 120 (. Interestingly, due to the lack of the side pocket, the binding poses of 6n in the COX-1 active site was forced to rotate and the above binding interactions was also absent (.
Figure 8. (A) Overlay of docking poses of 6n (green sticks) and the co-crystallised ligand (orange sticks) in the binding site of p38α MAPK. (B) Docking and binding pattern of 6n (green sticks) into p38α MAPK active site. (C) Docking and binding pattern of 6n (green sticks) into COX-2 active site. (D) Docking and binding pattern of 6n (green sticks) into COX-1 active site. Dashed lines represent hydrogen bonds.
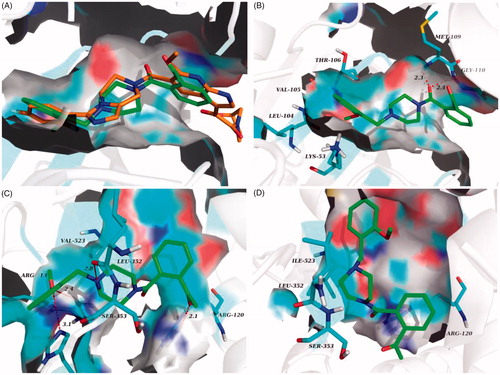
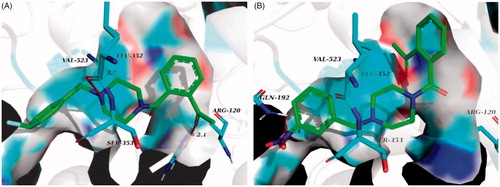
In comparison to compound 6n, 6j bound more strongly to the pivotal residue, that is, Leu 352 (. This may be the reason why 6j had lower binging energy value and better inhibitory activity against COX-2. Loss of two critical H-bonds with Leu 352 and Arg 120, the binding poses of 6f in COX-2 active site was reversed (), and it presented the lowest binging energy value (−7.55 kcal/mol) to other analogues. Correspondingly, the inhibitory activity of 6f against COX-2 was declining substantially. The result of hydrogen boding analyses may justify that the H-bonds with Leu 352 and Arg 120 were decisive factor for presence of COX-2 inhibitory activity in this series of analogues.
Figure 9. (A) Docking and binding pattern of 6j (green sticks) into COX-2 active site. (B) Docking and binding pattern of 6f (green sticks) into COX-2 active site. Dashed lines represent hydrogen bonds.
4. Conclusions
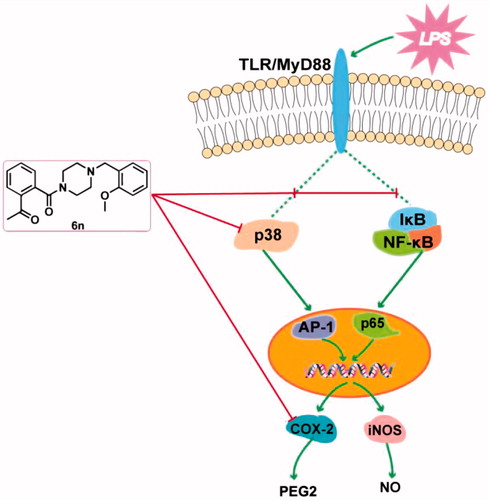
In conclusion, we have designed and synthesised a series of talmapimod analogues as the anti-inflammatory agents based on an unexpected product 6a obtained from an internal programme to prepare butylphthalide derivatives. Among these compounds, 6n exerted the best anti-inflammatory activity in vivo. As illustrated by the mechanism study, 6n-treatment culminated in a dose-dependent decrease in the LPS-induced expressions of iNOS and COX-2. Besides, 6n-treatment led to the dose-dependent down-regulations of NF-κB signalling pathway and the p38 MAPK phosphorylation, both of which may contribute to the decrease in LPS-induced expressions of iNOS and COX-2. The down-regulation of p38 MAPK phosphorylation indicated the inhibition of the upstream effector of p38 MAPK. Further in vitro enzymatic experiment identified 6n as a potent inhibitor against both p38α MAPK and COX-2. To our knowledge, this has been the first compound reported to exert p38α MAPK and COX-2 inhibitory activities. Importantly, the concomitant inhibition of p38α MAPK, its upstream effector, and COX-2, along with its confirmed capability to down-regulate NF-κB and MAPK-signalling pathways make 6n a promising polypharmacological anti-inflammatory agent (). The further investigation of 6n has been currently underway.
Figure 10. Anti-inflammatory molecular mechanism of 6n.
Acknowledgements
The authors thank the support of major projects of national science and technology on new drug creation and development (2018ZX09739-001). Besides, the authors thank the support of University-Enterprise Cooperative Project (No. 2018HZ6 and No. 2019HZ078), and the cooperation is among Anhui University of Chinese Medicine, Hefei Industrial Pharmaceutical Institute Co., Ltd., and Hefei Enruite Pharmaceutical Co., Ltd.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Schultze JL, Rosenstiel P, The SYSCID consortium. Systems medicine in chronic inflammatory diseases. Immunity 2018;48:608–13.
- Mazarakis N, Snibsond K, Licciardi PV, Karagiannis TC. The potential use of l-sulforaphane for the treatment of chronic inflammatory diseases: a review of the clinical evidence. Clin. Nutr 2019;38:pii: S0261-5614(19)30136-0.
- Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett 2014;345:164–73.
- Hanke T, Merk D, Steinhilber D, et al. Small molecules with anti-inflammatory properties in clinical development. Pharmacol. Ther 2016;157:163–87.
- PatronoRocca CB. Nonsteroidal antiinflammatory drugs: past, present and future. Pharm Res 2009;59:285–9.
- Pratsinis YH, Papadopoulou YA, Neidlinger Wilke C, et al. Cyclic tensile stress of human annulus fibrosus cells induces MAPK activation: involvement in proinflammatory gene expression. Osteoarthritis Cartilage 2016;24:679–87.
- Navarrete CM, Pérez M, de Vinuesa AG, et al. Endogenous N-acyl-dopamines induce COX-2 expression in brain endothelial cells by stabilizing mRNA through a p38 dependent pathway. Biochem Pharmacol 2010;79:1805–14.
- Lötsch J, Geisslinger G. Low-dose drug combinations along molecular pathways could maximize therapeutic effectiveness while minimizing collateral adverse effects. Drug Discov Today 2011;16:1001–6.
- Ma X, Lv X, Zhang J. Exploiting polypharmacology for improving therapeutic outcome of kinase inhibitors (KIs): an update of recent medicinal chemistry efforts. Eur J Med Chem 2018;143:449–63.
- Schierle S, Flauaus C, Heitel P, et al. Boosting anti-inflammatory potency of zafirlukast by designed polypharmacology. J Med Chem 2018;61:5758–64.
- Proschak E, Stark H, Merk D. Polypharmacology by design: a medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem 2019;62:420–44.
- Bolognesi ML. Harnessing polypharmacology with medicinal chemistry. ACS Med Chem Lett 2019;10:273–5.
- Gilberg E, Bajorath J. Recent progress in structure-based evaluation of compound promiscuity. ACS Omega 2019;4:2758–65.
- Murali Dhar TG, Wrobleski ST, Lin S, et al. Synthesis and SAR of p38a MAP kinase inhibitors based on heterobicyclic scaffolds. Bioorg Med Chem Lett 2007;17:5019–24.
- Laufer S, Lehmann F. Investigations of SCIO-469-like compounds for the inhibition of p38 MAP kinase. Bioorg Med Chem Lett 2009;19:1461–4.
- (a) Sloan KB, Koch SAM. Effect of nucleophilicity and leaving group ability on the sn2 reactions of amines with (acyloxy)alkyl α-halides: a product distribution study. J Org Chem 1983;48:635–40.; (b) Reich SH, Melnick M, Pino MJ, et al. Structure-based design and synthesis of substituted 2-butanols as nonpeptidic inhibitors of HIV protease: secondary amide series. J Med Chem 1996;39:2781–94.
- Xu X, Xiao W, Zhang Z, et al. Anti-pruritic and anti-inflammatory effects of oxymatrine in a mouse model of allergic contact dermatitis. J Dermatol Sci 2018;91:134–41.
- Beck PL, Li Yan, Wong J, et al. Inducible nitric oxide synthase from bone marrow-derived cells plays a critical role in regulating colonic inflammation. Gastroenterology 2007;132:1778–90.
- Lind M, Hayes A, Caprnda M, et al. Inducible nitric oxide synthase: good or bad? Biomed Pharmacothe 2017;93:370–5.
- Capece D, Verzella D, Tessitore A, et al. Cancer secretome and inflammation: the bright and the dark sides of NF-κB. Semin Cell Dev Biol 2018;78:51–61.
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2003;2:717–26.
- Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol 2014;26:237–45.
- Kulmacz RJ, Lands WEM. Requirements for hydroperoxide by the cyclooxygenase and peroxidase activities of prostaglandin H synthase. Prostaglandins 1983;25:531–40.
- Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8.
- Rowlinson SW, Kiefer JR, Prusakiewicz JJ, et al. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem 2003;278:45763–9.
- Mavunkel BJ, Perumattam JJ, Tan X, et al. Piperidine-based heterocyclic oxalyl amides as potent p38α MAP kinase inhibitors. Bioorg Med Chem Lett 2010;20:1059–62.
- Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics 2008;8:8–14.
- Lill MA, Danielson ML. Computer-aided drug design platform using PyMOL. J Comput Aid Mol Des 2011;25:13–9.