
Abstract
A small series of 2,4-dioxothiazolidinyl acetic acids was prepared from thiourea, chloroacetic acid, aromatic aldehydes, and ethyl-2-bromoacetate. They were assayed for the inhibition of four physiologically relevant carbonic anhydrase (CA, EC 4.2.1.1) isoforms of human (h) origin, the cytosolic hCA I and II, and the transmembrane hCA IX and XII, involved among others in tumorigenesis (hCA IX and XII) and glaucoma (hCA II and XII). The two cytosolic isoforms were not inhibited by these carboxylates, which were also rather ineffective as hCA IX inhibitors. On the other hand, they showed submicromolar hCA XII inhibition, with KIs in the range of 0.30–0.93 µM, making them highly CA XII-selective inhibitors.
1. Introduction
In most living organisms, the equilibrium between metabolically generated CO2 and bicarbonate is slow and needs a catalyst for supporting the metabolic requirements. This catalyst is the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1)Citation1–5, of which a multitude of genetically distinct families have been described so farCitation6–9. The CO2 hydration reaction also leads to the formation of a weak base (bicarbonate) and a strong acid (H+ ions) from two neutral molecules, being thus highly useful for pH regulation and several metabolic pathwaysCitation10–16. Furthermore, CAs are among the most efficient catalysts known in nature, being able to catalyse the hydration of >106 molecules of CO2 per secondCitation1–5. In vertebrates, including humans, only α-class CAs are present, with a high number of isoforms possessing a diverse subcellular/tissue localisation, catalytic activity and presumably physiologic roles were described so farCitation1–3,Citation8,Citation11. The 15 human (h) CA isoforms are in fact involved in a multitude of diseases, and mainly their inhibitors have pharmacologic applications for the treatment of a range of diseases including glaucoma and other ophthalmologic problems, oedema, epilepsy, obesity, tumours, arthritis, etcCitation17–25. Only sulphonamides and sulphamate CA inhibitors (CAIs) are in clinical use at this momentCitation1–3,Citation5,Citation23–25, although many other different chemotypes were discovered in the last period to exert such an action, among which coumarins and sulphocoumarinsCitation26–31, phenolsCitation20,Citation22, mono-/dithiocarbamatesCitation32, and carboxylatesCitation33–38. What is notable for these new chemotypes is the fact that they possess rather different inhibition mechanisms from the sulphonamides, which coordinate in deprotonated form to the metal ion from the CA active siteCitation1–5. On the contrary, many carboxylates, the coumarins and the sulphocoumarins (which act as prodrug CAIsCitation26–29), inhibit CAs by diverse mechanismsCitation5: they either anchor to the zinc-coordinated water molecule/hydroxide ionCitation35–37, occlude the active site entranceCitation26, or bind out of the active siteCitation38. Thus, this chemotype started to be quite investigated in the last period also because such derivatives are much more isoform-selective compared to the classical sulphonamide/sulphamate inhibitorsCitation1–5. This is mainly due to the fact that the binding sites not directly associated with the metal ion are less conserved among the many hCA isoforms, and as such non-classical inhibitors bind towards the exit of the active site, they interact with the non-conserved regions of the various isoforms, showing in this way a more selective inhibition profile compared to the sulphonamides and their isosteresCitation3,Citation5,Citation8–10. Considering our interest in developing novel classes of isoform-selective CAIs, we report here a new class of carboxylates which show a selective inhibition profile of the tumor-associated isoform CA XII.
2. Experimental
2.1. Materials and methods
All starting materials, chemicals, reagents, and solvents were purchased from commercially known reputable sources and were used without further purifications. IR spectra (KBr, cm−1) were recorded on Shimadzu 8201 PC FTIR spectrophotometer (Shimadzu Ltd., Japan). 1H and 13C-NMR spectrum were recorded using (400 MHZ) JEOL-NMR spectrometer (JEOL Ltd., Tokyo, Japan), and the chemical shifts are reported in δ ppm. Elemental analyses were performed on PerkinElmer 2400 elemental analyser (PerkinElmer Inc., 940 Winter Street, Waltham, MA, USA), and the values found were within ±0.3% of the theoretical values. TLC silica (Type 60 GF254, Merck) and was visualised by UV light at 254 nm were used to check the purity of the compounds and monitoring the progress of the reaction. Melting points were recorded in open capillary tubes and are uncorrected (Sigma-Aldrich Chemie GmbH, 82024 Taufkirchen, Germany).
2.2. General procedure for the synthesis of 2,4-dioxothiazolidin acid derivatives 3a–g
The target products 3a–g were prepared in three steps as follow:
Synthesis of thiazolidine-2,4-dione (TZD) was prepared according to the reported methodCitation39–41: A mixture of chloroacetic acid (0.1 mol) and thiourea (0.1 mol) in water (10 ml) were placed in a 100 ml round bottom flask, the reaction mixture was stirred at rt for 30 min and then cooled down to 0 °C. To the reaction mixture, 8 ml of conc. HCl was added dropwise and after complete addition, the reaction mixture was refluxed for 16–18 h. The white solid was obtained after cooling, filtered and washed with water several times to remove the acid traces, dried and then the product TZD 1 was recrystallised from ethanol to afford white crystals mp 123–124 °C, in 91% yield (literature m.p. 123–125 °C)Citation39.
A solution of TZD 1 was treated with various appropriate aldehydes via refluxing in ethanol for 24 h in the presence of piperidine as a catalyst. The reaction mixture was poured into water followed by acidification with acetic acid to afford the products 2a–g. The compounds 2a–g were used directly to the next step without further purification for preparation of 3a–g.
A mixture of 2a–g (1 mmol) and ethyl 2-bromoacetate (2 mmol) was refluxed for 24 h in acetone in presence of potassium carbonate (2 mmol) to furnish the target products obtained as white solid after evaporation of the solvent. The crude product was used directly to next step for preparation of the free carboxylic acid derivatives 3a–g where the solid product was refluxed with glacial acetic acid and HCl in ratio (4:1) for 2 h to afford the pure (2,4-dioxothiazolidin-3-yl) acetic acid derivatives 3a–g after evaporation of the solvent and then crystallised with ethanol. The spectral data for compounds 3a, 3f, and 3g were in agreement with the reported onesCitation42–45.
2.2.1. General procedure for the synthesis of 2,4-dioxothiazolidin-acetic acid derivatives
2-(5-benzylidene-2,4-dioxothiazolidin-3-yl)acetic acid (3a)
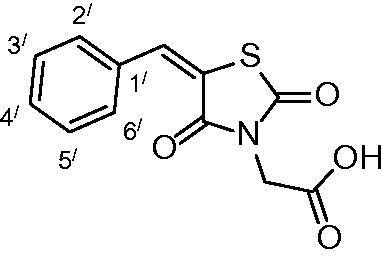
The product was obtained as a white crystal in 93% yield, mp: 214–216 °C. IR (KBr, cm−1): 3423 (OH of COOH), 2940 (CH-aliphatic), 1744, 1686, 1602 (CO). 1H-NMR (DMSO-d6, δ ppm): 4.40 (2H, s, CH2COOH), 7.51–8.00 (5H, m, Ar-H), 8.05 (1H, s, CH = C). 13C-NMR (DMSO-d6, δ ppm): 42.0 (CH2COOH), 114.3, 120.8, 130.0, 130.6, 133.7, 141.6, 163.1, 166.6, 167.4 (CO). Anal. Calc. for C19H9NO4S (263.27): C, 54.75; H, 3.45; N, 5.32. Found C, 54.96; H, 3.61; N, 5.53.
The product was obtained as light-yellow crystals in 96% yield, mp: 226–228 °C. IR (KBr, cm−1): 3371 (OH of COOH), 2950 (CH-aliphatic), 1733, 1685, 1600 (CO). 1H-NMR (DMSO-d6, δ ppm): 2.34 (3H, s, CH) , 4.36 (2H, s, CH2COOH), 7.33 (2H, d, J = 6.6 Hz, Ar-H, H3` & H5`), 7.51 (2H, d, J = 7.2 Hz, Ar-H, H2` & H6`), 7.92 (1H, s, CH = C).13C-NMR (DMSO-d6, δ ppm): 21.6 (CH3), 42.7 (CH2-COOH), 113.9, 119.8, 130.5, 130.7, 134.4, 141.7, 165.5, 167.4, 168.4 (CO). Anal. Calc. for C13H11NO4S (277.3): C, 56.31; H, 4.00; N, 5.05. Found C, 56.44; H, 4.12; N, 5.26.
2-(5-(4-chlorobenzylidene)-2,4-dioxothiazolidin-3-yl)acetic acid (3c)
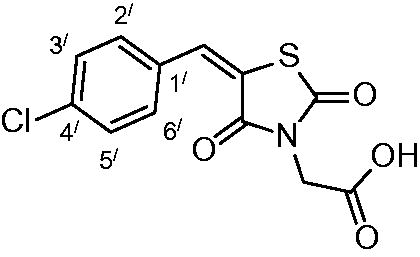
The product was obtained as light-yellow crystals in yield 92%, mp: 250–252 °C. IR (KBr, cm−1): 3383 (OH of COOH), 3008 (CH-aromatic), 1738, 1690, 1607 (CO). 1H-NMR (DMSO-d6, δ ppm): 4.42 (2H, s, CH2-COOH), 7.59–8.00 (4H, m, Ar-H), 8.03 (1H, s, CH = C).13C-NMR (DMSO-d6, δ ppm): 42.2 (CH2-COOH), 114.6, 121.3, 129.3, 131.5, 131.7, 132.4, 135.3, 142.1, 164.8, 166.5, 167.8 (CO). Anal. Calc. for C12H8ClNO4S (297.7): C, 48.40; H, 2.70; N, 4.70. Found C, 48.61; H, 2.83; N, 4.81
2-(5-(2-chlorobenzylidene)-2,4-dioxothiazolidin-3-yl)acetic acid (3d)
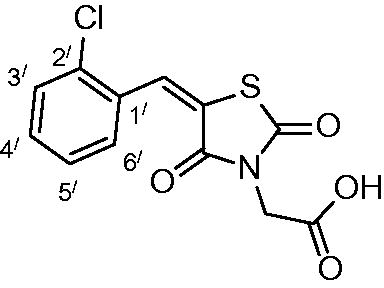
The product was obtained as white crystals in 90% yield, mp: 223–225 °C. IR (KBr, cm−1): 3364 (OH of COOH), 3064 (CH-aromatic), (2940) CH-aliphatic, 1490 (C = C), 1722, 1691, 1608 (CO). 1H-NMR (DMSO-d6, δ ppm): 4.43 (2H, s, CH2COOH), 7.55–7.69 (4H, m, Ar-H), 8.09 (1H, s, CH = C).13C-NMR (DMSO-d6, δ ppm): 42.0 (CH2COOH), 114, 124.1, 128.0, 130.8, 132.0, 137.4, 147.4, 164.3, 166.3, 167.6 (CO). Anal. Calc. for C12H8ClNO4S (297.7): C, 48.41; H, 2.71; N, 4.70. C, 48.65; H, 2.84; N, 4.95.
2-(5-(4-bromobenzylidene)-2,4-dioxothiazolidin-3-yl)acetic acid (3e)
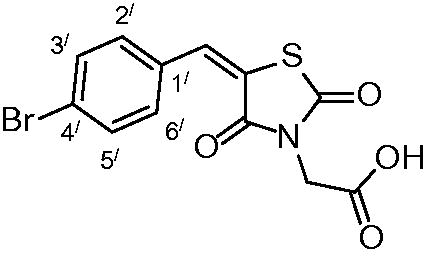
The product was obtained as yellowish white crystals in yield 94%, mp: 260–262 °C. IR (KBr, cm−1): 3371 (OH of COOH), 2948 (CH-aliphatic), 1696, 1606 (CO).1H-NMR (DMSO-d6, δ ppm): 4.37 (2H, s, CH2COOH), 7.56, (2H, d, J = 6.6 Hz, Ar-H, H2`& H6`), 7.73 (2H, d, J = 6 Hz, Ar-H, H3` & H5`); 7.95(1H, s, CH = C). 13C-NMR (DMSO-d6, δ ppm): 42.8 (CH2-COOH), 114.2, 121.9, 124.9, 132.4, 132.8, 133.1, 142.4, 165.3, 167.1, 168.4 (CO). Anal. Calc. for C12H8BrNO4S (342.16): C, 42.12; H, 2.36; N, 4.09. Found C, 42.45; H, 2.59; N, 4.25.
2-(5-(4-methoxybenzylidene)-2,4-dioxothiazolidin-3-yl) acetic acid (3f)
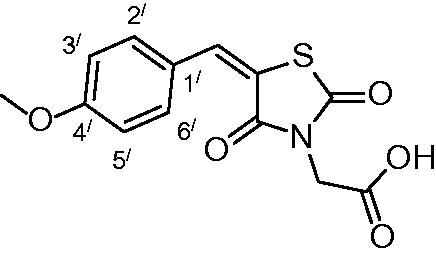
The product was obtained as yellowish white crystals in yield 91%, mp: 223–225 °C. IR (KBr, cm−1): 3368 (OH of COOH), 2926(CH-aliphatic), 1684, 1589 (CO). 1H-NMR (DMSO-d6, δ ppm): 3.81 (3H, s, OCH3), 4.35 (2H, 2, CH2COOH), 7.10 (2H, d, J = 7.2 Hz, Ar-H, H3` & H5`), 7.60 (2H, d, J= 6.6 Hz, Ar-H, H2`& H6`), 7.92 (1H, s, CH = C). 13C-NMR (DMSO-d6, δ ppm): 42.7 (CH2COOH), 56.0 (OCH3), 115.5, 117.8, 125.7, 132.9, 134.3, 142.7, 161.8, 165.6, 167.4, 168.5(CO). Anal. Calc. for C13H11NO5S (293.29): C, 53.24; H, 3.78; N, 4.78. Found C, 53.09; H, 3.85; N, 4.93.
2-(5-(3-methoxybenzylidene)-2,4-dioxothiazolidin-3-yl) acetic acid (3g)
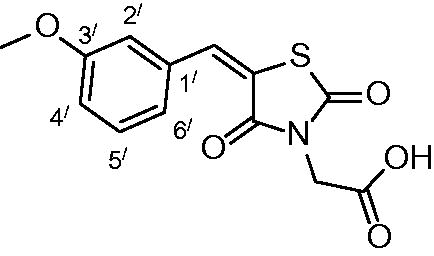
The product was obtained as light-yellow crystals in yield 93%, mp: 184–186 °C.
IR (KBr, cm−1): 3372 (OH of COOH), 2940 (CH-aliphatic), 1734, 1691, 1609 (CO). 1H-NMR (DMSO-d6, δ ppm): 3.78 (3H, s, OCH3), 4.36 (2H, s, CH2-COOH), 7.06 (1H, d, J = 7.8 Hz, Ar-H, H4`), 7.17 (2H, s, Ar-H, H2` & H6`), 7.42 (1H, dd, J = 7.8 & 4.2 Hz, Ar-H, H5`), 7.93(1H, s, CH=C). 13C-NMR (DMSO-d6, δ ppm): 42.7 (CH2-COOH), 55.7 (OCH3), 116.0, 117.2, 121.5, 122.4, 130.9, 134.3, 134.5.142.7, 160.1, 165.4, 167.3, 168.4 (CO). Anal. Calc. for C13H11NO5S (293.29): C, 53.24; H, 3.78; N, 4.78. Found C, 53.09; H, 3.85; N, 4.93.
2.3. CA inhibition assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation46. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled–deionised water, and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay in order to allow for the formation of the E–I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation47–50, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation51–53.
3. Results and discussion
3.1. Chemistry
Condensation of thiourea with chloroacetic acid afforded 2,4-dioxothiazolidine 1, which was reacted with aromatic aldehydes leading to the alkenyl key intermediates 2. These were further N-alkylated with methyl bromoacetate followed by removal of the methyl ester protection, which afforded the 2,4-dioxothiazolidinyl acetic acids 3a–g (Scheme 1).
Scheme 1. Preparation of 2,4-dioxothiazolidinyl acetic acids 3a–3g.
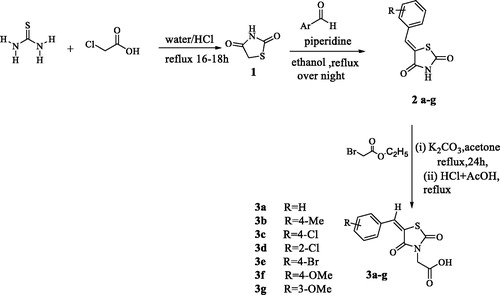
The nature of groups R attached to the aromatic ring was chosen in such a way as to induce chemical diversity, with both electron-attracting and electron-donating moieties being included in the new derivatives 3a–3g generated by the above-described approach.
3.2. Carbonic anhydrase inhibition
Carboxylic acid derivatives 3 reported here were assayed for the in vitro inhibition of four major human CA isoforms, the cytosolic hCA I and II (widespread isoforms in a multitude of tissues and organs)Citation1–5, and the tumor-associated, transmembrane ones hCA IX and XII, recently validated antitumor/antimetastatic targetsCitation6,Citation7 ().
Table 1. CA inhibitory activity of carboxylates 3a–3g and standard sulphonamide inhibitor acetazolamide AAZ, by a stopped-flow CO2 hydrase assayCitation45.
As shown from data of , unlike the standard sulphonamide acetazolamide, which is an efficient, nanomolar hCA I and II inhibitor, the carboxylic acids 3 did not inhibit these two isoforms (KIs > 100 µM), a situation also seen with other carboxylates such as the 2-hydroxy-cinnamic acids formed by the CAs catalysed hydrolysis of coumarinsCitation26. hCA IX was on the other hand inhibited in the high micromolar range by most of these derivatives, except 3b and 3g which had KIs > 100 µM. The best hCA IX inhibitors were 3c and 3f which have KIs of 3.1–3.2 µM and incorporate 4-chloro and 4-methoxy moieties in the aromatic part of the molecule. Structurally related derivatives such as 3a, 3d, and 3e had inhibition constants in the range of 22.2–33.3 µM, being thus an order of magnitude less effective compared to 3c and 3f. Thus, very minor structural changes lead from a low micromolar to a high micromolar and to an ineffective hCA IX inhibitor ().
Surprisingly, hCA XII was effectively inhibited by all carboxylates 3, in the submicromolar range, with KIs of 030–0.93 µM. The structure-activity relationship is quite flat, since the difference in activity between these compounds is quite low. What is really remarkable is the fact that some of these CAIS are highly CA XII-selective, such as for example 3b and 3g, which do not significantly inhibit hCA I, II and IX, but are submicromolar inhibitors of CA XII, a profile not seen with other classes of compounds until now.
4. Conclusions
A small series of 2,4-dioxothiazolidinyl acetic acids was prepared from thiourea, chloroacetic acid, aromatic aldehydes and ethyl-2-bromoacetate. They were assayed for the inhibition of four physiologically relevant CA isoforms, the cytosolic hCA I and II, and the transmembrane hCA IX and XII, involved among others in tumorigenesis (hCA IX and XII) and glaucoma (hCA II and XII). The two cytosolic isoforms were not inhibited by these carboxylates, which were also rather ineffective as hCA IX inhibitors. On the other hand, they showed submicromolar hCA XII inhibition, with KIs in the range of 0.30–0.93 µM, making them highly CA XII-selective inhibitors.
Acknowledgements
The authors would like to extend their sincere appreciation to Researchers Supporting Project Number (RSP-2019/50), King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97.
- Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836.
- Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48.
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32.
- Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704.
- Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018;28:745–54.
- Supuran CT, Ilies MA, Scozzafava A. Carbonic anhydrase inhibitors. Part 29. Interaction of isozymes I, II and IV with benzolamide-like derivatives. Eur J Med Chem 1998;33:739–52.
- Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors: perfluoroalkyl/aryl-substituted derivatives of aromatic/heterocyclic sulfonamides as topical intraocular pressure-lowering agents with prolonged duration of action. J Med Chem 2000;43:4542–51.
- Sentürk M, Gülçin I, Daştan A, et al. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–11.
- Scozzafava A, Briganti F, Mincione G, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, aminoacyl/dipeptidyl sulfonamides possessing long-lasting intraocular pressure-lowering properties via the topical route. J Med Chem 1999;42:3690–700.
- Sarikaya SBÖ, Topal F, Şentürk M, et al. In vitro inhibition of α-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–62.
- Supuran CT, Clare BW. Carbonic anhydrase inhibitors. Part 57. Quantum chemical QSAR of a group of 1,3,4-thiadiazole and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem 1999;34:41–50.
- Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8.
- Akocak S, Lolak N, Bua S, Supuran CT. Discovery of novel 1,3-diaryltriazene sulfonamides as carbonic anhydrase I, II, VII, and IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1575–80.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Temperini C, Innocenti A, Scozzafava A, et al. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: the antiepileptic lacosamide as an example. J Med Chem 2010;53:850–4.
- Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75.
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine 2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- Zengin Kurt B, Sonmez F, Durdagi S, et al. Synthesis, biological activity and multiscale molecular modeling studies for coumaryl-carboxamide derivatives as selective carbonic anhydrase IX inhibitors. J Enzyme Inhib Med Chem 2017;32:1042–52.
- Bozdag M, Bua S, Osman SM, et al. Carbonic anhydrase I, II, IV and IX inhibition with a series of 7-amino-3,4-dihydroquinolin-2(1H)-one derivatives. J Enzyme Inhib Med Chem 2017;32:885–92.
- Bua S, Bozdag M, Del Prete S, et al. Mono- and di-thiocarbamate inhibition studies of the δ-carbonic anhydrase TweCAδ from the marine diatom Thalassiosira weissflogii. J Enzyme Inhib Med Chem 2018;33:707–13.
- Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–9.
- Rotondi G, Guglielmi P, Carradori S, et al. Design, synthesis and biological activity of selective hCAs inhibitors based on 2-(benzylsulfinyl)benzoic acid scaffold. J Enzyme Inhib Med Chem 2019;34:1400–13.
- Cau Y, Vullo D, Mori M, et al. Potent and selective carboxylic acid inhibitors of tumor-associated carbonic anhydrases IX and XII. Molecules 2017;23:17.
- Cadoni R, Pala N, Lomelino C, et al. Exploring heteroaryl-pyrazole carboxylic acids as human carbonic anhydrase XII inhibitors. ACS Med Chem Lett 2017;8:941–6.
- Langella E, D’Ambrosio K, D’Ascenzio M, et al. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of carbonic anhydrases. Chemistry 2016;22:97–100.
- D’Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun 2015;51:302–15.
- Asati V, Bharti SK. Design, synthesis and molecular modeling studies of novel thiazolidine-2,4-dione derivatives as potential anti-cancer agents. J Mol Struct 2018;1154:406–17.
- Kumar BRP, Karvekar MD, Adhikary L, et al. Microwave induced synthesis of the thiazolidine-2,4-dione motif and the efficient solvent free-solid phase parallel syntheses of 5-benzylidene-thiazolidine-2,4-dione and 5-benzylidene-2-thioxo-thiazolidine-4-one compounds. J Heteroc Chem 2006;43:897–903.
- Maccari R, Ottana R, Curinga C, et al. Structure–activity relationships and molecular modelling of 5-arylidene-2,4-thiazolidinediones active as aldose reductase inhibitor. Bioorg Med Chem 2005;13:2809–23.
- Mori M, Takagi M, Noritake C, Kagabu 2. 4-Dioxo-1,3-thiazolidine derivatives as a lead for new fungicides. J Pestic Sci 2008;33:357–63.
- Alegaon SG, Alagawadi KR, Pawar SM, et al. Synthesis, characterization, and biological evaluation of thiazolidine-2,4-dione derivatives. Med Chem Res 2014;23:987–94.
- Maccari R, Vitale RM, Ottanà R, et al. Structure–activity relationships and molecular modelling of new5-arylidene-4-thiazolidinone derivatives as aldose reductase inhibitors and potential anti-inflammatory agent. Eur J Med Chem 2014;81:1–14.
- Bruno G, Costantino L, Curing C, et al. Synthesis and aldose reductase inhibitory activity of 5-arylidene-2,4-thiazolidinediones. Bioorg Med Chem 2002;10:1077–84.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Awadallah FM, Bua S, Mahmoud WR, et al. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J Enzyme Inhib Med Chem 2018;33:629–38.
- El-Gazzar MG, Nafie NH, Nocentini A, et al. Carbonic anhydrase inhibition with a series of novel benzenesulfonamide-triazole conjugates. J Enzyme Inhib Med Chem 2018;33:1565–74.
- Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorg Med Chem Lett 2010;20:5050–3.
- Maresca A, Scozzafava A, Supuran C. 7,8-disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8.
- Boztas M, Cetinkaya Y, Topal M, et al. Synthesis and carbonic anhydrase isoenzymes I, II, IX, and XII inhibitory effects of dimethoxybromophenol derivatives incorporating cyclopropane moieties. J Med Chem 2014;58:640–50.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun 2012;48:1868–70.
- Carta F, Vullo D, Maresca A, et al. Mono-/dihydroxybenzoic acid esters and phenol pyridinium derivatives as inhibitors of the mammalian carbonic anhydrase isoforms I, II, VII, IX, XII and XIV. Bioorg Med Chem 2013;21:1564–9.