
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
NorA is the most studied efflux pump of Staphylococcus aureus and is responsible for high level resistance towards fluoroquinolone drugs. Although along the years many NorA efflux pump inhibitors (EPIs) have been reported, poor information is available about structure-activity relationship (SAR) around their nuclei and reliability of data supported by robust assays proving NorA inhibition. In this regard, we focussed efforts on the 2-phenylquinoline as a promising chemotype to develop potent NorA EPIs. Herein, we report SAR studies about the introduction of different aryl moieties on the quinoline C-2 position. The new derivative 37a showed an improved EPI activity (16-fold) with respect to the starting hit 1. Moreover, compound 37a exhibited a high potential in time-kill curves when combined with ciprofloxacin against SA-1199B (norA+). Also, 37a exhibited poor non-specific effect on bacterial membrane polarisation and showed an improvement in terms of “selectivity index” in comparison to 1.
Introduction
Antimicrobial resistance (AMR) is a complex threat for human health and represents a hot topic in drug discoveryCitation1. The use of large amounts of antibiotics to control human and animal infections and in animal breeding has created unprecedented conditions for the rising and spread of antibiotic resistance among bacterial populations. The “right drug for the right bug” approach remains a distant perspective, currently replaced by an empirically-guided consumption. Recently, the World Health Organisation (WHO) has included AMR in the “ten threats to global health in 2019”, forecasting an imminent return to a time when we were unable to easily treat common infectionsCitation2. Considering the microbial promptness in achieving successful machinery escaping antibiotic activity also towards new drugsCitation3,Citation4, the use of non-antibiotic adjuvant molecules targeting resistance mechanisms, in co-administration with antibacterials, is a valid approach to recover drug sensitivity in resistant strainsCitation5,Citation6. The fascinating idea to “freeze” resistance would allow antibiotics, for which resistance occurred, to recover their activity thereby renewing our armamentarium to fight microbial infections. Amongst the wide range of resistance mechanisms developed by bacteria, one of the most common is the drug extrusion from the cell, which can reduce intracellular drugs to sub-inhibitory concentrations allowing bacteria to grow in the presence of routinely adopted therapeutic dosesCitation7. Indeed, for some drugs, microorganisms can only acquire resistance in the presence of efflux pump activity. Most likely, efflux pumps play a non-specific role in the early stages of antibiotic exposure, thereby allowing microorganisms to develop more specific and effective mechanisms of resistanceCitation4,Citation8,Citation9. Therefore, the use of efflux pump inhibitors (EPIs) in combination with extruded drugs may be a major strategy in the development of effective antimicrobial treatments. To date, little has been done in terms of EPI development and no inhibitors have ever reached the clinical useCitation10.
Among the six multi-drug resistant bacterial species termed ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species), S. aureus and its methicillin-resistant strain (MRSA) represent a serious problem worldwide, due to its acquired resistance to several classes of antibiotics, encoded by the SCC-mec cassettesCitation11. Moreover, the trans-membrane protein NorA, belonging to the Major Facilitator Superfamily, is commonly overexpressed in S. aureus resistant strains and strongly upregulated in response to fluoroquinolones treatment. NorA can extrude different toxic compounds, including the fluoroquinolone ciprofloxacin (CPX) and the dye ethidium bromide (EtBr), by an antiporter mechanism exploiting the proton motive forceCitation12.
Along the years many NorA EPIs have been discovered by three different approaches: i) screening libraries of natural or synthetic molecules; ii) repurposing molecules with known biological activity and iii) designing and synthesising new compoundsCitation10,Citation13–16. The lack of NorA structural information has strongly hampered the identification of potent NorA EPIs. No examples of structure-based drug design have been so far reported for EPI identification, which, therefore, relies on ligand-based drug design approaches or classical medicinal chemistry strategies. In addition, the risk for a strategy aimed at identifying an EPI relies on the poor availability of quick and smart biological screenings able to early identify active molecules. On the contrary, due to the poor knowledge of the NorA efflux mechanisms and the lack of biophysical experiments validating a true NorA inhibition, too frequently molecules have been described in literature as NorA EPIs when they are not. The ability of a compound to act in synergy with CPX against S. aureus resistant strains seems not to be sufficient to consider that molecule as NorA EPI. Additional experiments are needed to rule out non-specific effects of compounds boosting antibiotic activity. Since many efflux pumps work through the proton motive force, its disruption by depolarising the bacterial membrane is the most common example of a non-specific effect still resulting in efflux pump inhibition. However, selective membrane depolarisation in microorganisms appears challenging and often compounds result very toxic by eliciting the same effect on eukaryotic cells. Conversely, an actual NorA EPI must exert a strong synergistic effect with CPX (reducing its MIC at least 4-fold) against overexpressing norA S. aureus strains while resulting poorly active or ineffective against both wild-type and norA knock-out strains. In addition, NorA efflux inhibition activity should be demonstrated by phenotypic assays (i.e. EtBr efflux assays) in norA overexpressing S. aureus strains and non-specific EPI effect needs to be excluded by performing bacterial membrane polarisation experiments. Moreover, the identified EPIs should not possess any intrinsic antibacterial effect at the concentrations needed to reach synergism with antibiotics in order to prevent a potential interference throughout the synergism with antibacterials.
In this direction, we are currently working on 2-phenylquinoline nucleus giving rise to a really promising class of NorA EPIs. 2-Phenylquinoline derivatives have shown an excellent NorA EPI activity, widely restoring CPX antibacterial activity against resistant S. aureus strains. Efforts aimed at delineating a robust structure-activity relationship (SAR) investigation around this scaffold highlighted some significant findings: i) an alkylamino chain on the oxygen at quinoline C-4 position is needed to retain NorA EPI activity and preferred over N-1 positionCitation17,Citation18; ii) the introduction of a methoxy group on the C-6 position of the 2-phenylquinoline core strongly improved NorA EPI activity while lowering host cell toxicityCitation19; iii) the replacement of the methoxy group at C-6 position with a benzyloxy moiety retained NorA inhibition activity (although increasing human cell toxicity), while a free hydroxy group or different O-alkylamino chains at C-6 yielded less potent analoguesCitation20; iv) the shift of the aryl portion from C-2 to C-3 position of the quinoline core increased EPI activity towards nontuberculous mycobacteria (NTM) while decreasing that against S. aureus ()Citation21,Citation22.
Figure 1. Known SAR around the 2-phenylquinoline scaffold and new designed compounds.
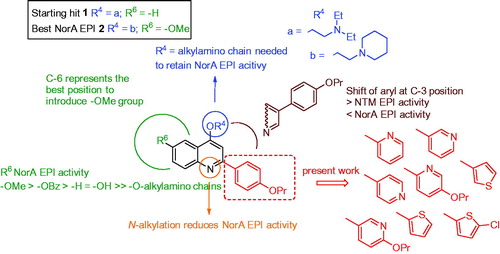
In this work, we focus efforts on the exploration of the C-2 position by replacing the 4’-proproxyphenyl substituent with differently substituted pyridine or thiophene moieties in order to identify potential isosteric replacements (). Indeed, previous SAR studies did not cover such a portion on the quinoline scaffold. Although more potent 2-phenylquinoline-based NorA EPIs have been identified by us, we selected the previously reported compound 1 as starting hitCitation23, because the lack of substituents on the quinoline benzene ring allows for a better comparison with the new C-2 modified analogues (). Considering the role of O-alkylamino chains on the quinoline C-4 position to retain NorA EPI activity, we introduced two different chains (a, O-ethyl-N,N-diethylamino and b, O-ethylpiperidine) for each new C-2 aryl quinoline scaffold. In particular, chain “a” was selected because present in the starting hit 1 and chain “b” since it resulted the best chain in the last work, where the most potent methoxy 2-phenylquinoline derivative 2 was identified ()Citation19.
Materials and methods
Chemistry
All starting materials, reagents and solvents were purchased from common commercial suppliers and were used as such, unless otherwise indicated. Organic solutions were dried over anhydrous Na2SO4 and concentrated with a rotary evaporator at low pressure. All reactions were routinely checked by thin-layer chromatography (TLC) on silica gel 60F254 (Merck) and visualised by using UV or iodine. Flash chromatography separations were carried out on Merck silica gel 60 (mesh 230–400). Melting points were determined in capillary tubes (Stuart SMP30) and are uncorrected. Yields were of purified products and were not optimised. 1H NMR spectra were recorded at 200 or 400 MHz (Bruker Avance DRX-200 or 400, respectively) while 13C NMR spectra were recorded at 101 MHz (Bruker Avance DRX-400). Chemical shifts are given in ppm (δ) relative to TMS. Spectra were acquired at 298 K. Data processing was performed with standard Bruker software XwinNMR and the spectral data are consistent with the assigned structures. The purity of the tested compounds was evaluated by combustion analysis using a Fisons elemental analyser, model EA1108CHN, and data for C, H, and N are within 0.4% of the theoretical values (≥95% sample purity).
N-(2-acetylphenyl)-5-propoxypyridine-2-carboxamide (14)
SOCl2 (1.2 mL, 16.44 mmol) was slowly added at 0 °C to 5-propoxypyridine-2-carboxylic acid 3 (0.39 g, 2.20 mmol), then the reaction mixture was stirred at 60 °C for 30 min. The excess of SOCl2 was removed under reduced pressure to give 5-propoxypyridine-2-carbonyl chloride (5) as a yellow oil that immediately was dissolved in dry THF (6 mL) and added to a mixture of aminoacetophenone 13 (0.29 g, 2.20 mmol) and Et3N (0.9 mL, 6.60 mmol) in dry THF (14 mL). The reaction was stirred at rt for 2 h, then it was poured in ice/water and the pH was adjusted to ≃8 with 2 N HCl. The mixture was extracted by EtOAc and the organic layers were washed by brine, dried over Na2SO4 and evaporated to dryness. After purification by flash chromatography column (CH2Cl2/acetone 98/2), derivative 14 was obtained as a yellow solid (0.41 g, 63% yield), m.p. 132.0–134.0 °C. 1H NMR (200 MHz, DMSO-d6): δ = 1.02 (t, J = 7.5 Hz, 3H, OCH2CH2CH3), 1.73–1.87 (m, 2H, OCH2CH2CH3), 2.59 (s, 3H, CH3), 4.00 (t, J = 6.6 Hz, 2H, OCH2CH2CH3), 7.11 (t, J = 7.6 Hz, 1H, H-4’), 7.23–7.28 (m, 1H, H-4), 7.56 (t, J = 8.4 Hz, 1H, H-5’), 7.90 (d, J = 7.9 Hz, 1H, H-6’), 8.15 (d, J = 8.5 Hz, 1H, H-3’), 8.39 (d, J = 2.1 Hz, 1H, H-6), 8.96 (d, J = 8.7 Hz, 1H, H-3), 13.37 ppm (s, 1H, NH).
N-(2-acetylphenyl)-6-propoxynicotinamide (15)
SOCl2 (0.6 mL, 8.22 mmol) was slowly added at 0 °C to 6-propoxynicotinic acid 4 (0.20 g, 1.10 mmol), then the reaction mixture was stirred at 60 °C for 30 min. The excess of SOCl2 was removed under reduced pressure to give 6-propoxynicotinoyl chloride (6) as a yellow oil that immediately was dissolved in dry THF (4 mL) and added to a mixture of aminoacetophenone 13 (0.15 g, 1.10 mmol) and Et3N (0.7 mL, 5.50 mmol) in dry THF (10 mL). The reaction was stirred at rt for 2 h, then it was poured in ice/water and the pH was adjusted to ≃8 with 2 N HCl. The mixture was extracted by EtOAc and the organic layers were washed by brine, dried over Na2SO4 and evaporated to dryness. After purification by flash chromatography column (CH2Cl2/acetone 98/2), derivative 15 was obtained as a brown oil (0.18 g, 56% yield). 1H NMR (200 MHz, DMSO-d6): δ = 0.81 (t, J = 7.3 Hz, 3H, OCH2CH2CH3), 1.57–1.72 (m, 2H, OCH2CH2CH3), 2.60 (s, 3H, CH3), 3.89 (t, J = 7.3 Hz, 2H, OCH2CH2CH3), 6.48 (d, J = 8.7 Hz, 1H, H-6’), 7.18 (t, J = 7.5 Hz, 1H, H-4’), 7.59 (t, J = 7.7 Hz, 1H, H-5’), 7.78–7.84 (m, 1H, H-5), 8.00 (d, J = 8.0 Hz, 1H, H-3’), 8.38–8.41 (m, 2H, H-2 and H-4), 11.91 ppm (s, 1H, NH).
General procedure A for the synthesis of compounds 16–21
A solution of acyl chlorides 7–12 (1 equiv.) in dry THF (4 ml per mmol) was added to a solution of aminoacetophenone 13 (1 equiv) and Et3N (5 equiv.) in dry THF (5 ml per mmol of 13). The reaction was stirred at rt for 90 min–72 h, then it was poured in ice/water. The pH was adjusted up to ≃8 with 2 N HCl, the mixture was extracted by EtOAc and the organic layer was washed by brine, dried over Na2SO4 and evaporated under vacuum to a solid that was purified by flash column chromatography.
N-(2-acetylphenyl)pyridine-2-carboxamide (16)
General procedure A: time, 90 min; used chloride, pyridine-2-carbonyl chloride 7 (0.57 g, 4.06 mmol); purification, (EP/EtOAc 60/40). Derivative 16Citation24 was obtained as a grey solid (0.71 g, 73% yield), mp 107.5–108.0 °C. 1H NMR (400 MHz, CDCl3): δ = 2.69 (s, 3H, CH3), 7.27 (t, J = 7.3 Hz, 1H, H-4’), 7.67–7.71 (m, 2H, H-5’ and H-6’), 8.06–8.20 (m, 3H, H-3, H-5 and H-3’), 8.76–8.87 (m, 2H, H-4 and H-6), 13.31 ppm (s, 1H, NH).
N-(2-acetylphenyl)nicotinamide (17)
General procedure A: time, 72 h; used chloride, nicotinoyl chloride 8 (0.57 g, 4.06 mmol); purification, (EP/EtOAc 60/40). Derivative 17Citation24 was obtained as a white solid (0.96 g, 99% yield), mp 114.0–115.0 °C. 1H NMR (200 MHz, CDCl3): δ = 2.69 (s, 3H, CH3), 7.12 (m, 1H, H-4’), 7.37–7.42 (m, 1H, H-4), 7.61 (dt, J = 1.5 and 8.7 Hz, 1H, H-5’), 7.95 (dd, J = 1.4 and 8.3 Hz, 1H, H-3’), 8.27–8.33 (m, 1H, H-5), 8.75 (dd, J = 1.8 and 8.5 Hz, 1H, H-6’), 8.90 (d, J = 8.4 Hz, 1H, H-6), 9.27 (d, J = 1.9 Hz, 1H, H-2), 12.73 ppm (s, 1H, NH).
N-(2-acetylphenyl)isonicotinamide (18)
General procedure A: time, 20 h; used chloride, isonicotinoyl chloride 9 (3.45 g, 24.40 mmol); purification, (EP/EtOAc 50/50). Derivative 18Citation24 was obtained as a white solid (3.52 g, 60% yield), mp 111.0–112.0 °C. 1H NMR (200 MHz, CDCl3): δ = 2.66 (s, 3H, CH3), 7.16 (dt, J = 1.2 and 7.9 Hz, 1H, H-4’), 7.61 (dt, J = 1.6 and 7.4 Hz, 1H, H-5’), 7.83–7.86 (m, 2H, H-3 and H-5), 7.94 (dd, J = 1.5 and 8.0 Hz, 1H, H-6’), 8.73–8.80 (m, 2H, H-2 and H-6), 8.89 (dd,J = 1.1 and 8.5 Hz, 1H, H-3’), 12.85 ppm (s, 1H, NH).
N-(2-acetylphenyl)thiophene-2-carboxamide (19)
General procedure A: time, 20 h; used chloride, thiophene-2-carbonyl chloride 10 (0.34 g, 2.34 mmol); purification, (EP/EtOAc 50/50). Derivative 19Citation24 was obtained as a yellow solid (0.33 g, 58% yield), mp129.0–131.0 °C. 1H NMR (200 MHz, CDCl3): δ = 2.68 (s, 3H, CH3), 7.04–7.15 (m, 2H, H-4 and H-4’), 7.51–7.61 (m, 2H, H-3 and H-5’), 7.79 (dd, J = 1.1 and 5.0 Hz, 1H, H-5), 7.91 (dd, J = 1.6 and 8.0, 1H, H-6’), 8.83 (dd, J = 1.2 and 8.5 Hz, 1H, H-3’), 12.70 ppm (s, 1H, NH).
N-(2-acetylphenyl)thiophene-3-carboxamide (20)
General procedure A: time, 12 h; used chloride, thiophene-3-carbonyl chloride 11 (0.41 g, 2.80 mmol); purification (EP/EtOAc 50/50). Derivative 20Citation24 was obtained as a white solid (0.34 g, 50% yield), mp 83.0–84.0 °C. 1H NMR (200 MHz, CDCl3): δ = 2.67 (s, 3H, CH3), 7.10 (dt, J = 1.1 and 7.2 Hz, 1H, H-4’), 7.35 (dd, J = 3.0 and 5.5 Hz, 1H, H-4), 7.57 (dt, J = 1.6 and 8.6 Hz, 1H, H-5’), 7.63 (dd, J = 1.1 and 5.3 Hz, 1H, H-2), 7.91 (dd, J = 1.6 and 8.0 Hz, 1H, H-6’), 8.11 (dd, J = 1.1 and 3.0 Hz, 1H, H-5), 8.88 (dd, J = 1.1 and 8.4 Hz, 1H, H-3’), 12.57 (s, 1H, NH).
N-(2-acetylphenyl)-5-chlorothiophene-2-carboxamide (21)
General procedure A: time, 12 h; used chloride, 5-chlorothiophene-2-carbonyl chloride 12 (0.56 g, 3.08 mmol); purification, (EP/EtOAc 50/50). Derivative 21 was obtained as a white solid (0.73 g, 85% yield), mp 136.0–137.0 °C. 1H NMR (200 MHz, CDCl3): δ = 2.67 (s, 1H, CH3), 6.93 (d, J = 6.1 Hz, 1H, H-3), 7.11 (t, J = 7.6 Hz, 1H, H-4’), 7.52–7.62 (m, 2H, H-4 and H-5’), 7.91 (d, J = 8.0 Hz, 1H, H-6’), 8.77 (d, J = 8.4 Hz, H-3’), 12.66 ppm (s, 1H, NH).
General procedure B for the synthesis of compounds 22–29
Under N2 atmosphere in a pressure tube, NaOH powder (3 equiv) was added to a solution of derivatives 14–21 (1 equiv) in dry dioxane (2 ml per mmol). The reaction mixture was stirred at 110 °C for 2–8 h, then it was poured in ice/water. The pH was acidified up to 6 with 2 N HCl and the obtained precipitate was filtered to give the desired compounds as solids. After crystallization by Et2O/EtOH, compounds were used for the next reactions.
2–(5-Propoxypyridin-2-yl)quinolin-4-ol (22)
General procedure B: time, 6 h; starting material, N-(2-acetylphenyl)-5-propoxypyridine-2-carboxamide 14 (0.23 g, 0.73 mmol). Compound 22 was obtained as a white solid (0.20 g, 100% yield), mp 190.0–191.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.95 (t, J = 7.4 Hz, 3H, OCH2CH2CH3), 3.42–3.60 (m, 2H, OCH2CH2CH3), 4.09 (t, J = 6.6 Hz, 2H, OCH2CH2CH3), 6.80 (s, 1H, H-3), 7.29 (t, J = 7.4 Hz, 1H, H-6), 7.56–7.64 (m, 2H, H-7 and H4’), 8.00 (d, J = 8.4 Hz, 1H, H-3’), 8.05 (d, J = 7.4 Hz, 1H, H-8), 8.20 (d, J = 8.8 Hz, 1H, H-5), 8.45 (d, J = 2.7 Hz, 1H, H-6’), 11.81 ppm (s, 1H, OH).
2–(6-Propoxypyridin-3-yl)quinolin-4-ol (23)
General procedure B: time, 6 h; starting material, N-(2-acetylphenyl)-6-propoxynicotinamide 15 (0.75 g, 2.51 mmol). Compound 23 was obtained as a white solid (0.40 g, 57% yield), mp 212.5–215.0 °C.1H NMR (400 MHz, DMSO-d6): δ = 0.86 (t, J = 7.5 Hz, 3H, OCH2CH2CH3), 1.65–1.74 (m, 2H, OCH2CH2CH3), 3.90 (t, J = 7.4 Hz, 2H, OCH2CH2CH3), 6.28 (s, 1H, H-3), 6.53 (d, J = 8.8 Hz, 1H, H-5’), 7.27–7.30 (m, 1H, H-6), 7.60–7.66 (m, 2H, H-7 and H-8), 7.87 (dd, J = 1.5 and 9.4 Hz, 1H, H-4’), 8.03 (d, J = 7.9 Hz, 1H, H-5), 8.38 (d, J = 2.5 Hz, 1H, H-2’), 11.45 ppm (s, 1H, OH).
2-Pyridin-2-ylquinolin-4-ol (24)
General procedure B: time, 2 h; starting material, N-(2-acetylphenyl)pyridine-2-carboxamide 16 (0.50 g, 2.08 mmol). Compound 24Citation24 was obtained as a white solid (0.28 g, 61% yield), mp 236.5–237.0 °C. 1H NMR (200 MHz, DMSO-d6): δ = 6.83 (s, 1H, H-3), 7.27 (t, J = 6.8 Hz, 1H, H-6), 7.51–7.65 (m, 2H, H-7 and H-5’), 7.90–8.05 (m, 3H, H-8, H-3’ and H-4’), 8.22 (d, J = 7.5 Hz, 1H, H-5), 8.77 (d, J = 4.9 Hz, 1H, H-6’), 11.90 ppm (s, 1H, OH).
2-Pyridin-3-ylquinolin-4-ol (25)
General procedure B: time, 7 h; starting material, N-(2-acetylphenyl)nicotinamide 17Citation25 (0.20 g, 0.83 mmol). Compound 25Citation24 was obtained as a yellow solid (0.13 g, 70% yield), mp 243.0–244.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 6.37 (s, 1H, H-3), 7.32 (t, J = 7.0 Hz, 1H, H-6), 7.58 (dd, J = 4.8 and 7.9 Hz, 1H, H-4’), 7.63–7.71 (m, 2H, H-7 and H-5’), 8.07 (d, J = 7.9 Hz, 1H, H-8), 8.20 (d, J = 7.9 Hz, 1H, H-5), 8.72 (d, J = 4.5 Hz, 1H, H-6’), 8.99 (d, J = 1.8 Hz, 1H, H-2’), 11.81 ppm (s, 1H, OH).
2-Pyridin-4-ylquinolin-4-ol (26)
General procedure B: time, 8 h; starting material, N-(2-acetylphenyl)isonicotinamide18Citation26 (1.00 g, 4.16 mmol). Compound 26Citation24 was obtained as a white solid (0.53 g, 57% yield), mp > 300 °C. 1H NMR (400 MHz, DMSO-d6): δ = 6.64 (s, 1H, H-3), 7.35 (t, J = 7.3 Hz, 1H, H-6), 7.68 (dt, J = 1.4 and 8.1 Hz, 1H, H-7), 7.76 (d, J = 8.2 Hz, 1H, H-8), 7.85 (d, J = 6.2 Hz, 2H, H-3’ and H-5’), 8.08 (dd, J = 1.3 and 8.1 Hz, 1H, H-5), 8.76 (d, J = 5.1 Hz, 2H, H-2’ and H-6’), 11.94 ppm (s, 1H, OH).
2–(2-Thienyl)quinolin-4-ol (27)
General procedure B: time, 8 h; starting material, N-(2-acetylphenyl)thiophene-2-carboxamide 19Citation27 (0.20 g, 0.82 mmol). Compound 27Citation24 was obtained as a pink solid (0.11 g, 62% yield), mp > 300 °C. 1H NMR (400 MHz, DMSO-d6): δ = 6.43 (s, 1H, H-3), 7.23–7.25 (m, 1H, H-5’), 7.30 (t, J = 7.5 Hz, 1H, H-6), 7.63 (t, J = 7.1 Hz, 1H, H-7), 7.77–7.91 (m, 3H, H-8, H-3’ and H-4’), 8.03 (d, J = 8.0 Hz, 1H, H-5), 11.74 ppm (s, 1H, OH).
2–(3-Thienyl)quinolin-4-ol (28)
General procedure B: time, 8 h; starting material, N-(2-acetylphenyl)thiophene-3-carboxamide 20 (0.20 g, 0.82 mmol). Compound 28Citation24 was obtained as a white solid (0.13 g, 72% yield), mp > 300 °C. 1H NMR (400 MHz, DMSO-d6): δ = 6.47 (s, 1H, H-3), 7.30 (t, J = 7.1 Hz, 1H, H-6), 7.63 (dd, J = 1.2 and 7.0 Hz, 1H, H-7), 7.68–7.77 (m, 3H, H-8, H-4’ and H-5’), 8.01 (d, J = 8.1 Hz, 1H, H-5), 8.24–8.31 (m, 1H, H-4’), 11.64 ppm (s, 1H, OH).
2–(5-Chloro-2-thienyl)quinolin-4-ol (29)
General procedure B: time, 8 h; starting material, N-(2-acetylphenyl)-5-chlorothiophene-2-carboxamide 21 (0.50 g, 1.79 mmol). Compound 29 was obtained as a yellow solid (0.32 g, 73% yield), mp > 300 °C. 1H NMR (200 MHz, DMSO-d6): δ = 6.74 (s, 1H, H-3), 7.28 (d, J = 4.1 Hz, 1H, H-3’), 7.40 (t, J = 7.1 Hz, 1H, H-6), 7.70 (dt, J = 2.0 and 7.1 Hz, 1H, H-7), 7.81–7.92 (m, 2H, H-8 and H-4’), 8.06 (d, J = 8.2 Hz, 1H, H-5), 11.64 ppm (s, 1H, OH).
General procedure C for the synthesis of compounds 30a,b–37a,b
To a solution of derivatives 22–29 (1 equiv) in dry DMF (10 per mmol), K2CO3 (4 equiv) and 2-chloro-N,N-dimethylethylamine hydrochloride or 1–(2-chloroethyl)piperidine hydrochloride (3 equiv) were added. The reaction mixture was stirred at 80 °C for 2–12 h, then it was poured in ice/water and extracted with EtOAc. The organic layers were washed with brine, dried over Na2SO4 and evaporated under vacuum to give crude oils. After purification by flash chromatography column, compounds (30b, 32b, 33b, 34a, 35a, 36a, 36b and 37b) were obtained as solids. Differently, to a solution of the compounds (30a, 31a, 31b, 32a, 33a, 34b, 35b and 37a) in Et2O, HClgas was bubbled and, after filtration, compounds were collected as hydrochloride solids.
N,N-diethyl-2-{[2–(5-propoxypyridin-2-yl)quinolin-4-yl]oxy}ethanamine hydrochloride (30a)
General procedure C: time, 4 h; starting materials, 2–(5-propoxypyridin-2-yl)quinolin-4-ol 22 (0.20 g, 0.71 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 97/3) and hydrochlorination, compound 30a was obtained as a white solid (0.04 g, 17% yield), mp 212.0–213.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 0.99 (t, J = 7.4 Hz, 3H, OCH2CH2CH3), 1.29 (t, J = 7.4 Hz, 6H, NCH2CH3 x 2), 1.73–1.82 (m, 2H, OCH2CH2CH3), 3.21–3.28 (m, 4H, NCH2CH3 x 2), 3.71–3.75 (m, 2H, OCH2CH2N), 4.14 (t, J = 6.6 Hz, 2H, OCH2CH2CH3), 4.93–4.97 (m, 2H, OCH2CH2N), 7.70–7.75 (m, 2H, H-6 and H-4’), 7.97 (t, J = 7.7 Hz, 1H, H-7), 8.11 (s, 1H, H-3), 8.39–8.42 (m, 2H, H-8 and H-3’), 8.53 (d, J = 2.6 Hz, 1H, H-6’), 8.80 (d, J = 8.6 Hz, 1H, H-5), 11.32 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 8.93, 10.74, 22.32, 47.32, 49.64, 66.26, 70.51, 99.93, 120.29, 122.01, 123.27, 125.30, 125.43, 125.57, 127.96, 127.99, 133.39, 133.72, 138.98, 154.37, 157.76 ppm. Anal calcd for C23H30ClN3O2: C, 66.41; H, 7.27; N, 10.10; found: C, 66.38; H, 7.28; N, 10.13.
4–(2-Piperidin-1-ylethoxy)-2–(5-propoxypyridin-2-yl)quinoline (30b)
General procedure C: time, 4 h; starting materials, 2–(5-propoxypyridin-2-yl)quinolin-4-ol 22 (0.23 g, 0.81 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CH2Cl2/MeOH 95/5), compound 30b was obtained as a white solid (0.11 g, 33% yield), mp 90.0–91.0 °C. 1H NMR (400 MHz, CDCl3): δ = 1.05 (t, J = 7.4 Hz, 3H, OCH2CH2CH3), 1.39–1.45 (m, 2H, piperidine CH2), 1.52–1.63 (m, 4H, piperidine CH2 x 2), 1.78–1.97 (m, 2H, OCH2CH2CH3), 2–49-2.61 (m, 4H, piperidine NCH2 × 2), 2.97 (t, J = 5.9 Hz, 2H, OCH2CH2N), 4.02 (t, J = 6.5 Hz, 2H, OCH2CH2CH3), 4.46 (t, J = 5.9 Hz, 2H, OCH2CH2N), 7.31 (dd, J = 2.8 and 8.7 Hz, 1H, H-4’), 7.45 (t, J = 7.3 Hz, 1H, H-6), 7.66 (t, J = 7.1 Hz, 1H, H-7), 7.89 (s, 1H, H-3), 8.03 (d, J = 8.2 Hz, 1H, H-8), 8.17 (d, J = 8.1 Hz, 1H, H-5), 8.36 (d, J = 2.5 Hz, 1H, H-6’), 8.56 ppm (d, J = 8.8 Hz, 1H, H-3’). 13C NMR (101 MHz, CDCl3): δ = 10.47, 22.51, 24.17, 26.07, 54.99, 57.63, 66.75, 69.96, 97.85, 121.09, 121.55, 121.91, 122.49, 125.34, 129.00, 129.72, 136.91, 148.95, 155.98, 157.37, 161.99 ppm. Anal calcd for C24H29N3O2: C, 73.63; H, 7.47; N, 10.73;found: C, 73.70; H, 7.46; N, 10.71.
N,N-diethyl-2-{[2–(6-propoxypyridin-3-yl)quinolin-4-yl]oxy}ethanamine hydrochloride (31a)
General procedure C: time, 2 h; starting materials, 2–(6-propoxypyridin-3-yl)quinolin-4-ol 23 (0.20 g, 0.71 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 95/5) and hydrochlorination, compound 31a was obtained as a white solid (0.19 g, 65% yield), mp 188.0–190.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 0.89 (t, J = 7.6 Hz, 3H, OCH2CH2CH3), 1.29 (t, J = 7.1 Hz, 6H, NCH2CH3 x 2), 1.72–1.82 (m, 2H, OCH2CH2CH3), 3.21–3.31 (m, 4H, NCH2CH3 x 2), 3.61–3.68 (m, 2H, OCH2CH2N), 4.00 (t, J = 7.6 Hz, 2H, OCH2CH2CH3), 4.87–5.01 (m, 2H, OCH2CH2N), 6.60 (d, J = 9.7 Hz, 1H, H-5’), 7.72 (t, J = 7.5 Hz, 1H, H-6), 7.77 (s, 1H, H-3), 7.98 (t, J = 7.8 Hz, 1H, H-7), 8.37 (d, J = 8.3 Hz, 1H, H-5), 8.41 (dd, J = 2.4 and 9.6 Hz, 1H, H-4’), 8.52–8.54 (m, 1H, H-8), 9.26 (s, 1H, H-2’), 11.16 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 9.01, 11.25, 22.49, 47.58, 49.82, 51.31, 65.19, 100.03, 112.58, 119.61, 123.10, 123.56, 123.72, 127.45, 133.25, 138.72, 142.69, 143.02, 153.97, 161.44, 164.62 ppm. Anal calcd for C23H30ClN3O2: C, 66.41; H, 7.27; N, 10.10;found: C, 66.37; H, 7.30; N, 10.12.
4–(2-Piperidin-1-ylethoxy)-2–(6-propoxypyridin-3-yl)quinoline hydrochloride (31b)
General procedure C: time, 5 h; starting materials, 2–(6-propoxypyridin-3-yl)quinolin-4-ol 23 (0.30 g, 1.09 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CH2Cl2/MeOH 99/1) and hydrochlorination, compound 31 b was obtained as a white solid (0.25 g, 54% yield), mp 223.0–224.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 0.89 (t, J = 7.4 Hz, 3H, OCH2CH2CH3), 1.32–1.42 (m, 1H, piperidine CH), 1.67–1.90 (m, 7H, piperidine CH, piperidine CH2 x 2 and OCH2CH2CH3), 3.03–3.10 (m, 2H, piperidine NCH2), 3.37–3.40 (m, 2H, piperidine NCH2), 3.67–3.68 (m, 2H, OCH2CH2N), 3.99 (t, J = 8.1 Hz, 2H, OCH2CH2CH3), 4.95–5.00 (m, 2H, OCH2CH2N), 6.60 (d, J = 9.2 Hz, 1H, H-5’), 7.71 (t, J = 7.4 Hz, 1H, H-6), 7.75 (s, 1H, H-3), 7.97 (t, J = 7.5 Hz, 1H, H-7), 8.40–8.43 (m, 2H, H-8 and H-4’), 8.45–8.51 (m, 1H, H-5), 9.22 (s, 1H, H-2’), 11.19 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 11.25, 21.63, 22.50, 22.76, 51.30, 52.89, 54.49, 64.87, 99.72, 119.64, 123.10, 124.06, 124.26, 127.26, 132.97, 133.15, 138.69, 142.37, 142.59, 154.07, 161.47, 164.12. Anal calcd for C24H30ClN3O2: C, 67.36; H, 7.07; N, 9.82;found: C, 67.42; H, 7.06; N, 9.80.
N,N-diethyl-2-[(2-pyridin-2-ylquinolin-4-yl)oxy]ethanamine hydrochloride (32a)
General procedure C: time, 12 h; starting materials, 2-pyridin-2-ylquinolin-4-ol 24 (0.30 g, 1.35 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 95/5) and hydrochlorination, compound 32a was obtained as a white solid (0.10 g, 21% yield), mp 173.0–174.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.28 (t, J = 7.2 Hz, 6H, NCH2CH3 x 2), 3.21–3.29 (m, 4H, NCH2CH3 x 2), 3.67–3.71 (m, 2H, OCH2CH2N), 4.76 (t, J = 4.9 Hz, 2H, OCH2CH2N), 7.52 (ddd, J = 1.1, 4.8 and 9.1 Hz, 1H, H-3’), 7.60 (dt, J = 1.1 and 7.0 Hz, 1H, H-6), 7.79 (dt, J = 1.4 and 7.0, 1H, H-7), 7.99 (dt, J = 1.8 and 8.0 Hz, 1H, H-5’), 7.81–8.06 (m, 2H, H-3 and H-4’), 8.25 (dd, J = 1.0 and 8.2 Hz, 1H, H-8), 8.59 (d, J = 7.9 Hz, 1H, H-5), 8.73 (dd, J = 1.5 and 5.0 Hz, 1H, H-6’), 10.70 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 9.06, 47.52, 49.92, 63.66, 120.90, 121.72, 122.35, 125.41, 126.91, 129.29, 130.99, 137.94, 148.52, 149.58, 155.25, 156.97, 161.51 ppm. Anal calcd for C20H24ClN3O: C, 67.12; H, 6.76; N, 11.74;found: C, 67.21; H, 6.76; N, 11.70.
4–(2-Piperidin-1-ylethoxy)-2-pyridin-2-ylquinoline (32b)
General procedure C: time, 4 h; starting materials, 2-pyridin-2-ylquinolin-4-ol 24 (0.21 g, 0.93 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CHCl3/MeOH 97/3), compound 32 b was obtained as a white solid (0.21 g, 69% yield), mp 77.0–79.5 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.33–1.35 (m, 2H, piperidine CH2), 1.44–1.50 (m, 4H, piperidine CH2 x 2), 2.46–2.49 (m, 4H, piperidine NCH2 2), 2.84 (t, J = 6.4 Hz, 2H, OCH2CH2N), 4.41 (t, J = 6.8 Hz, 2H, OCH2CH2N), 7.48 (ddd, J = 1.2, 5.1 and 8.9 Hz, 1H, H-3’), 7.55 (dt, J = 1.9 and 7.6 Hz, 1H, H-6), 7.75 (dt, J = 1.8 and 7.3 Hz, 1H, H-7), 7.97 (dt, J = 2.5 and 8.4 Hz, 1H, H-5’), 7.99–8.01 (m, 2H, H-3 and H-4’), 8.11 (d, J = 7.9 Hz, 1H, H-8), 8.56 (dd, J = 2.0 and 7.9 Hz, 1H, H-5), 8.70 ppm (dd, J = 1.2 and 5.4 Hz, 1H, H-6’). 13C NMR (101 MHz, DMSO-d6): δ = 24.30, 26.07, 54.79, 57.41, 67.17, 98.46, 121.19, 121.60, 122.01, 125.19, 126.67, 129.44, 130.69, 137.77, 148.71, 149.57, 155.66, 157.19, 162.04 ppm. Anal calcd for C21H23N3O: C, 75.65; H, 6.95; N, 12.60;found: C, 75.74; H, 6.93; N, 12.54.
N,N-diethyl-2-[(2-pyridin-3-ylquinolin-4-yl)oxy]ethanamine hydrochloride (33a)
General procedure C: time, 5 h; starting materials, 2-pyridin-3-ylquinolin-4-ol 25 (0.30 g, 1.35 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 90/10) and hydrochlorination, compound 33a was obtained as a white solid (0.17 g, 35% yield), mp 181.0–182.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.29 (t, J = 7.1 Hz, 6H, NCH2CH3 x 2), 3.12–3.25 (m, 4H, NCH2CH3 2), 3.70–3.71 (m, 2H, OCH2CH2N), 4.90–4.93 (m, 2H, OCH2CH2N), 7.71 (t, J = 7.5 Hz, 1H, H-6), 7.91 (t, J = 7.7 Hz, 1H, H-7), 7.98 (s, 1H, H-3), 8.09 (t, J = 6.1 Hz, 1H, H-5’), 8.25 (d, J = 8.3 Hz, 1H, H-8), 8.35 (d, J = 8.2 Hz, 1H, H-5), 8.98 (d, J = 7.9 Hz, 1H, H-6’), 9.26 (d, J = 8.1 Hz, 1H, H-4’), 9.68 (s, 1H, H-2’), 11.31 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 24.30, 26.07, 54.79, 57.41, 67.17, 98.46, 121.19, 121.60, 122.01, 125.19, 126.67, 129.44, 130.69, 137.77, 148.71, 149.57, 155.66, 157.19, 162.04 ppm. Anal calcd for C20H24ClN3O: C, 67.12; H, 6.76; N, 11.74;found: C, 67.31; H, 6.75; N, 11.70.
4–(2-Piperidin-1-ylethoxy)-2-pyridin-3-ylquinoline (33b)
General procedure C: time, 4 h; starting materials, 2-pyridin-3-ylquinolin-4-ol 25 (0.30 g, 1.35 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CH2Cl2/MeOH 95/5), compound 33 b was obtained as a white solid (0.26 g, 58% yield), mp 101.5–102.5 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.33–1.36 (m, 2H, piperidine CH2), 1.47–1.52 (m, 4H, piperidine CH2 x 2), 2.47–2.51 (m, 4H, piperidine NCH2 x 2), 2.81–2.85 (m, 2H, OCH2CH2N), 4.47 (t, J = 5.8 Hz, 1H, OCH2CH2N), 7.52–7.58 (m, 2H, H-6 and H-5’), 7.64 (s, 1H, H-3), 7.74 (t, J = 7.4 Hz, 1H, H-7), 7.99 (d, J = 8.4 Hz, 1H, H-8), 8.10 (d, J = 8.2 Hz, 1H, H-5), 8.61 (d, J = 7.9 Hz, 1H, H-6’), 8.65 (d, J = 4.7 Hz, 1H, H-4’), 9.43 ppm (s, 1H, H-2’). 13C NMR (101 MHz, DMSO-d6): δ = 24.29, 26.05, 54.76, 57.50, 67.37, 99.59, 120.49, 121.97, 124.13, 126.39, 129.36, 130.73, 134.96, 135.17, 148.94, 149.06, 150.73, 155.87, 162.26 ppm. Anal calcd for C21H23N3O: C, 75.65; H, 6.95; N, 12.60;found: C, 75.71; H, 6.93; N, 12.57.
N,N-diethyl-2-[(2-pyridin-4-ylquinolin-4-yl)oxy]ethanamine (34a)
General procedure C: time, 4 h; starting materials, 2-pyridin-4-ylquinolin-4-ol 26 (0.30 g, 1.35 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 90/10), compound 34a was obtained as a white solid (0.22 g, 51% yield), mp 73.5–74.0 °C. 1H NMR (400 MHz, CDCl3): δ = 1.09 (t, J = 6.1 Hz, 6H, NCH2CH3 x 2), 2.70 (q, J = 7.3 Hz, 4H, NCH2CH3 x 2), 3.05 (t, J = 6.0 Hz, 2H, OCH2CH2N), 4.34 (t, J = 6.2 Hz, 2H, OCH2CH2N), 7.19 (s, 1H, H-3), 7.50 (t, J = 7.2 Hz, 1H, H-6), 7.71 (t, J = 6.8 Hz, 1H, H-7), 7.97–7.99 (m, 2H, H-3’ and H-5’), 8.08 (d, J = 8.9 Hz, 1H, H-8), 8.18 (d, J = 8.3 Hz, 1H, H-5), 8.71–8.77 ppm (m, 2H, H-2’ and H-6’). 13C NMR (101 MHz, CDCl3): δ = 12.08, 48.06, 51.40, 67.68, 98.19, 120.92, 121.66, 121.80, 126.15, 129.43, 130.29, 147.29, 149.20, 150.41, 155.83, 162.52 ppm. Anal calcd for C20H23N3O: C, 74.74; H, 7.21; N, 13.07;found: C, 74.81; H, 7.19; N, 13.01.
4–(2-Piperidin-1-ylethoxy)-2-pyridin-4-ylquinoline hydrochloride (34b)
General procedure C: time, 4 h; 2-pyridin-4-ylquinolin-4-ol 26 (0.30 g, 1.35 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CH2Cl2/MeOH 99/1) and hydrochlorination, compound 34 b was obtained as a white solid (0.30 g, 61% yield), mp 183.5–185.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.33–1.42 (m, 2H, piperidine CH2), 1.66–1.93 (m, 4H, piperidine CH2 × 2), 3.04–3.09 (m, 2H, piperidine NCH2), 3.51–3.54 (m, 2H, piperidine NCH2), 3.63–3.67 (m, 2H, OCH2CH2N), 4.93 (t, J = 5.0 Hz, 2H, OCH2CH2N), 7.69 (t, J = 7.3 Hz, 1H, H-6), 7.87 (t, J = 7.0 Hz, 1H, H-7), 8.02 (s, 1H, H-3), 8.15 (d, J = 8.4 Hz, 1H, H-8), 8.35 (d, J = 8.1 Hz, 1H, H-5), 8.92 (d, J = 6.7 Hz, 2H, H-3’ and H-5’), 9.08 (d, J = 6.7 Hz, 2H, H-2’ and H-6’), 11.42 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 21.66, 22.72, 52.74, 54.52, 64.34, 101.11, 121.09, 122.73, 124.99, 128.38, 129.29, 131.92, 143.00, 148.19, 152.38, 153.36, 162.55 ppm. Anal calcd for C21H24ClN3O: C, 68.19; H, 6.54; N, 11.36;found: C, 68.01; H, 6.56; N, 11.40.
N,N-diethyl-2-{[2–(2-thienyl)quinolin-4-yl]oxy}ethanamine (35a)
General procedure C: time, 4 h; starting materials, 2–(2-thienyl)quinolin-4-ol 27 (0.30 g, 1.32 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 98/2), compound 35a was obtained as a white solid (0.26 g, 60% yield), mp 84.0–86.0 °C. 1H NMR (400 MHz, CDCl3): δ = 0.99 (t, J = 7.0 Hz, 6H, NCH2CH3 2), 2.58 (q, J = 7.0 Hz, 4H, NCH2CH3 x 2), 2.93 (t, J = 5.5 Hz, 2H, OCH2CH2N), 4.37 (t, J = 5.4 Hz, 2H, OCH2CH2N), 7.17–7.19 (m, 1H, H-4’), 7.47 (t, J = 7.3 Hz, 1H, H-6), 7.54 (s, 1H, H-3), 7.65–7.69 (m, 2H, H-7 and H-5’), 7.84 (d, J = 8.4 Hz, 1H, H-8), 8.02–8.04 ppm (m, 2H, H-5 and H-3’). 13C NMR (101 MHz, CDCl3): δ = 12.50, 47.53, 51.37, 68.08, 98.05, 120.52, 121.94, 125.77, 127.52, 128.61, 128.68, 129.89, 130.70, 145.74, 148.71, 153.64, 161.91 ppm. Anal calcd for C19H22N2OS: C, 69.90; H, 6.79; N, 8.58;found: C, 69.99; H, 6.77; N, 8.58.
4–(2-piperidin-1-ylethoxy)-2–(2-thienyl)quinoline hydrochloride (35b)
General procedure C: time, 5 h; 2–(2-thienyl)quinolin-4-ol 27 (0.30 g, 1.32 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CH2Cl2/MeOH 95/5) and hydrochlorination, compound 35 b was obtained as a white solid (0.29 g, 58% yield), mp 208.0–209.5 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.66–1.69 (m, 2H, piperidine CH2), 1.76–1.89 (m, 4H, piperidine CH2 2), 3.01–3.14 (m, 2H, piperidine NCH2), 3.42–3.55 (m, 2H, piperidine NCH2), 3.62–3.71 (m, 2H, OCH2CH2N), 4.87–4.89 (m, 2H, OCH2CH2N), 7.28 (t, J = 4.1 Hz, 1H, H-4’), 7.59 (t, J = 7.4 Hz, 1H, H-6), 7.62 (s, 1H, H-3), 7.83 (t, J = 7.6 Hz, 1H, H-7), 7.88 (d, J = 4.5 Hz, 1H, H-5’), 8.16 (d, J = 6.9 Hz, 1H, H-8), 8.30 (d, J = 8.2 Hz, 1H, H-5), 8.34 (bs, 1H, H-3’), 11.33 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 21.61, 22.81, 52.82, 54.64, 64.21, 99.15, 120.07, 122.80, 126.68, 129.13, 129.63, 130.14, 131.68, 132.03, 144.46, 146.35, 152.64, 162.50 ppm. Anal calcd for C20H23ClN2OS: C, 64.07; H, 6.18; N, 7.47;found: C, 64.11; H, 6.17; N, 7.45.
N,N-diethyl-2-{[2–(3-thienyl)quinolin-4-yl]oxy}ethanamine (36a)
General procedure C: time, 4 h; starting materials, 2–(3-thienyl)quinolin-4-ol 28 (0.30 g, 1.32 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 95/5), compound 36a was obtained as a white solid (0.13 g, 30% yield), mp 66.0–67.0 °C. 1H NMR (400 MHz, CDCl3): δ = 1.12 (t, J = 7.3 Hz, 6H, NCH2CH3 2), 2.71 (q, J = 7.0 Hz, 4H, NCH2CH3
2), 3.07 (t, J = 6.1 Hz, 2H, OCH2CH2N), 4.34 (t, J = 6.1 Hz, 2H, OCH2CH2N), 7.09 (s, 1H, H-3), 7.39–7.47 (m, 2H, H-6 and H-8), 7.66 (dt, J = 1.4 and 8.4 Hz, 1H, H-7), 7.80 (dd, J = 1.3 and 5.0 Hz, 1H, H-5’), 7.98 (dd, J = 1.1 and 4.4 Hz, 1H, H-4’), 8.02 (d, J = 8.5 Hz, 1H, H-5), 8.12 ppm (dd, J = 1.1 and 8.3 Hz, 1H, H-2’). 13C NMR (101 MHz, CDCl3): δ = 11.92, 47.48, 51.29, 67.16, 98.55, 120.39, 121.64, 124.44, 125.18, 126.27, 126.85, 128.95, 129.96, 143.12, 149.19, 154.51, 161.85 ppm. Anal calcd for C19H22N2OS: C, 69.90; H, 6.79; N, 8.58;found: C, 69.94; H, 6.78; N, 8.56.
4–(2-Piperidin-1-ylethoxy)-2–(3-thienyl)quinoline (36b)
General procedure C: time, 3 h; starting materials, 2–(3-thienyl)quinolin-4-ol 28 (0.30 g, 1.32 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CH2Cl2/MeOH 98/2), compound 36b was obtained as a white solid (0.27 g, 61% yield), mp 109.5–111.0 °C. 1H NMR (400 MHz, CDCl3): δ = 1.44–1.48 (m, 2H, piperidine CH2), 1.60–1.66 (m, 4H, piperidine CH2 2), 2.55–2.61 (m, 4H, piperidine CH2
2), 2.98 (t, J = 6.8 Hz, 2H, OCH2CH2N), 4.40 (t, J = 6.0 Hz, 2H, OCH2CH2N), 7.08 (s, 1H, H-3), 7.40–7.48 (m, 2H, H-6 and H-8), 7.66 (dt, J = 1.5 and 8.3 Hz, 1H, H-7), 7.80 (dd, J = 1.4 and 5.0 Hz, 1H, H-5’), 7.97 (dd, J = 1.1 and 4.2 Hz, 1H, H-4’), 8.01 (d, J = 8.2 Hz, 1H, H-5), 8.13 ppm (dd, J = 1.1 and 8.3 Hz, 1H, H-2’). 13C NMR (101 MHz, CDCl3): δ = 24.01, 25.94, 55.13, 57.52, 66.67, 98.55, 120.41, 121.68, 124.43, 125.18, 126.27, 126.85, 128.94, 129.97, 143.12, 149.18, 154.52, 161.78 ppm. Anal calcd for C20H22N2OS: C, 70.97; H, 6.55; N, 8.28;found: C, 70.85; H, 6.56; N, 8.30.
(2-{[2–(5-Chloro-2-thienyl)quinolin-4-yl]oxy}ethyl)diethylamine hydrochloride (37a)
General procedure C: time, 4 h; starting materials, 2–(5-chloro-2-thienyl)quinolin-4-ol 29 (1.00 g, 4.14 mmol) and (2-chloroethyl)diethylamine hydrochloride. After purification (CH2Cl2/MeOH 95/5) and hydrochlorination, compound 37a was obtained as a white solid (0.50 g, 32% yield), mp 189.0–191.0 °C. 1H NMR (400 MHz, DMSO-d6): δ = 1.28 (t, J = 7.2 Hz, 6H, NCH2CH3 × 2), 3.19–3.26 (m, 4H, NCH2CH3 × 2), 3.65–3.83 (m, 2H, OCH2CH2N), 4.76–4.78 (m, 2H, OCH2CH2N), 9.76 (d, J = 4.1 Hz, 1H, H-3’), 7.54 (t, J = 7.0 Hz, 1H, H-6), 7.63 (s, 1H, H-3), 7.74 (dt, J = 1.1 and 7.0 Hz, 1H, H-7), 7.92 (d, J = 8.3 Hz, 1H, H-8), 8.05 (d, J = 4.0 Hz, 1H, H-4’), 8.18 (d, J = 8.1 Hz, 1H, H-5), 11.07 ppm (s, 1H, HCl). 13C NMR (101 MHz, DMSO-d6): δ = 9.01, 47.40, 49.68, 64.01, 98.10, 120.29, 122.47, 126.48, 127.91, 128.10, 128.81, 131.44, 132.64, 141.11, 145.34, 152.34, 161.72 ppm. Anal calcd for C19H22Cl2N2OS: C, 57.43; H, 5.58; N, 7.05;found: C, 57.50; H, 5.56; N, 7.04.
2–(5-Chloro-2-thienyl)-4–(2-piperidin-1-ylethoxy)quinoline (37b)
General procedure C: time, 3 h; starting materials, 2–(5-chloro-2-thienyl)quinolin-4-ol 29 (0.29 g, 1.20 mmol) and 1–(2-chloroethyl)piperidine hydrochloride. After purification (CH2Cl2/MeOH 95/5), compound 37b was obtained as a white solid (0.14 g, 35% yield), mp 99.0–101.0 °C. 1H NMR (400 MHz, CDCl3): δ = 1.47–1.51 (m, 2H, piperidine CH2), 1.66–1.71 (m, 4H, piperidine CH2 × 2), 2.58–2.65 (m, 4H, piperidine NCH2 × 2), 3.03 (t, J = 5.7 Hz, 2H, OCH2CH2N), 4.44 (t, J = 5.7 Hz, 2H, OCH2CH2N), 6.92 (d, J = 4.0 Hz, 1H, H-3’), 7.01 (s, 1H, H-3), 7.39–7.43 (m, 2H, H-6 and H-4’), 7.64 (dt, J = 1.5 and 8.6 Hz, 1H, H-7), 7.94 (d, J = 8.2 Hz, 1H, H-8), 8.07 ppm (d, J = 7.3 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3): δ = 23.72, 25.52, 55.00, 57.27, 66.34, 93.14, 120.55, 121.62, 124.45, 125.40, 127.07, 128.72, 130.22, 133.31, 144.40, 148.91, 152.48, 161.60 ppm. Anal calcd for C20H21ClN2OS: C, 64.42; H, 5.68; N, 7.51;found: C, 64.50; H, 5.66; N, 7.50.
Microbiological procedures
The strains of S. aureus used in this study included SA-1199B, which overexpresses norA and possesses an A116E GrlA substitution, and its isogenic parent SA-1199 (norA wt). MICs assays were performed using broth microdilution method in 96 wells-microtiter plates, following the CLSI guidelinesCitation28.
Chequerboard assays and time-kill curves were performed as previously describedCitation29. The formers were performed using 2-fold increasing concentrations of both antibiotic (from 0.001 to 20 µg/mL) and compounds (from 0.39 to 25 µg/mL), while time-kill curves were performed testing CPX concentrations ranging from ¼x to 1x MIC alone and in combination with the compound 37a at 3.13 and 6.25 µg/mL. The dynamic of the bactericidal activity of the combination CPX-Compound was evaluated by CFU counts after 2, 4, 6, 8 and 24 h incubation at 37 °C.
FIC values for 37a and CPX were calculated according to the following equation: MIC in combination/MIC alone. FICI values were calculated by the sum of FIC values of 37a and CPX.
The loss of EtBr from S. aureus SA-1199B was determined fluorometrically as previously describedCitation30. The effect of various concentrations of tested compounds on the EtBr efflux of SA-1199B was compared to that in their absence, allowing the calculation of the percentage reduction in efflux.
Haemolysis assays were performed as previously describedCitation31.
Cell viability assays for compounds 1 and 37a were performed on human leukemic monocyte cell line (THP-1). THP-1 cells were grown in RPMI 1640 supplemented with 10% heat-inactivated foetal calf serum, 10,000 units of penicillin, and 10 µg of streptomycin/mL overnight to confluence. Monolayers were treated for 24 h at 37 °C with scalar concentration of tested compounds (0 − 250 µg/mL). Cell viability was then evaluated using an ATP bioluminescence kit (Via Light kit; Cambrex). Results are expressed as 50% cytotoxic concentration (CC50). The CC50 was defined as the concentration required to reduce the cell number by 50% compared to that for the untreated controls. Each concentration was tested in triplicate.
Membrane potential assays were carried out by measuring the effect of 1 and 37a on the membrane potential using the BacLight Bacterial Membrane Potential Kit (Molecular Probes, Life Technologies) according to the manufacturer’s instructions. Briefly, SA-1199B was grown in MHB at 37 °C until reaching an OD600 of 0.6. Bacterial cells were then washed in PBS and diluted to 1 × 106 CFU/mL with filtered PBS (filter 0.22 µm) in flow cytometry tubes. Ten microliters of 3 mM 3,3-diethyloxacarbocyanine iodide (DiOC2(3) in DMSO) was added to each tube (final concentration 30 µM) and mixed. Then, 1 or 37a from a stock solution in DMSO (10 mg/mL) was added to reach final concentrations of 1, 5, and 10 µg/mL. As positive control, 10 µL of 500 µM carbonyl cyanide m-chlorophenylhydrazone (CCCP, final concentration 5 µM) was used to eradicate the proton gradient by eliminating the membrane potential. The samples were analysed after 30 min by measuring the fluorescence using a cytometer Attune NxT (ThermoFisher Scientific) with a laser emitting at 488 nm and collecting in the green and red channels. The red to green fluorescence ratio was determined and normalised against the emission from the DiOC2(3) blank tube having 1 ml of the bacterial suspension and a final concentration of DiOC2(3) of 30 µM. The results are presented as the percentage of depolarised membranes compared with the drug-free control.
In silico ADME
Quinoline derivatives 1, 2 and 37a were built using the Schrodinger Maestro InterfaceCitation32 and then imported in MetaSiteCitation33. The protonation state of the compounds was normalised at a pH of 7.5. The MetaSite analysis was performed using the liver enzymes model. Compond 37a was built in SeeSARCitation34 which automatically assigns the proper geometry, protonation state and tautomeric form of the ligands using ProToss methodCitation35. The Optibrium models for PK properties predictions were downloaded from Optibrium webpageCitation36.
Results and discussion
Chemistry
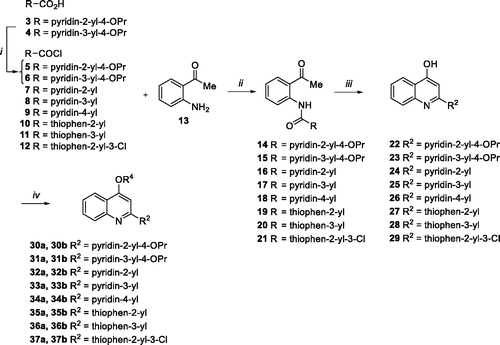
All designed compounds 30a,b–37a,b were synthesised according to the procedure depicted in Scheme 1. Chlorination of acids 3 and 4 with SOCl2 afforded acyl chlorides 5 and 6 that were immediately reacted with the commercially available aminoacetophenone 13 in presence of Et3N to give the amide derivatives 14 and 15. Similarly, coupling reaction of commercially available acyl chlorides 7–12 with 13 afforded amides 16–20Citation24 and 21. Following the same procedure reported by Brouwer et al.Citation37, ring closure of 14–21 through NaOH in dioxane at 110 °C in a pressure tube gave the C-2 aryl quinoline derivatives 22, 23, 24–28Citation24 and 29. O-alkylation of 4-hydroxyquinolines 22–29 with 2-chloro-N,N-dimethylethylamine hydrochloride or 1–(2-chloroethyl)piperidine hydrochloride afforded desired quinoline analogues 30a,b–37a,b (Scheme 1).
Scheme 1. (i) SOCl2, 60 °C, 30 min; (ii) Et3N, dry THF, rt, 90 min-6 h, 50–99%; (iii) NaOH, dry dioxane, 110 °C, 2–8 h, 57–100%; (iv) chloroalkylamine hydrochloride, K2CO3, dry DMF, 80 °C, 2–12 h, 17–69%.
Synergistic assays
In order to quickly identify derivatives having significant synergistic activity, the 16 compounds were firstly assayed at 25 µg/mL in combination with scalar concentrations of CPX against the norA overexpressing S. aureus strain SA-1199B (norA+/GrlA mutation)Citation38 (data not shown). Four compounds (30a, 30b, 35b and 36b) showed a significant synergistic effect producing a 4-fold reduction of the antibiotic MIC while derivative 37a exhibited an impressive CPX MIC reduction (32-fold). Worthy of note, among the five active compounds, three were thiophene derivatives (35b, 36b and 37a) displaying their activity regardless of the position of the sulphur atom on the thiophene ring. On the contrary, only pyridine-2-yl-5-OPr derivatives (30a and 30b) exhibited a significant synergistic effect whereas all the remaining pyridine analogues (31a, 31b, 32a, 32b, 33a, 33b, 34a and 34b) lost EPI activity regardless of the nitrogen atom position within the pyridine moiety.
Chequerboard assays
The five derivatives showing the best synergistic activity were tested by chequerboard assays in combination with CPX against S. aureus SA-1199B (), also including reference compounds 1 and 2. The two quinoline analogues (35b and 36b) with the C-2 unsubstituted thiophene portion did not retain any synergistic effect with CPX when their concentration did fall below 25 µg/mL. On the other hand, pyridine derivatives 30a and 30b at concentrations of 6.25 and 12.5 µg/mL, respectively, reduced the CPX MIC by 4-fold, thereby retaining an EPI activity comparable to 1. Interestingly, the chlorothiophene derivative 37a showed the best results being able, up to 0.39 µg/mL, to decrease by 4-fold the CPX MIC against SA-1199B, resulting about 16-fold more active than the starting hit 1. Actually, compound 37a exhibited a synergistic activity with CPX greater than that shown by compound 2, which produced a weaker and less significant effect (2-fold MIC reduction) at the same concentration (0.39 µg/mL).
Figure 2. Chequerboard assays of compounds 30a, 30b, 35b, 36b, 37a and reference compounds 1 and 2 in combination with CPX against SA-1199B.
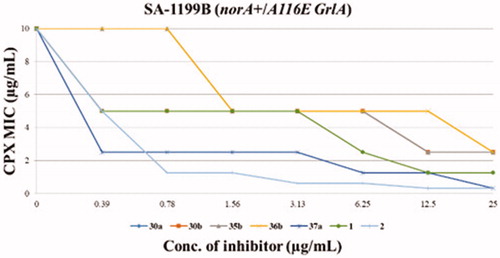
Thus, when rationalising SAR information, it appears clear as the replacement of the phenyl portion at quinoline C-2 with a thiophene ring was well tolerated when both O-ethylamino chains were present at the quinoline C-4 position. Interestingly, in the case of derivative 37a, the substitution of the H-bond acceptor –OPr group with a chlorine atom yielded very good results, paving the way for future modifications. On the contrary, the replacement of the phenyl portion at the quinoline C-2 position with pyridine moieties, regardless the presence of –OPr group, afforded less potent EPI derivatives. 30a and 30b compounds, when tested at the concentration of 25 µg/mL (synergistic assays) showed a potent synergistic effect with CPX while at lower concentrations (chequerboard assays), they displayed a poor ability to decrease the CPX MIC.
Focussing on the best identified compound 37a, to indirectly rule out that its synergism with CPX against SA-1199B was due to non-specific effects, this derivative was tested at 25 µg/mL (a concentration 64-fold higher than that needed to reduce by 4-fold CPX MIC) in combination with scalar concentrations of CPX against the S. aureus strain SA-1199 (norA wild-type). Interestingly, compound 37a did not exhibit any significant synergistic effect with CPX in presence of a norA basal expression (data not shown). Whether the synergistic activity of 37a with CPX was due to a mechanism different from that of the NorA inhibition, we would have likely to observe a synergistic effect with CPX also against SA-1199 wild-type strain. Consequently, the observed high difference in the synergistic activity of 37a with CPX against SA-1199B and SA-1199 strongly supports a 37a-mediated NorA inhibition.
MIC evaluation
Considering the very promising results of derivative 37a, it was tested alone against SA-1199B in order to prove that the synergistic activity with CPX was not influenced by a direct antibacterial effect. Worthy of note, up to 25 µg/mL, this compound did not show any antibacterial activity. This result is essential for a potential EPI compound; indeed, a poor or absent antibacterial effect is a key requirement to avoid the evolutionary pressure on microorganisms that can evolve resistance only towards compounds endowed with an antimicrobial activityCitation39. In this case, compound 37a reduces CPX MIC (from 10 µg/mL to 2.5 µg/mL) at a concentration ≥64-fold lower than its MIC. As a confirmation of the synergistic effect of 37a with CPX, we calculated the FIC values for both compounds: FIC37a < 0.0156 and FICCPX = 0.25. The sum of both FIC values led to a FIC index (FICI) < 0.27 showing a significant synergistic effect as widely recognised for FICI values <0.5Citation40. Therefore, although FICI evaluation is mainly used to determine the synergism between two antimicrobial agents, the EPI 37a, devoid of any antibacterial effect, exhibited a clear synergistic activity with the fluoroquinolone CPX.
EtBr efflux assays
Since EtBr is a known NorA substrate resulting fluorescent only when inside bacterial cells, monitoring bacterial fluorescence of SA-1199B, overexpressing norA gene, through a fluorimeter is a fast method to indirectly evaluate NorA inhibition. Thus, to demonstrate that the synergistic effect of 37a with CPX was due to the NorA efflux inhibition, we performed EtBr efflux assays against SA-1199B. Interestingly, at 50 µM compound 37a exhibited 74.3 ± 3.5% of EtBr efflux inhibition, thus confirming its ability to inhibit NorA efflux pump. Indeed, we commonly consider as active NorA EPIs those compounds having an EtBr efflux inhibition ≥70%Citation17,Citation18.
Time-kill curves
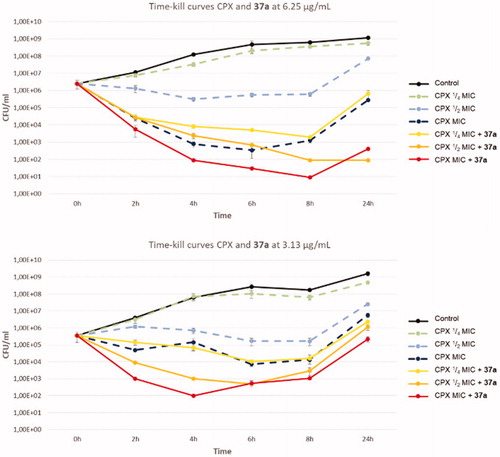
Subsequently, we decided to test the synergistic effect of compound 37a (at 3.13 and 6.25 µg/mL) when combined with CPX in time-kill curve analysis against SA-1199B (). Interestingly, at both concentrations, derivative 37a exhibited a strong effect on the bactericidal activity of CPX, thereby proving its efficacy when combined with CPX over 24 h. Indeed, the combination of compound 37a (at both the used concentrations) and CPX at ¼ MIC yielded the same bactericidal effect as CPX tested at its MIC. Moreover, the same concentrations of 37a combined with CPX at its MIC showed a strongly significant and definitely higher bactericidal activity than that shown by CPX alone over the all 24 h. These findings highlight the high potential of the chlorothiophene derivative 37a in potentiating CPX activity against resistant strains.
Figure 3. Time-kill curves of CPX and combination of compound 37a with different concentrations of CPX against SA-1199B.
Cytotoxicity assays
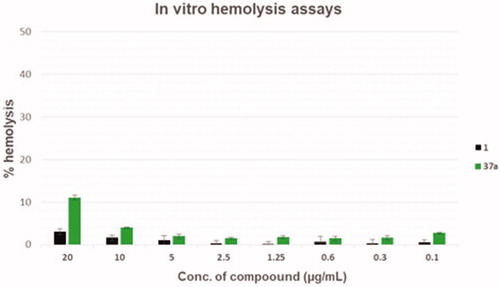
In order to evaluate the toxic effects of the derivative 37a, haemolysis assays were performed at scalar compound concentrations in comparison with the starting hit 1 (). Although derivative 37a showed a slightly higher haemolytic effect than the starting hit 1, when considering the activity-toxicity relationship, the chlorothiophene derivative 37a exhibited an improved safety profile. In particular, 37a at 20 µg/mL exhibited about 10% of haemolytic effect, a value 3-fold higher than the starting hit 1, when tested at the same concentration. However, at concentrations ≤ 5 µg/mL (about 12-fold higher than that needed to reach synergism with CPX), compound 37a reduced its haemolytic activity below 2%.
Figure 4. Haemolysis assays of compounds 37a and starting hit 1.
In addition, to further evaluate the toxic profile of compound 37a, we determined its CC50 against human monocytic cells (THP-1); compound 1 was also tested for comparison (). To note, on THP-1 cells derivative 37a exhibited a CC50 of 6.33 µg/mL, a concentration slightly lower than the CC50 of starting hit 1. However, when compared with compound 2, previously tested by usCitation19, the toxicity of the chlorothiophene derivative 37a resulted significantly increased. Therefore, it appeared evident that the replacement of the C-2 pOPr phenyl moiety (compound 1) with a 5-chloro-2-thiophene portion (compound 37a) led to a significant increase in the synergistic activity with CPX against SA-1199B while retaining the same toxic profile. Therefore, comparing both the concentrations needed to reduce by 4-fold CPX MIC and the respective CC50 values on THP-1 of 1 and 37a, it was clear that 37a exhibited a significant improvement in terms of “selectivity index” (16.2 for 37a and 1.6 for 1—). On the other hand, the presence of a –OMe substituent at the C-6 and an O-ethylpiperidine chain at C-4 of the quinoline core resulted essential to reduce the cytotoxicity against THP-1 cells as well as to confer a potent NorA EPI activity. Thus, thinking in terms of EPI activity, the introduction of the chlorothiophene moiety at the quinoline C-2 position is strongly recommended to boost the synergistic effect with CPX against SA-1199B; yet the increase in toxicity towards human cells should be considered. Therefore, these results could suggest further chemical modifications leading to new potent and safe quinoline-based NorA EPIs.
Table 1. Cytotoxicity assays against THP-1 cells of compounds 1, 2 and 37a and their calculated “selectivity index.”
Membrane polarisation assays
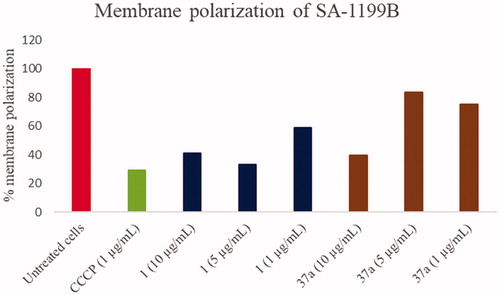
Since efflux pumps need a proton gradient along the bacterial membrane, its modification/disruption can lead to an efflux inhibition. However, this effect cannot be related to a specific EPI activity; on the contrary, it relies on a non-specific interruption of the energy source (protons) that efflux pumps use to extrude their substrates by an antiport mechanism. A typical example of a compound inhibiting efflux pumps activity by this non-specific mechanism is represented by carbonyl cyanide m-chlorophenyl hydrazone (CCCP). Therefore, in order to know whether compound 37a might counteract the NorA-mediated CPX efflux by disrupting proton motive force, we performed membrane polarisation assays against SA-1199B treated with 37a at three different concentrations (1, 5, and 10 µg/mL); starting hit 1 (at the same concentrations) for comparison and CCCP at 1 µg/mL as positive control were included. Membrane polarisation was assessed by cytofluorimeter analysis using the fluorescent probe 3,3-diethyloxacarbocyanine iodide (DiOC2(3)) as its distribution is proton gradient-sensitive. DiOC2(3) in the presence of a bacterial membrane potential exhibits red fluorescence that shifts to green emission as the membrane potential is lost, thereby allowing calculation of the percentage of membrane polarisation by a red/green fluorescence ratioCitation41.
Overall, the effect of the concentrations of 1 and 37a on S. aureus membrane polarisation was dose-dependent. Indeed, both 1 and 37a at 10 µg/mL extensively (>50%) depolarised S. aureus membrane. On the contrary, when 1 and 37a were tested at 5 and 1 µg/mL, starting 1 retained a significant depolarising effect on the bacterial membrane while 37a reduced its influence on the proton motive force. Indeed, at 5 and 1 µg/mL, compound 37a was able to depolarise only about 20% of the bacterial membrane similarly to 2, as previously reported by usCitation19. Since 37a retained a significant synergistic effect with CPX at a concentration as low as 0.39 µg/mL, we can observe that most of its activity is due to the NorA inhibition and not to a non-specific effect on the S. aureus membrane.
Thus, the replacement of the pOPr-phenyl moiety of compound 1 with the chlorothiophene portion (compound 37a), could reduce the depolarising effect on the bacterial membrane while increasing the synergistic effect with CPX; interestingly, this improvement in EPI activity can be related to a significant NorA inhibition thereby excluding an extensive disruption of the proton motive force ().
Figure 5. Membrane polarisation assays of compounds 1 and 37a against SA-1199B at three different concentrations (1, 5 and 10 µg/mL) using the BacLight Bacterial Membrane Potential Kit. CCCP was used as positive control at 1 µg/mL (5 µM). % of membrane polarisation was calculated from the red/green fluorescence ratio by comparing bacterial cells in the presence of compounds with untreated cells.
In silico ADME studies
We have previously demonstrated that our potent quinoline-based NorA EPI 2 exhibited a good metabolic stability in both in silico and in vitro experiments. However, demethylation of the C-6 methoxy group of 2 produced the most abundant metabolite after 2 h of incubation with mouse liver microsomesCitation19.
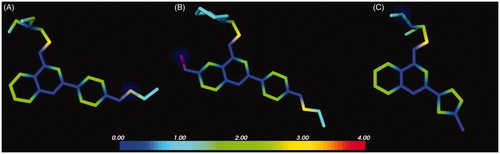
Therefore, given the comparable EPI activity between derivatives 2 and 37a, a prediction of the metabolic stability for the latter compound was planned by means of MetaSite softwareCitation33, with the inclusion of the initial hit 1 and the potent derivative 2 for comparison. In agreement with the previous resultsCitation19, the in silico protocol correctly predicted the methoxy moiety as the most reactive group as well as the most probable site of metabolism of quinoline 2 (). By contrast, no highly reactive atoms were foreseen for derivatives 1 and 37a (). These results suggested that the chlorothiophene portion of compound 37a represented a less reactive moiety than the pOPr chain present in derivatives 1 and 2, underlining that the new potent EPI 37a could couple the good biological activity of 2 with an enhanced metabolic stability. Indeed, the comparison between the NorA EPI activity and predicted metabolic stability of analogues 1, 2 and 37a showed that the use of the chlorothiophene moiety could lead to potent NorA quinoline-based EPIs missing the metabolic reactivity problems encountered with the introduction of the C-6 methoxy group as in derivative 2.
Figure 6. Predicted reactivity for compounds 1 (A), 2 (B) and 37a (C). The atoms are colour-coded based on their predicted reactivity (red: high reactivity; blue: low reactivity). The blue sphere highlights the most probable site of metabolism.
In addition, it is noteworthy that 37a obeyed the Lipinski’s rule of five, showing a lower molecular weight when compared to derivatives 1 and, in particular, 2 (361, 379 and 431 Da, respectively) and had computed physicochemical and pharmacokinetic descriptors in line with the recommended guidelines for orally dosed compounds ()Citation42.
Table 2. Predicted physicochemical and ADME descriptors for derivative 37a.
Conclusions
In order to identify new potent EPIs to counteract the rapid insurgence of bacterial resistance towards common antibiotics, we made a further effort to build a robust SAR delineation around the quinoline-based NorA inhibitors. Herein, we have described the design, synthesis and biological evaluation of new 2-arylquinoline derivatives. In particular, a new chlorothiophene analogue (37a) endowed with high synergistic effect with CPX against SA-1199B, was identified. To balance the lack of biophysical experiments proving compound binding to NorA pump and in an attempt to exclude potential non-specific effects, we carried out further experiments supporting that a NorA inhibition could produce the observed synergistic activity of 37a. Indeed, the demonstration that our compound did inhibit EtBr efflux in a phenotypic assay and did not extensively depolarise S. aureus membrane strongly underpinned that 37a can specifically inhibit NorA. In addition, time-kill curves of this EPI combined with CPX displayed the high potential of an EPI in boosting CPX bactericidal effect.
Data collected on 2-arylquinoline derivatives will be useful to definitely obtain a set of compounds having potent NorA EPI activity, poor non-specific effects on bacterial membrane and an acceptable cytotoxic profile in order to justify the use of these EPIs in animal models of infections.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- WHO. Antimicrobial resistance. Bull World Health Organ 2014; 61:383–94.
- Ten threats to global health in 2019. Available from: https://www.who.int/emergencies/ten-threats-to-global-health-in-2019 [last accessed 7 Apr 2019].
- Opperman TJ, Nguyen ST. Recent advances toward a molecular mechanism of efflux pump inhibition. Front Microbiol 2015;6:1–16.
- Piddock LJV. Understanding the basis of antibiotic resistance: a platform for drug discovery. Microbiology 2014;160:2366–73.
- Wright GD. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol 2016;24:862–71.
- Brown D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat Rev Drug Discov 2015;14:821–32.
- Piddock LJV. Multidrug-resistance efflux pumps - not just for resistance. Nat Rev Microbiol 2006; 4:629–36.
- Ricci V, Tzakas P, Buckley A, et al. Ciprofloxacin-resistant Salmonella enterica serovar typhimurium strains are difficult to select in the absence of AcrB and TolC. Antimicrob Agents Chemother 2006;50:38–42.
- Zhang Q, Lambert G, Liao D, et al. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science 2011;333:1764–7.
- Schillaci D, Spanò V, Parrino B, et al. Pharmaceutical approaches to target antibiotic resistance mechanisms. J Med Chem 2017;60:8268–97.
- Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin Infect Dis 2009;48:1–12.
- Schindler BD, Kaatz GW. Multidrug efflux pumps of Gram-positive bacteria. Drug Resist Updat 2016;27:1–13.
- Singh S, Kalia NP, Joshi P, et al. Boeravinone B, a novel dual inhibitor of NorA bacterial efflux pump of Staphylococcus aureus and human P-glycoprotein, reduces the biofilm formation and intracellular invasion of bacteria. Front Microbiol 2017;8:1868.
- Lepri S, Buonerba F, Goracci L, et al. Indole based weapons to fight antibiotic resistance: a structure-activity relationship study. J Med Chem 2016;59:867–91.
- Schindler BD, Jacinto P, Kaatz GW. Inhibition of drug efflux pumps in Staphylococcus aureus: current status of potentiating existing antibiotics. Future Microbiol 2013;8:491–507.
- Rath SK, Singh S, Kumar S, et al. Synthesis of amides from (E)-3-(1-chloro-3,4-dihydronaphthalen-2-yl)acrylic acid and substituted amino acid esters as NorA efflux pump inhibitors of Staphylococcus aureus. Bioorg Med Chem 2019;27:343–53.
- Sabatini S, Gosetto F, Iraci N, et al. Re-evolution of the 2-phenylquinolines: ligand-based design, synthesis, and biological evaluation of a potent new class of Staphylococcus aureus NorA efflux pump inhibitors to combat antimicrobial resistance. J Med Chem 2013;56:4975–89.
- Astolfi A, Felicetti T, Iraci N, et al. Pharmacophore-based repositioning of approved drugs as novel Staphylococcus aureus NorA efflux pump inhibitors. J Med Chem 2017;60:1598–604.
- Felicetti T, Cannalire R, Pietrella D, et al. 2-Phenylquinoline S. aureus NorA efflux pump inhibitors: evaluation of the importance of methoxy group introduction. J Med Chem 2018;61:7827–48.
- Felicetti T, Cannalire R, Nizi MG, et al. Studies on 2-phenylquinoline Staphylococcus aureus NorA efflux pump inhibitors: new insights on the C-6 position. Eur J Med Chem 2018;155:428–33.
- Cannalire R, Machado D, Felicetti T, et al. Natural isoflavone biochanin A as a template for the design of new and potent 3-phenylquinolone efflux inhibitors against Mycobacterium avium. Eur J Med Chem 2017; 140:321–30.
- Felicetti T, Machado D, Cannalire R, et al. Modifications on C6 and C7 positions of 3-phenylquinolone efflux pump inhibitors led to potent and safe antimycobacterial treatment adjuvants. ACS Infect Dis 2019; 5:982–1000.
- Sabatini S, Gosetto F, Manfroni G, et al. Evolution from a natural flavones nucleus to obtain 2-(4-propoxyphenyl)quinoline derivatives as potent inhibitors of the S. aureus NorA efflux pump. J Med Chem 2011; 54:5722–36.
- Jones CP, Anderson KW, Buchwald SL. Sequential Cu-catalyzed amidation-base-mediated camps cyclization: a two-step synthesis of 2-aryl-4-quinolones from o-halophenones. J Org Chem 2007; 72:7968–73.
- Chanda T, Chowdhury S, Ramulu BJ, et al. Regioselective quadruple domino aldolization/aldol condensation/Michael/SNAr-cyclization: construction of hexacyclic indeno-fused C-nor-D-homo-steroid frameworks. Tetrahedron 2014; 70:2190–4.
- Kumar S, Kumar D. Polystyrene-supported iodobenzene diacetate (PSIBD)-mediated synthesis of 1,2-diacylbenzenes from 2-hydroxyaryl aldehyde/ketone acylhydrazones. Synth Commun 2008; 38:3683–99.
- Santhi J, Baire B. Carbonyl directed regioselective hydration of alkynes under Ag-catalysis. ChemistrySelect 2017; 2:4338–42.
- CLSI. M07-A10: methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; Approved Standard – Tenth edition. Wayne (PA): Clinical and Laboratory Standards Institute; 2015.
- Isenberg HD. Clinical microbiology procedures handbook. Washington (DC): ASM; 1992.
- Kaatz GW, Seo SM, O'Brien L, et al. Evidence for the existence of a multidrug efflux transporter distinct from NorA in Staphylococcus aureus. Antimicrob Agents Chemother 2000; 44:1404–6.
- Chongsiriwatana NP, Patch JA, Czyzewski AM, et al. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci USA 2008; 105:2794–9.
- Schrödinger Release 2016-2: Maestro, version 10.6. New York (NY): Schrödinger, LLC; 2016.
- Cruciani G, Carosati E, De Boeck B, et al. MetaSite: understanding metabolism in human cytochromes from the perspective of the chemist. J Med Chem 2005; 48:6970–9.
- See SAR version 5.5-2017. Sankt Augustin (Germany): BioSolveIT GmbH. Available from: www.biosolveit.de.
- Bietz S, Urbaczek S, Schulz B, et al. Protoss: a holistic approach to predict tautomers and protonation states in protein-ligand complexes. J Cheminform 2014; 6:12.
- Optibrium. Available from: http://www.optibrium.com/stardrop.
- Brouwer C, Jenko K, Zoghbi SS, et al. Development of N-methyl-(2-arylquinolin-4-yl)oxypropanamides as leads to PET radioligands for translocator protein (18 kDa). J Med Chem 2014; 57:6240–51.
- Kaatz GW, Seo SM. Mechanisms of fluoroquinolone resistance in genetically related strains of Staphylococcus aureus. Antimicrob Agents Chemother 1997; 41:2733–7.
- Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov 2010; 9:117–28.
- Odds FC. Editorial synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 2003; 52:1.
- Novo DJ, Perlmutter NG, Hunt RH, et al. Multiparameter flow cytometric analysis of antibiotic effects on membrane potential, membrane permeability, and bacterial counts of Staphylococcus aureus and Micrococcus luteus. Antimicrob Agents Chemother 2000; 44:827–834.
- Segall MD, Barber C. Addressing toxicity risk when designing and selecting compounds in early drug discovery. Drug Discov Today 2014; 19:688–693.