
Abstract
A series of 3H-1,2-benzoxathiepine 2,2-dioxides incorporating 7-acylamino moieties were obtained by an original procedure starting from 5-nitrosalicylaldehyde, which was treated with propenylsulfonyl chloride followed by Wittig reaction of the bis-olefin intermediate. The new derivatives, belonging to the homosulfocoumarin chemotype, were assayed as inhibitors of the zinc metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1). Four pharmacologically relevant human (h) isoforms were investigated, the cytosolic hCA I and II and the transmembrane, tumour-associated hCA IX and XII. No relevant inhibition of hCA I and II was observed, whereas some of the new derivatives were effective, low nanomolar hCA IX/XII inhibitors, making them of interest for investigations in situations in which the activity of these isoforms is overexpressed, such as hypoxic tumours, arthritis or cerebral ischaemia.
1. Introduction
Sulfocoumarins (1,2-benzoxathiine 2,2-dioxides) and homosulfocoumarins (3H-1,2-benzoxathiepine 2,2-dioxides)Citation1–5 are among the most investigated new classes of carbonic anhydrase (CA, EC 4.2.1.1) inhibitors, which have been designed considering the structurally similar coumarinsCitation6–8 as lead molecules. Indeed, Cas are widely spread enzymes in organisms of all types, from simple to complex onesCitation9–15, and are involved in crucial physiological processes, among which carbon fixation in diatoms and other marine organisms in which several genetic families of such metalloenzymes were reportedCitation9. In protozoans, Cas are involved in biosynthetic reactionsCitation9 whereas in bacteria, where at least three genetic families were described (α-, β-, and γ-Cas) these enzymes play crucial roles related both to metabolism but also virulence and survival in various nichesCitation10. In vertebrates, including humans, a high number of different CA isoforms belonging to the α-CA class were describedCitation11,Citation12, which by hydrating CO2 to a weak base (bicarbonate) and a strong acid (hydronium ions), are involved in a multitude of processes, starting with pH regulation and ending with metabolismCitation13,Citation14. As thus, Cas are drug targets for decades, with their inhibitors having pharmacological applications in a multitude of fieldsCitation11–16. The primary sulphonamides were discovered as CA inhibitors (CAIs) in the ‘40 s, and most of the drugs that were launched in the next decades as diuretics, antiepileptics, or antiglaucoma agents belonged to this class of compounds or to their isosteres such as the sulfamates and sulfamidesCitation11. An important drawback of such first generation CA inhibitors (CAIs) was their lack of isoform selectivity, considering the fact that in humans at least 12 catalytically active and three acatalytic isoforms are presentCitation11,Citation12. However, the new generation CAIs to which coumarins and sulfocoumarins belong, show significant isoform-selective inhibition profiles, as demonstrated in a considerable number of studiesCitation1–8. This is principally due to the fact that these compounds possess a distinct inhibition mechanism compared to the sulphonamides, which coordinate to the zinc ion from the CA active site as anionsCitation11,Citation12. In fact, coumarins and sulfocoumarins act as prodrug inhibitors, undergoing an active site mediated hydrolysis, which leads to the formation of 2-hydroxy-cinnamic acids in the case of the coumarins, and ethane-sulphonates in the case of the sulfocoumarins, which subsequently bind in different active site regions, different of those where the classical sulphonamide CAIs bindCitation1–8. As shown by X-ray crystallography, the hydrolysed coumarins occlude the entrance of the CA active site cavityCitation6, whereas the sulfocoumarins bind deeper within the active site, but still do not coordinate to the metal ion. Instead, the formed sulphonates anchor to the zinc-coordinated water molecule, as shown again by means of X-ray crystallographic techniquesCitation2. As these regions of the CA active site are the most variable ones, a straightforward explanation of the isoform selectivity of these new generation CAIs was furnished by using a combination of crystallographic and kinetic studies, which also allowed the development of compounds showing a higher degree of selectivityCitation15,Citation16. This allowed for the development of inhibitors useful for new pharmacological applications such as antitumor/antimetastatic compoundsCitation13, CAIs useful for the management of arthritisCitation17, neuropathic painCitation18, and cerebral ischaemiaCitation19.
Considering our interest in designing non-sulphonamide CAIs with various potential applications, we report here a new series of homosulfocoumarins and their inhibitory profiles against the major human (h) CA isoforms, hCA I, II, IX, and XII, involved in many pathologies, including cancer.
2. Experimental part
2.1. Chemistry
Reagents, starting materials/intermediates 1–7 and solvents were obtained from commercial sources (Sigma-Aldrich, St. Louis, MO) and used as received. Anhydrous CH2Cl2 and toluene were obtained by passing commercially available solvents through activated alumina columns. Thin-layer chromatography was performed on silica gel, spots were visualised with UV light (254 and 365 nm). Melting points were determined on an OptiMelt automated melting point system. IR spectra were recorded on Shimadzu FTIR IR Prestige-21 spectrometer. NMR spectra were recorded on Bruker Avance Neo (400 MHz) spectrometer with chemical shifts values (δ) in ppm relative to TMS using the residual DMSO-d6 signal (1H 2.50; 13 C 39.52) or CDCl3 signal (1H 7.26; 13 C 77.16) as an internal standard. High-resolution mass spectra (HRMS) were recorded on a mass spectrometer with a Q-TOF micro mass analyser using the ESI technique.
General procedure for synthesis of acyl compound 8–17
To a solution of amino derivative 7 (1.0 eq.) in dry CH2Cl2 (20 ml per mmol of compound 7) at 0 °C appropriate acyl chloride (1.1 eq.) and Net3 (1.1 eq.) were added. The resulting mixture was stirred at room temperature under an argon atmosphere for 2 h. Water was added (20 ml per mmol of compound 7). Layers were separated, water layer was washed with EtOAc (2 × 40 ml). Combined organic layers were washed with brine, dried over anh. Na2SO4, filtered, evaporated. The crude solids were recrystallised form EtOAc/petrol ether mixture to afford product.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)acetamide (8)
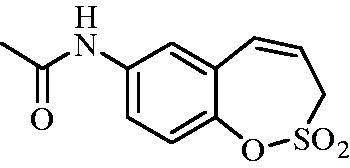
Compound 8a was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), acetyl chloride (56 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (127 mg; 70%). Mp 164–165 °C.
IR (film, cm−1) νmax= 3276 (N–H), 1670 (C = O), 1370 (S = O), 1361 (S = O), 1166 (S = O), 1162 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 2.06 (s, 3H), 4.37–4.41 (m, 2H), 5.96–5.6.04 (m, 1H), 6.89 (d, 1H, J = 11.3 Hz), 7.28 (d, 1H, J = 8.9 Hz), 7.58 (dd, 1H, J = 8.9, 2.5 Hz), 7.69 (d, 1H, J = 2.5 Hz), 10.16 (s, 1H) ppm.
13C NMR (100 MHz, DMSO-d6) δ = 24.0, 51.0, 120.6, 120.8, 122.7, 128.4, 131.5, 138.0, 142.2, 168.6 ppm.
HRMS (ESI) [M + H]+: m/z calcd for (C11H12NO4S) 254.0487. Found 254.0498.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)benzamide (9)
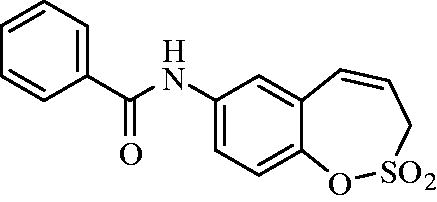
Compound 9 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), benzoyl chloride (90 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (162 mg; 72%). Mp 174–175 °C.
IR (film, cm−1) νmax= 3289 (N–H), 1652 (C = O), 1370 (S = O), 1363 (S = O), 1163 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.43 (dd, 2H, J = 6.0, 0.9 Hz), 5.99–6.06 (m, 1H), 6.93 (d, 1H, J = 11.2 Hz), 7.35 (d, 1H, J = 8.8 Hz), 7.52–7.58 (m, 2H), 7.59–7.64 (m, 1H), 7.82 (dd, 1H, J = 8.8, 2.5 Hz), 7.91 (d, 1H, J = 2.5 Hz), 7.94–7.99 (m, 2H), 10.46 (s, 1H) ppm.
13C NMR (100 MHz, DMSO-d6) δ = 51.1, 120.9, 122.0, 122.1, 122.6, 127.7, 128.3, 128.5, 131.4, 131.8, 134.6, 137.9, 142.7, 165.7 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C16H14NO4S) 316.0644. Found 316.0654.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-4-methy benzamide (10)
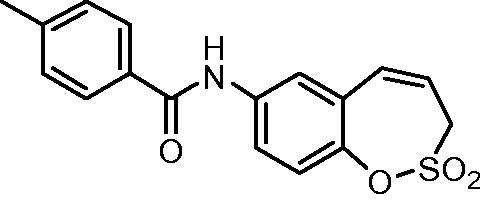
Compound 10 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 4-methylbenzoyl chloride (103 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white crystals (170 mg; 73%). Mp 197–198 °C.
IR (film, cm−1) νmax = 3324 (N–H), 1646 (C = O), 1378 (S = O), 1363 (S = O), 1177 (S = O), 1169 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 2.39 (s, 3H), 4.41–4.45 (m, 2H), 5.99–6.06 (m, 1H), 6.92 (d, 1H, J = 11.2 Hz), 7.32–7.37 (m, 3H), 7.82 (dd, 1H, J = 8.9, 2.6 Hz), 7.86–7.92 (m, 3H), 10.37 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 21.0, 51.1, 120.8, 121.9, 122.1, 122.6, 127.7, 128.3, 129.0, 131.4, 131.6, 138.0, 141.9, 142.6, 165.5 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C17H16NO4S) 330.0800. Found 330.0815.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)4-bromobenzamide (11)
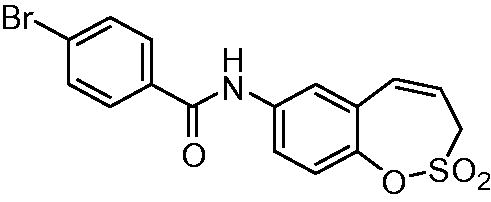
Compound 11 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 4-bromobenzoyl chloride (171 mg; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (166 mg; 59%). Mp 185–186 °C.
IR (film, cm−1) νmax= 3260 (N–H), 1653 (C = O), 1375 (S = O), 1363 (S = O), 1167 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.42–4.46 (m, 2H), 5.99–6.06 (m, 1H), 6.92 (d, 1H, J = 11.3 Hz), 7.35 (d, 1H, J = 8.8 Hz), 7.74–7.83 (m, 3H), 7.88–7.94 (m, 3H), 10.52 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 51.2, 120.9, 122.0, 122.2, 122.7, 125.6, 128.3, 129.8, 131.4, 131.5, 133.6, 137.7, 142.8, 164.7 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C16H13BrNO4S) 393.9749. Found 393.9736.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-2-iodobenzamide (12)
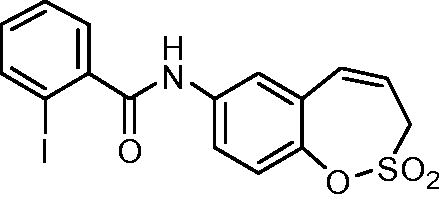
Compound 12 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 2-iodobenzoyl chloride (208 mg; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (276 mg; 88%). Mp 188–189 °C.
IR (film, cm−1) νmax= 3240 (N–H), 1641 (C = O), 1374 (S = O), 1362 (S = O), 1156 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.41–4.45 (m, 2H), 6.00–6.08 (m, 1H), 6.94 (d, 1H, J = 11.2 Hz), 7.22–7.28 (m, 1H), 7.36 (d, 1H, J = 8.8 Hz), 7.47–7.55 (m, 2H), 7.72 (dd, 1H, J = 8.8, 2.5 Hz), 7.87 (d, 1H, J = 2.5 Hz), 7.9–7.97 (m, 1H), 10.67 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 51.0, 93.6, 121.0, 121.2, 121.3, 122.9, 128.1, 128.2, 128.5, 131.2, 131.5, 137.7, 139.1, 142.7, 142.8, 167.7 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C16H13INO4S) 441.9610 Found 441.9609.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-2-bromobenzamide (13)
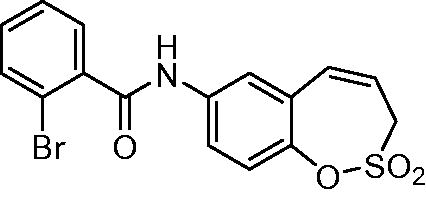
Compound 13 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 2-bromobenzoyl chloride (102 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (230 mg; 82%). Mp 177–178 °C.
IR (film, cm−1) νmax= 3288 (N–H), 1653 (C = O), 1371 (S = O), 1176 (S = O), 1156 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.43 (dd, 2H, J = 6.0, 0.9 Hz), 6.00–6.07 (m, 1H), 6.94 (d, 1H, J = 11.2 Hz), 7.36 (d, 1H, J = 8.9 Hz), 7.41–7.47 (m, 1H), 7.51 (dt, 1H, J = 7.4, 1.1 Hz), 7.55–7.59 (m, 1H), 7.69–7.76 (m, 2H), 7.87 (d, 1H, J = 2.6 Hz), 10.73 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 51.0, 118.9, 121.1, 121.2, 122.9, 127.8, 128.6, 128.9, 131.4, 131.5, 132.8, 137.6, 138.8, 142.8, 166.0 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C16H13BrNO4S) 393.9749 Found 393.9766.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-2-fluorobenzamide (14)
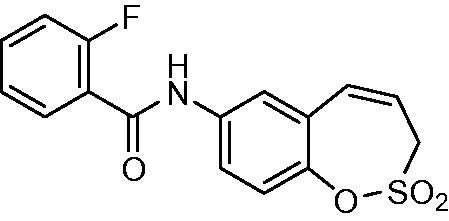
Compound 14 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 2-fluorobenzoyl chloride (93 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (188 mg; 79%). Mp 173–174 °C.
IR (film, cm−1) νmax= 1671 (C = O), 1371 (S = O), 1365 (S = O), 1164 (S = O) 1156 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.41–4.45 (m, 2H), 6.00–6.07 (m, 1H), 6.93 (d, 1H, J = 11.2 Hz), 7.32–7.40 (m, 3H), 7.56–7.63 (m, 1H), 7.65–7.71 (m, 1H), 7.74 (dd, 1H, J = 8.8, 2.5 Hz), 7.86 (d, 1H, J = 2.5 Hz), 10.65 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 51.1, 116.2 (d, J = 21.7 Hz), 121.0, 121.4, 121.5, 122.8, 124.6 (d, J = 5.5 Hz), 124.7 (d, J = 6.3 Hz), 128.5, 129.9 (d, J = 2.6 Hz), 131.4, 132.8 (d, J = 8.5 Hz), 137.5, 142.8, 159.9 (d, J = 249 Hz), 163.0 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C16H13FNO4S) 334.0549 Found 334.0554.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)-2-(trifluoromethyl)benzamide (15)
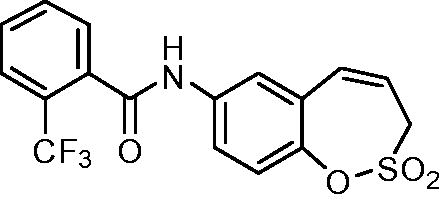
Compound 15 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 2-(trifluoromethyl)benzoyl chloride (115 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (236 mg; 87%). Mp 192–193 °C.
IR (film, cm−1) νmax= 3195 (N–H), 1666 (C = O), 1377 (S = O), 1316 (S = O), 1164 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.42–4.45 (m, 2H), 6.00–6.08 (m, 1H), 6.94 (d, 1H, J = 11.2 Hz), 7.36 (d, 1H, J = 8.8 Hz), 7.67–7.76 (m, 3H), 7.78–7.89 (m, 3H), 10.81 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 51.0, 121.1, 121.3, 122.9, 123.8 (q, J = 274 Hz), 125.8 (q, J = 31.2 Hz), 126.4 (q, J = 4.6 Hz), 128.5, 128.6, 130.3, 131.4, 132.7, 135.8 (q, J = 2.3 Hz), 137.6, 142.8, 165.8 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C17H13NO4 F3S) 384.0517 Found 384.0519.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)thiophene-2-carboxamide (16)
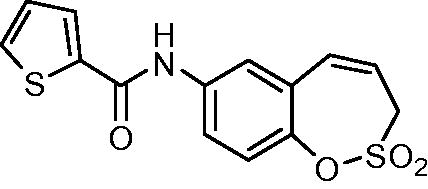
Compound 16 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 2-thiophenecarbonyl chloride (84 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (185 mg; 81%). Mp 162–163 °C.
IR (film, cm−1) νmax= 3357 (N–H), 1648 (C = O), 1372 (S = O), 1356 (S = O), 1178 (S = O), 1165 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.44 (dd, 2H, J = 6.0, 1.1 Hz), 5.99–6.06 (m, 1H), 6.92 (d, 1H, J = 11.2 Hz), 7.23–7.26 (m, 1H), 7.35 (d, 1H, J = 8.8 Hz), 7.78 (dd, 1H, J = 8.8, 2.6 Hz), 7.84 (d, 1H, J = 2.6 Hz), 7.88 (dd, 1H, J = 5.0, 1.1 Hz), 8.04 (dd, 1H, J = 3.8, 1.1 Hz), 10.43 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 51.2, 120.9, 121.9, 122.1, 122.7, 128.2, 128.4, 129.5, 131.3, 132.3, 137.5, 139.5, 142.7, 160.0 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C14H12NO4S2) 322.0208 Found 322.0221.
N-(2,2-Dioxido-3H-1,2-benzoxathiepin-7-yl)furan-2-carboxamide (17)
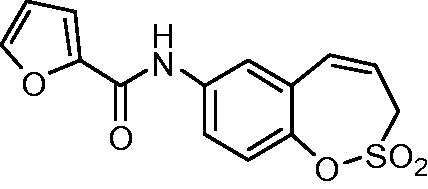
Compound 17 was prepared according to the general procedure from amino derivative 7 (150 mg; 0.71 mmol), 2-furoyl chloride (84 µL; 0.78 mmol) and Et3N (110 µL; 0.78 mmol) as white solid (185 mg; 81%). Mp 162–163 °C.
IR (film, cm−1) νmax= 3299 (N–H), 1663 (C = O), 1367 (S = O), 1363 (S = O), 1165 (S = O), 1158 (S = O);
1H NMR (400 MHz, DMSO-d6) δ = 4.41–4.45 (m, 2H), 5.98–6.06 (m, 1H), 6.70–6.74 (m, 1H), 6.90 (d, 1H, J = 11.2 Hz), 7.32–7.38 (m, 2H), 7.80 (dd, 1H, J = 8.8, 2.6 Hz), 7.87 (d, 1H, J = 2.6 Hz), 7.95–7.97 (m, 1H), 10.41 (s, 1H) ppm
13C NMR (100 MHz, DMSO-d6) δ = 51.2, 112.3, 115.2, 120.9, 122.0, 122.2, 122.6, 128.3, 131.4, 137.3, 142.7, 146.0, 147.2, 156.3 ppm
HRMS (ESI) [M + H]+: m/z calcd for (C14H12NO5S) 306.0436 Found 306.0463.
2.2. CA inhibitory assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation20. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10 − 100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5 − 10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled − deionised water, and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 6 h at room temperature prior to assay in order to allow for the formation of the E − I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng − Prusoff equation, as reported earlierCitation21–23, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation21,Citation24.
3. Results and discussion
3.1. Chemistry
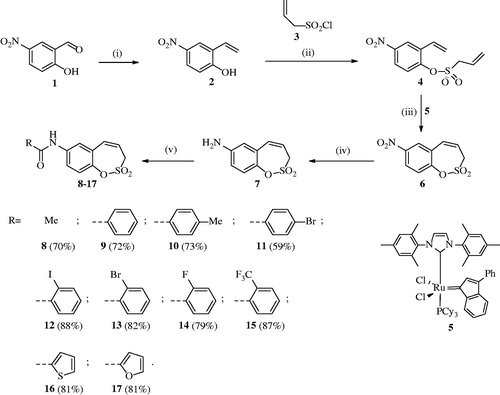
Starting from the benzaldehyde derivative 1, the synthesis of the key intermediate 7 was reported earlier by our groupsCitation1. Briefly, the synthesis of 7-amino-3H-1,2-benzoxathiepine 2,2-dioxide (7) was started with a Wittig reaction in which 5-nitro-salicylic aldehyde 1 was converted to the corresponding mono-olefin 2 in 65% yield (Scheme 1). Treatment of compound 2 with allyl sulphonyl chloride (3) provided the bisolefin 4 in 65% yield. In the next step, the olefin metathesis reaction with Ru-catalyst 5 was employed, leading to the conversion of compound 4 to 7-nitro-3H-1,2-benzoxathiepine 2,2-dioxide 6 in 96% yield. The nitro derivative 6 was thereafter reduced with iron in acidic medium to the corresponding amine 7 in nearly quantitative yield (98%). The key intermediate 7 was subsequently reacted with a series of acyl chlorides to afford the desired compounds 8–17 in good to excellent yields (see Experimental for details). The nature of moieties R was chosen in such a way to assure chemical diversity. Apart R = Me in compound 8, the remaining derivatives 9–17 incorporated aromatic or heterocyclic moieties, such as phenyl, 2- or 4-substituted phenyls, thienyl and furyl. We found out in previous papersCitation1–3 that aryl or hetaryl moieties on the sulfocoumarin, homosulfocoumarin or coumarin ringCitation6 systems lead to compounds with an effective inhibition profile against CA isoforms of pharmacologic interest, such as the tumour-associated ones CA IX and XII.
Scheme 1. Reagents and conditions: (i) MePPh3Br, tBuOK, THF, RT, 18 h, 65%; (ii) Net3, CH2Cl2, 0 °C to RT, 4 h, 57%; (iii) 5, toluene, 70 °C, 4 h, 96%; (iv) Fe, AcOH, EtOH, H2O, 75 °C, 1 h, 98%; (v) RCOCl, Net3, CH2Cl2, 0 °C to RT, 4 h.
3.2. Carbonic anhydrase inhibition
The obtained homosulfocoumarins 8–17 were investigated for their CA inhibitory properties by using a stopped-flow CO2 hydrase assayCitation20 and four human CA isoforms (hCA I, II, IX, and XII) known to be drug targetsCitation1 ().
Table 1. Inhibition data of human CA isoforms CA I, II, IX and XII with 3H-1,2-benzoxathiepines 2,2-dioxide 8–17 using acetazolamide (AAZ) as a standard drug.
As seen from data of , derivatives 8–17 did not significantly inhibit the cytosolic isoforms hCA I and II, similar to other homosulfocoumarins, sulfocouamrins or coumarins investigated earlierCitation1–8. On the other hand, the transmembrane, tumour-associated isoforms hCA IX and XII were inhibited by all these compounds in the nanomolar range. For hCA IX the KIs were in the range of 19.7–353.3 nM whereas for hCA XII in the range of 8.7–643.7 nM (). The nature of the R moiety on the carboxamide functionality greatly influenced the inhibitory power. For hCA IX/XII the optimal substitution was that present in compound 15, 2-trifluoromethylphenyl, whereas the one leading to the least effective inhibitor was the one with 4-bromophenylcarboxamide moiety (compound 9) for hCA IX and 2-iodophenylcarboxamide (compound 12) for hCA XII. Overall, all these new homosulfocoumarins act as isoform IX/XII selective CAIs over hCA I and II, which is highly desirable for these new chemotypes with enzyme inhibitory properties.
4. Conclusions
A series of 3H-1,2-benzoxathiepine 2,2-dioxides incorporating 7-acylamino moieties were obtained by an original procedure starting from 5-nitrosalicylaldehyde which was treated with propenylsulfonyl chloride followed by cyclisation through a Wittig reaction of the bis-olefin intermediate. The new derivatives, belonging to the homosulfocoumarin chemotype, were assayed as inhibitors of the zinc metalloenzyme CA. Four pharmacologically relevant human (h) isoforms were investigated, the cytosolic hCA I and II, and the transmembrane, tumour-associated hCA IX and XII. No relevant inhibition of hCA I and II was observed, whereas some of the new derivatives were effective, low nanomolar hCA IX/XII inhibitors, making them of interest for investigations in situations in which the activity of these isoforms is overexpressed, such as hypoxic tumours, arthritis or cerebral ischaemia.
Disclosure statement
The author(s) do not declare any conflict of interest.
Additional information
Funding
References
- (a) Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75.; (b) Pustenko A, Nocentini A, Balašova A, et al. Aryl derivatives of 3H-1,2-benzoxathiepine 2,2-dioxide as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2020;35:245–54.
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine 2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- Tanc M, Carta F, Bozdag M, et al. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem 2013;21:4502–10.
- Nocentini A, Ceruso M, Carta F, Supuran CT. 7-Aryl-triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrase IX and XII. J Enzyme Inhib Med Chem 2016;31:1226–33.
- Grandane A, Tanc M, Mannelli LDC, et al. Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83.
- (a) Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62; (b) Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44; (c) Temperini C, Innocenti A, Scozzafava A, et al. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: the antiepileptic lacosamide as an example. J Med Chem 2010;53:850–4; (d) Touisni N, Maresca A, McDonald PC, et al. Glycosylcoumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7.
- Zengin Kurt B, Sonmez F, Durdagi S, et al. Synthesis, biological activity and multiscale molecular modeling studies for coumaryl-carboxamide derivatives as selective carbonic anhydrase IX inhibitors. J Enzyme Inhib Med Chem 2017;32:1042–52.
- (a) Maresca A, Scozzafava A, Supuran CT. 7,8-disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8; (b) Maresca A, Supuran CT. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett 2010;20:4511–4.
- (a) Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61; (b) Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum-the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96; (c) Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J 2019;13:2094–106.
- (a) Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704; (b) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32; (c) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704; (d) Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018;28:745–54; (e) Nishimori I, Minakuchi T, Morimoto K, et al. Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. J Med Chem 2006;49:2117–26.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Discov 2008;7:168–81; (b) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68; (c) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32; (d) Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorg Med Chem Lett 2010;20:5050–3.
- (a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60; (b) Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97; (c) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88; ( d) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- (a) Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70; (b) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12; (c) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21; (d) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nature Rev Drug Discov 2011;10:767–77; (e) Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836.
- (a) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25; (b) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48; (c) Da’dara AA, Angeli A, Ferraroni M, et al. Crystal structure and chemical inhibition of essential schistosome host-interactive virulence factor carbonic anhydrase SmCA. Commun Biol 2019;2:333.
- (a) Supuran CT, Ilies MA, Scozzafava A. Carbonic anhydrase inhibitors. Part 29. Interaction of isozymes I, II and IV with benzolamide-like derivatives. Eur J Med Chem 1998;33:739–52; (b) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Ang ew Chem Int Ed Engl 2007;46:7697–9; (c) Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors: perfluoroalkyl/aryl-s ubstituted derivatives of aromatic/heterocyclic sulfonamides as topical intraocular pressure-lowering agents with prolonged duration of action. J Med Chem 2000;43:4542–51; (d) Sentürk M, Gülçin I, Daştan A, et al. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–11.
- (a) Supuran CT, Altamimi ASA, Carta F. Carbonic anhydrase inhibition and the management of glaucoma: a literature and patent review 2013-2019. Expert Opin Ther Pat 2019;29:781–92; ( b) Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95; (c) Bilginer S, Gul HI, Erdal FS, et al. Synthesis, cytotoxicities, and carbonic anhydrase inhibition potential of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones. J Enzyme Inhib Med Chem 2019;34:1722–9.
- (a) Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31:60–3; (b) Bua S, Di Cesare Mannelli L, Vullo D, et al. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem 2017;60:1159–70; (c) Akgul O, Di Cesare Mannelli L, Vullo D, et al. Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the management of rheumatoid arthritis. J Med Chem 2018;61:4961–77.
- (a) Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;23:1828–40; (b) Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8.
- Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. Enzyme Inhib Med Chem 2016;31:894–9.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Vermelho AB, da Silva Cardoso V, Ricci Junior E, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46; (b) Nocentini A, Carta F, Tanc M, et al. Deciphering the mechanism of human carbonic anhydrases inhibition with sulfocoumarins: computational and experimental studies. Chemistry 2018;24:7840–4; (c) Awadallah FM, Bua S, Mahmoud WR, et al. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J Enzyme Inhib Med Chem 2018;33:629–38.
- (a) Bua S, Bozdag M, Del Prete S, et al. Mono- and di-thiocarbamate inhibition studies of the δ-carbonic anhydrase TweCAδ from the marine diatom Thalassiosira weissflogii. J Enzyme Inhib Med Chem 2018;33:707–13; (b) Ferraroni M, Gaspari R, Scozzafava A, et al. Dioxygen, an unexpected carbonic anhydrase ligand. J Enzyme Inhib Med Chem 2018;33:999–1005; (c) El-Gazzar MG, Nafie NH, Nocentini A, et al. Carbonic anhydrase inhibition with a series of novel benzenesulfonamide-triazole conjugates. J Enzyme Inhib Med Chem 2018;33:1565–74; (d) Akocak S, Lolak N, Bua S, Supuran CT. Discovery of novel 1,3-diaryltriazene sulfonamides as carbonic anhydrase I, II, VII, and IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1575–80.
- (a) Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–9; (b) Nocentini A, Trallori E, Singh S, et al. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms IX and XII showing hypoxia-enhanced antiproliferative profiles. J Med Chem 2018;61:10860–74; (c) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43; (d) Oztürk Sarikaya SB, Topal F, Sentürk M, et al. In vitro inhibition of α-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–62.
- (a) Awadallah FM, Bua S, Mahmoud WR, et al. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J Enzyme Inhib Med Chem 2018;33:629–38; (b) Supuran CT, Clare BW. Carbonic anhydrase inhibitors. Part 57. Quantum chemical QSAR of a group of 1,3,4-thiadiazole and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem 1999;34:41–50.