
Abstract
Thymidylate synthase (TS) has been an attention-grabbing area of research for the treatment of cancers due to their role in DNA biosynthesis. In the present study, we have synthesised a library of thiazolidinedione-1,3,4-oxadiazole hybrids as TS inhibitors. All the synthesised hybrids followed Lipinski and Veber rules which indicated good drug likeness properties upon oral administration. Among the synthesised hybrids, compound 9 and 10 displayed 4.5 and 4.4 folds activity of 5-Fluorouracil, respectively against MCF-7 cell line whereas 3.1 and 2.5 folds cytotoxicity against HCT-116 cell line. Furthermore, compound 9 and 10 also inhibited TS enzyme with IC50 = 1.67 and 2.21 µM, respectively. Finally, the docking studies of 9 and 10 were found to be consistent with in vitro TS results. From these studies, compound 9 and 10 has the potential to be developed as TS inhibitors.
Introduction
Despite the availability of various chemotherapeutic agents, cancer is the second highest cause of deaths next to cardiovascular diseases.Citation1,Citation2 Thymidylate synthase (TS) has become an area of interest in cancer chemotherapy due to their important role in DNA biosynthesis.Citation3,Citation4 TS is responsible for the formation of deoxythymidine monophosphate (dTMP) from deoxyuridine monophosphate (dUMP) which is further phosphorylated to triphosphate group (dTTP), a direct precursor for DNA synthesis.Citation5–7 TS inhibition causes inhibition of thymidylate biosynthesis which in turn causes cessation of cell growth and proliferation.Citation8,Citation9 Research on TS has been going on since many years, but still it is a challenge for medicinal chemists to develop new, safe and effective chemotherapeutic agents as TS inhibitor.
Thiazolidinediones (glitazones) are insulin sensitisers used for type II diabetes treatment. They are high-affinity ligands of PPAR-γ, which alleviates insulin resistance and effectively improves plasma glucose levels.Citation10–11 Beside hypoglycaemic agents, thiazolidinedione derivatives are reported as anti-inflammatory,Citation12 antimicrobialCitation13 and anticancer agents.Citation14–16 It has been mentioned that compounds which activate PPAR-γ might lead to differentiation induction of cancer cells.Citation17 For example, a TZD analogue, efatutazone (CS-7017) is a potent PPAR-γ full agonist as well as a cancer differentiation-inducing agent.Citation18 PPAR-γ agonists are reported to inhibit components of the insulin growth factor 1 (IGF1) pathway and modulate the activity of AMP-activated protein kinase (AMPK) pathway to reduce cancer risk.Citation19,Citation20 Thiazolidinediones derivatives have exerted anticancer effects by various mechanism of action viz. by inhibiting PI3K-α,Citation21 blockade of the Raf/MEK/ERK and PI3K/Akt signalling pathways.Citation22 On the other hand, 1,3,4-oxadiazoles are promising candidates in medicinal chemistry due to its wide applications. Zibotentan (ZD4054) containing 1,3,4-oxadiazole pharmacophore is an orally available selective antagonist of the ET-A receptor with potential antineoplastic effect for the treatment of various type of cancers.Citation23,Citation24 Recently, novel 1,3,4-oxadiazole bearing thioether derivatives has been reported as potential TS inhibitors.Citation25
Combination of the important bioactive pharmacophores under one construct plays an important role in medicinal chemistry for the development of biologically active molecules with novel entity.Citation26 To the best of our knowledge, anticancer effect of thiazolidinedione derivatives as TS inhibitors has not been reported till now. Therefore, we tried to conjugate thiazolidinedione and 1,3,4-oxadiazole under one construct to develop potential TS inhibitors. The present work describes the synthesis, in silico pharmacokinetic studies, in vitro antiproliferative and TS inhibitory activities. The docking studies have also been carried out for the most active compounds to understand the possible underlying molecular interactions.
Experimental
General
All the reagents and chemicals used in the present study were procured from Sigma Aldrich (Germany) and Loba (India). IR was recorded on Thermos scientific iS-50 by direct sampling method. NMR analysis was performed on Bruker 300 and 850 MHz instruments in either CDCl3 or DMSO-d6 solvents. Tetramethylsilane (TMS) was used as internal reference. Chemical shift and coupling constant are provided in Hertz and parts per million (ppm), respectively. Thermo scientific-LCQ Fleet (LCF10605) using electron spray ionisation method was used for recording the mass spectra and provided in m/z. Melting points were recorded on Stuart SMP40. Elemental analyses were performed on LEECO Elementar Elemental Analyser. The elemental analysis data were reported in % standard and found to be within ±0.4% of the calculated values. Purity of the compounds was checked on thin layer chromatography using silica gel G plate (Merck Germany). The spectral data and synthetic method of compounds 4–6 are provided in the Supplementary Material.
Chemistry
General procedure for the synthesis of thiazolidiene-2,4 dione-1,3,4-oxadiazole hybrids (7–21)
A mixture of compound 6 (0.01 mol) and different substituted aromatic hydrazides (0.01 mmol) in POCl3 (20 ml) was stirred and refluxed for 10–12 h. After reaction completion, the reaction mixture was poured onto crushed ice and neutralised with NaHCO3 solution. The resulting precipitate 7–21 was filtered, washed with excess cold water and dried. Purification of compounds 7–21 was done either by recrystallization in suitable solvents or by column chromatography using n-hexane and ethylacetate as eluents. The 1H NMR, 13 C NMR and mass spectra of the compounds are provided in Supplementary Material.
(Z)-5-(4-methoxybenzylidene)-3-((5-phenyl-1,3,4-oxadiazol-2-yl)methyl)thiazoli dine-2,4-dione (7). Yield 70% mp 212–216 °C, 1H NMR (850 MHz, DMSO-d6) δ: 3.84 (s, 3H), 4.48 (s, 2H), 7.13 (d, 2H, J = 8.5 Hz), 7.44–7.47 (m, 2H), 7.61–7.73 (m, 5H), 7.96 (s, 1H); 13C NMR (213 MHz, DMSO-d6) δ: 42.63, 56.03, 115.53, 117.60, 125.16, 125.64, 127.08, 129.10, 129.98, 132.98, 134.62, 161.53, 161.89, 165.53, 167.26, 167.46; ESI + ve MS (m/z): 394 (M + H)+. Anal. Calc. for C20H15N3O4S: C, 61.06; H, 3.84; N, 10.68; O, 16.27; S, 8.15. Found: C, 61.05; H, 3.85; N, 10.66; O, 16.29; S, 8.14.
(Z)-5-(4-methoxybenzylidene)-3-((5-(3-chlorophenyl)-1,3,4-oxadiazol-2-yl)methyl)thiazoli dine-2,4-dione (8). Yield 60% mp 155–156 °C, 1H NMR (850 MHz, DMSO-d6) δ: 3.82 (s, 3H), 5.19 (s, 2H), 7.11 (d, 2H, J = 9.3 Hz), 7.61–7.64 (m, 3H), 7.70–7.71 (m, 1H), 7.92–7.95 (m, 2H), 7.96 (s, 1H); 13C NMR (213 MHz, DMSO-d6) δ: 35.78, 55.63, 115.15, 117.38, 125.05, 125.31, 125.45, 126.17, 131.67, 132.13, 132.56, 134.11, 134.13, 161.47, 161.65, 163.62, 164.97, 166.99; ESI + ve MS (m/z): 428 (M + H)+, 430 (M + 2 + H)+. Anal. Calc. for C20H14ClN3O4S: C, 56.14; H, 3.30; N, 9.82; O, 14.96; S, 7.49. Found: C, 56.11; H, 3.32; N, 9.83; O, 14.95; S, 7.50.
(Z)-5-(4-methoxybenzylidene)-3-((5-(2-chlorophenyl)-1,3,4-oxadiazol-2-yl)methyl)thiazolid ine-2,4-dione (9). Yield 65% mp 276–277, 1H NMR (300 MHz, DMSO-d6) δ: 3.84 (s, 3H), 4.40 (s, 2H), 7.13 (d, 2H, J = 8.7 Hz), 7.42–7.57 (m, 4H), 7.63 (d, 2H, J = 8.7 Hz), 7.95 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ: 40.80, 56.01, 115.51, 118.19, 125.93, 127.59, 129.71, 130.29, 132.83, 134.02, 161.32, 161.78, 164.93, 165.68, 167.52; ESI + ve MS (m/z): 428 (M + H)+, 430 (M + 2 + H)+.Anal. Calc. for C20H14ClN3O4S: C, 56.14; H, 3.30; N, 9.82; O, 14.96; S, 7.49. Found: C, 56.11; H, 3.32; N, 9.83; O, 14.95; S, 7.50.
(Z)-5-(4-methoxybenzylidene)-3-((5-(4-bromophenyl)-1,3,4-oxadiazol-2-yl)methyl)thiazolid ine-2,4-dione (10). Yield 65% mp 263–264 °C, 1H NMR (300 MHz, DMSO-d6) δ: 3.83 (s, 3H), 4.41 (s, 2H), 7.12 (d, 2H, J = 8.7 Hz), 7.63 (d, 2H, J = 8.4 Hz), 7.71 (d, 2H, J = 8.7 Hz), 7.80 (d, 2H, J = 8.4 Hz), 7.94 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ: 36.50, 56.01, 115.51, 120.59, 125.78, 129.98, 132.06, 132.83, 155.22, 161.98, 165.76, 166.42, 166.76; ESI + ve MS (m/z): 472 (M + H)+, 474 (M + 2 + H)+.Anal. Calc. for C20H14BrN3O4S: C, 50.86; H, 2.99; N, 8.90; O, 13.55; S, 6.79. Found: C, 50.84; H, 2.98; N, 8.92; O, 13.57; S, 6.81.
(Z)-5-(4-methoxybenzylidene)-3-((5-p-tolyl-1,3,4-oxadiazol-2-yl)methyl)thiazolidine-2,4-dione (11). Yield 72% mp 197–198 °C,1H NMR (300 MHz, DMSO-d6) δ: 2.38 (s, 3H), 3.83 (s, 3H), 5.19 (s, 2H), 7.12 (d, 2H, J = 8.7 Hz), 7.40 (d, 2H, J = 7.8 Hz), 7.62 (d, 2H, J = 8.7 Hz), 7.85 (d, 2H, J = 8.1 Hz), 7.97 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ: 21.58, 36.21, 56.01, 115.50, 117.73, 120.77, 125.70, 127.01, 130.45, 132.91, 134.52, 142.81, 161.37, 161.86, 165.17, 165.32, 167.30; ESI + ve MS (m/z): 408 (M + H)+. Anal. Calc. for C21H17N3O4S: C, 61.90; H, 4.21; N, 10.31; O, 15.71; S, 7.87. Found: C, 61.89; H, 4.20; N, 10.29; O, 15.72; S, 7.85.
(Z)-5-(4-methoxybenzylidene)-3-((5-(2-hydroxyphenyl)-1,3,4-oxadiazol-2-yl)methyl)thiazol idine-2,4-dione (12). Yield 65% mp 217–218 °C, 1H NMR (850 MHz, CDCl3) δ: 3.88 (s, 3H), 5.24 (s, 2H), 6.99–7.02 (m, 3H), 7.11 (d, 1H, J = 8.5 Hz), 7.44–7.46 (m, 1H), 7.49 (d, 2H, J = 8.5 Hz), 7.72 (dd, 1H, J = 9.3, 1.7 Hz), 7.95 (s, 1H), 9.94 (s, 1H); 13C NMR (213 MHz, CDCl3) δ: 35.55, 55.59, 107.60, 114.95, 117.11, 117.67, 120.01, 125.48, 126.76, 132.57, 134.09, 135.61, 157.63, 159.19, 161.92, 165.21, 165.29, 167.27; ESI + ve MS (m/z): 410 (M + H)+. Anal. Calc. for C20H15N3O5S: C, 58.67; H, 3.69; N, 10.26; O, 19.54; S, 7.83. Found: C, 58.65; H, 3.68; N, 10.27; O, 19.55; S, 7.85.
(Z)-5-(4-methoxybenzylidene)-3-((5-o-tolyl-1,3,4-oxadiazol-2-yl)methyl)thiazolidine-2,4-dione (13). Yield 57% mp 149–150 °C, 1H NMR (300 MHz, DMSO-d6) δ: 2.49 (s, 3H), 3.83 (s, 3H), 5.22 (s, 2H), 7.12 (d, 2H, J = 8.7 Hz), 7.56–7.72 (m, 4H), 7.92–7.98 (m, 3H); 13C NMR (75 MHz, DMSO-d6) δ: 21.55, 36.23, 56.01, 115.53, 117.72, 121.78, 125.79, 127.21, 128.38, 131.55, 132.94, 161.39, 161.90, 162.26, 165.38, 167.38; ESI + ve MS (m/z): 408 (M + H)+. Anal. Calc. for C21H17N3O4S: C, 61.90; H, 4.21; N, 10.31; O, 15.71; S, 7.87. Found: C, 61.89; H, 4.20; N, 10.29; O, 15.72; S, 7.85.
(Z)-5-(4-methoxybenzylidene)-3-((5-(3-nitrophenyl)-1,3,4-oxadiazol-2-yl)methyl)thiazolidin e-2,4-dione (14). Yield 75% mp 178–179 °C, 1H NMR (300 MHz, DMSO-d6) δ: 3.84 (s, 3H), 5.24 (s, 2H), 7.12 (d, 2H, J = 8.4 Hz), 7.63 (d, 2H, J = 8.4 Hz), 7.88–8.07 (m, 2H), 8.34–8.64 (m, 2H), 8.75 (s, 1H); 13C NMR (213 MHz, DMSO-d6) δ: 36.16, 56.03, 114.87, 115.54, 117.77, 121.63, 124.98, 125.69, 127.07, 131.93, 132.96, 133.20, 134.54, 148.68, 161.87, 162.33, 163.69, 165.36, 167.38; ESI + ve MS (m/z): 439 (M + H)+. Anal. Calc. for C20H14N4O6S: C, 54.79; H, 3.22; N, 12.78; O, 21.90; S, 7.31. Found: C, 54.80; H, 3.24; N, 12.75; O, 21.89; S, 7.32.
(Z)-5-(4-methoxybenzylidene)-3-((5-m-tolyl-1,3,4-oxadiazol-2-yl)methyl)thiazolidine-2,4-dione (15). Yield 65% mp 184–185 °C, 1H NMR (300 MHz, DMSO-d6) δ: 2.40 (s, 3H), 3.84 (s, 3H), 5.13 (s, 2H), 7.13 (d, 2H, J = 8.1 Hz), 7.38–7.51 (m, 2H), 7.62–7.69 (m, 2H), 7.76–7.96 (m, 2H), 7.98 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ: 21.27, 36.23, 56.02, 115.52, 117.75, 124.28, 125.71, 127.31, 129.85, 132.92, 133.31, 134.54, 139.44, 161.60, 161.87, 165.16, 167.35, 167.82; ESI + ve MS (m/z): 408 (M + H)+. Anal. Calc. for C21H17N3O4S: C, 61.90; H, 4.21; N, 10.31; O, 15.71; S, 7.87. Found: C, 61.89; H, 4.20; N, 10.29; O, 15.72; S, 7.85.
(Z)-5-(4-methoxybenzylidene)-3-((5-(phenoxymethyl)-1,3,4-oxadiazol-2-yl)methyl)thiazolidine-2,4-dione (16). Yield 70% mp 152–153 °C, 1H NMR (300 MHz, DMSO-d6) δ: 3.85 (s, 3H), 5.17 (s, 2H), 5.39 (s, 2H), 7.0–7.06 (m, 3H), 7.13 (d, 2H, J = 9.0 Hz), 7.32 (t, 2H, J = 6.9 Hz), 7.63 (d, 2H, J = 8.7 Hz), 7.97 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ: 36.14, 56.02, 59.87, 115.34, 115.54, 117.58, 122.28, 125.67, 130.09, 132.94, 134.71, 157.70, 161.92, 162.44, 164.17, 167.23; ESI + ve MS (m/z): 424 (M + H)+. Anal. Calc. for C21H17N3O5S: C, 59.57; H, 4.05; N, 9.92; O, 18.89; S, 7.57. Found: C, 59.59; H, 4.04; N, 9.91; O, 18.90; S, 7.53.
(Z)-3-((5-((3-chlorophenoxy)methyl)-1,3,4-oxadiazol-2-yl)methyl)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione (17). Yield 72% mp 142–143 °C, 1H NMR (400 MHz, CDCl3) δ: 3.86 (s, 3H), 5.14 (s, 2H), 5.21 (s, 2H), 6.86–6.89 (m, 1H), 6.98–7.0 (m, 3H), 7.19–7.25 (m, 2H), 7.47 (d, 2H, J = 8.8 Hz), 7.91 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 35.63, 55.56, 59.90, 113.08, 114.87, 114.95, 115.69, 122.62, 125.54, 130.53, 132.38, 132.52, 135.20, 135.49, 158.07, 161.93, 162.87, 165.23, 167.19; ESI + ve MS (m/z): 458 (M + H)+, 460 (M + 2 + H)+. Anal. Calc. for C21H16ClN3O5S: C, 55.08; H, 3.52; N, 9.18; O, 17.47; S, 7.00. Found: C, 55.07; H, 3.54; N, 9.19; O, 17.45; S, 7.01.
(Z)-3-((5-((2,3-dichlorophenoxy)methyl)-1,3,4-oxadiazol-2-yl)methyl)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione (18). Yield 72% mp 209–210 °C, 1H NMR (300 MHz, CDCl3) δ: 3.87 (s, 3H), 5.15 (s, 2H), 5.31 (s, 2H), 6.98–7.01 (m, 3H), 7.12–7.15 (m, 2H), 7.48 (d, 2H, J = 6.6 Hz), 7.91 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 35.64, 55.54, 61.23, 112.90, 114.96, 124.42, 125.53, 127.54, 132.52, 135.50, 155.69, 161.94, 162.58, 167.15, 168.32; ESI + ve MS (m/z): 492 (M + H)+, 494 (M + 2 + H)+. Anal. Calc. for C21H15Cl2N3O5S: C, 51.23; H, 3.07; N, 8.53; O, 16.25; S, 6.51. Found: C, 51.21; H, 3.06; N, 8.51; O, 16.27; S, 6.50.
(Z)-5-(4-methoxybenzylidene)-3-((5-((naphthalen-1-yloxy)methyl)-1,3,4-oxadiazol-2-yl)methyl) thiazolidine-2,4-dione (19). Yield 50% mp 220–221 °C, 1H NMR (300 MHz, CDCl3) δ: 3.86 (s, 3H), 5.15 (s, 2H), 5.43 (s, 2H), 6.93–7.01 (m, 3H), 7.33–7.51 (m, 7H), 7.76–7.79 (m, 1H), 7.90 (s, 1H); 13C NMR (213 MHz, CDCl3) δ: 41.43, 55.52, 62.18, 115.12, 116.65, 117.90, 124.26, 125.59, 125.65, 128.12, 128.38, 132.40, 132.49, 134.80, 135.37, 161.52, 164.29, 165.56, 167.35, 167.19; ESI + ve MS (m/z): 474 (M + H)+. Anal. Calc. for C25H19N3O5S: C, 63.41; H, 4.04; N, 8.87; O, 16.89; S, 6.77. Found: C, 63.42; H, 4.03; N, 8.90; O, 16.86; S, 6.78.
(Z)-5-(4-methoxybenzylidene)-3-((5-((naphthalen-3-yloxy)methyl)-1,3,4-oxadiazol-2-yl)met hyl) thiazolidine-2,4-dione (20). Yield 58% mp 210–211 °C, 1H NMR (400 MHz, CDCl3) δ: 3.86 (s, 3H), 5.15 (s, 2H), 5.35 (s, 2H), 6.98–7.0 (m, 3H), 7.18–7.20 (m, 2H), 7.46–7.48 (m, 4H), 7.74–7.76 (m, 2H), 7.89 (s, 1H); 13C NMR (213 MHz, CDCl3) δ: 41.45, 55.55, 62.21, 114.92, 116.64, 117.70, 123.93, 125.45, 125.65, 128.12, 128.38, 132.44, 132.56, 134.89, 135.46, 155.51, 161.71, 165.12, 166.19, 167.23, 167.64; ESI + ve MS (m/z): 474 (M + H)+. Anal. Calc. for C25H19N3O5S: C, 63.41; H, 4.04; N, 8.87; O, 16.89; S, 6.77. Found: C, 63.42; H, 4.03; N, 8.90; O, 16.86; S, 6.78.
(Z)-5-(4-methoxybenzylidene)-3-((5-((quinolin-8-yloxy)methyl)-1,3,4-oxadiazol-2-yl)methyl) thiazolidine-2,4-dione (21). Yield 70% mp 161–162 °C, 1H NMR (300 MHz, DMSO-d6) δ: 3.84 (s, 3H), 5.19 (s, 2H), 5.62 (s, 2H), 7.11–7.14 (m, 2H), 7.36 (d, 1H, J = 7.8 Hz), 7.46–7.64 (m, 5H), 7.96 (s, 1H); 8.33 (d, 1H, J = 9.3 Hz), 8.85–8.86 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ: 35.69, 58.53, 60.48, 111.46, 115.04, 117.05, 121.07, 121.25, 125.80, 128.53, 129.42, 132.45, 134.23, 138.45, 138.87, 149.32, 152.58, 160.47, 162.44, 163.32, 163.73, 166.72; ESI + ve MS (m/z): 475 (M + H)+. Anal. Calc. for C24H18N4O5S: C, 60.75; H, 3.82; N, 11.81; O, 16.86; S, 6.76. Found: C, 60.74; H, 3.81; N, 11.80; O, 16.87; S, 6.77.
Cytotoxicity activity
The methods are provided in Supplementary material.
In vitro thymidylate synthase assay
TS activity was performed according to the method as described by Wahab and FriedkinCitation27 with a slight modification according to Santi.Citation28 (Refer to Supplementary material for the method.)
Molecular docking
Docking studies were performed at Intel(R) Core(TM) i3 CPU(2.3 GHz) with XP-based operating system (Windows 2007). 2 D structures of the compounds were drawn by Marvin Sketch and then converted into 3 D structures and saved in pdb file format. Ligand preparation was done by assigning Gastegier charges, merging non-polar hydrogen’s, and saving in PDBQT file format using AutoDock Tools (ADT) 1.5.4. X-ray crystal structure of DNA (PDB ID: 6QXG) was obtained from the Protein Data Bank (http://www.rcsb.org/pdb). Gastegier charges were assigned to DNA and saved in PDBQT file format using ADT. Preparation of parameter files for grid and docking was done using ADT. Docking was performed on AutoDock 4.0 (Scripps Research Institute, USA) considering all the rotatable bonds of the ligands as rotatable and DNA as rigid.Citation29 The grid centre was established by centring the grid box on whole DNA. Grid boxsize of 60 × 80 × 110 Å with 0.375 Å spacing was used. Macromolecule docking was performed using an empirical-free energy function and Lamarckian Genetic Algorithm, with an initial population of 150 randomly placed individuals, a maximum number of 2,500,000 energy evaluations, a mutation rate of 0.02, and cross-over rate of 0.80. Fifty independent docking runs were performed of each ligand and DNA–ligand complex for lowest free energy of binding conformation from the largest cluster, which was written and saved in PDBQT format. The results are summarised in and .
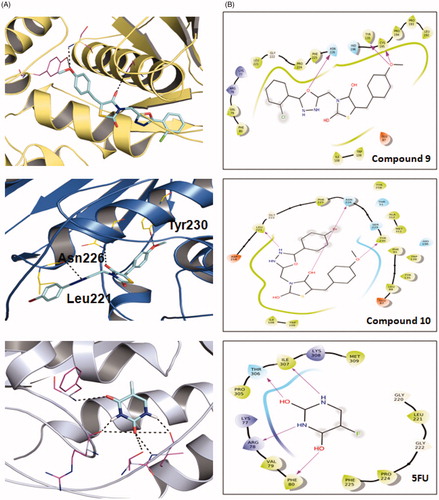
Figure 1. Molecular docking of the active compounds 9, 10 and 5FU against Thimidylate synthase (TS) protein 6QXG. A: Binding mode of 9, 10 and 5FU at TS binding site 3D plot. B: Binding mode of 9, 10 and 5FU at TS binding site 2D plot. 5FU: -5-fluorouracil.
Results and discussion
Chemistry
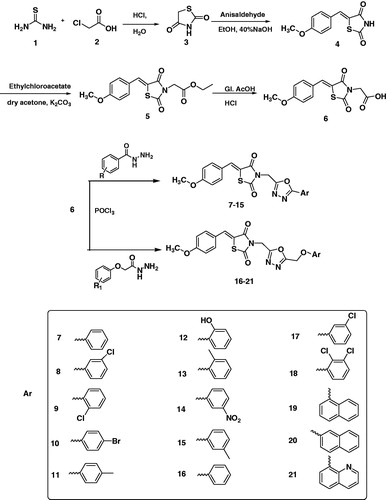
Thiazolidinediones 3 was prepared by refluxing thiourea and chloroacetic acid in hydrochloric acid and water which on Knovenagel condensation with anisaldehyde in ethanol and aqueous sodium hydroxide gives intermediate benzylidene thiazolidinediones 4. Alkylation of intermediate 4 with ethyl chloroacetate in the presence of anhydrous potassium carbonate and dry acetone followed by acidic hydrolysis using glacial acetic acid and hydrochloric acid gives the key intermediate 6. The key intermediate 6 was then refluxed with different aromatic acid hydrazides and substituted phenoxy acetic acid hydrazides in the presence of phosphorus oxychloride to give the target compounds 7–15 and 16–21, respectively in moderate to good yield (Scheme 1). The structures of the synthesised hybrids were confirmed by different analytical techniques such as 1H NMR, 13C NMR, IR spectroscopy, elemental analyser and mass spectrometry. Formation of the condensed product 4 was supported by the presence of two doublets at δ 7.09 (J = 8.7 Hz) and δ 7.56 (J = 8.7 Hz) which was assigned to four aromatic protons, a singlet at δ 7.75 for olefinic protons bridging phenyl and thiazolidinedione rings and δ 11.92 for NH of TZD ring in the 1H NMR. This structure was supported by 13 C NMR which revealed the downfield signals at δ 161.45 for olefinic C=C and two signals at δ 167.88 and δ 168.40 for carbonyl groups of TZD ring. The NH and C=O bands of compound 4 were observed at 3215 cm−1(broad) and 1728 and 1690 cm−1, respectively in the IR spectrum. The conversion of compound 4 to compound 5 was observed by the disappearance of NH signal at δ 11.92 and appearance of a singlet at δ 4.47 for N–CH2, quartette at δ 4.17 (J = 7.2, 14.1 Hz) for O–CH2 and a triplet at δ 1.21 (J = 6.9 Hz) for terminal methyl protons of the ester in the 1H NMR whereas in 13C NMR these signals appeared at δ 42.60, δ 62.15 and δ 14.38 for N–CH2, O–CH2 and terminal CH3, respectively. The absence of quartet and triplet and presence of broad signal at δ 13.44 in the 1H NMR of key intermediate 6 supported its formation. This data was further supported by 13 C NMR exhibiting one additional downfield signal for new carbonyl group of the acid and IR spectrum exhibited broad signal for hydroxyl proton at 3014 cm−1. Compound 6 was finally confirmed by the appearance of molecular ion peak at 292 (M − H)+ in mass spectrometry. The formation of the target compounds 7–21 was confirmed by the presence of additional aromatic protons in the range of δ 6.99–8.86 in their 1H NMR spectra. The absence of carboxyl proton signal in the target compounds 7–21 which was present in the 1H NMR spectrum of the key intermediate 6 also support their formation. Further structural confirmation of the target compounds 7–21 was provided by the presence of absorption bands in the range of 1509–1589 cm−1 for C=N stretching of oxadiazole ring in the IR spectra and the signals in the range of δ 159.19–166.19 for C=N of oxadiazole carbons in the 13 C NMR spectra of these compounds. The target compounds 16–21 exhibited additional signal as singlet in the range of δ 5.21–5.62 for O–CH2 bridged methylene aromatic ring and 1,3,4-oxadiazole ring. Finally, confirmatory evidence was done by their mass spectral data.
Scheme 1. Synthesis of thiazolidinedione-1,3,4-oxadiazole hybrids.
Pharmacokinetics studies/absorption, distribution, metabolism elimination (ADME) predictions
The capability of a drug to exhibit a pharmacological or therapeutic effect depends upon various physiochemical properties of the drug. Nowadays, ADME predictions of molecules are studied in initial phases of drug development by in silico approaches to generate potential lead molecules. Drug-likeness in drug development gives an idea whether the particular synthesised compound is comparable to already existing drug molecules in the market. The synthesised molecule expected to be an orally active drug should obey Lipinski ruleCitation30 which states the following four criteria: molecular weight should not be more than 500, hydrogen bond acceptor should not be more than 10, hydrogen bond donor should not be more than 5 and partition coefficient (Clog P) should not be more than 5. Violation of any of these criteria would result in problems of bioavailability upon oral administration. According to Veber et al.,Citation31 number of rotatable bonds should be ≤10 which is an indicator for good bioavailability. In the present study, we calculated several parameters for predicting drug-likeness properties of synthesised compounds. The thiazolidinedione linked 1,3,4-oxadiazole hybrids (7–21) were subjected to following in silico physicochemical studies: number of rotatable bonds (nROTB), hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), lipophilicity (iLogP) and topological polar surface area (TPSA). In silico %age absorption were calculated using the reported formula [(%ABS = 109–(0.345 X TPSA)]. From the results, it was observed that % absorption was found to be in the range of 54.98–70.76%. Compound 7, 8, 9, 10, 11, and 13 showed highest in silico % absorption 70.76% whereas compound 14 revealed 54.98 in silico % absorption. All the synthesised compounds follow Lipinski rule of 5. Molecular weight ranges from 394 to 492 (<500), HBA range 6–8 (≤10), HBD range 0–1 (≤5), iLogP (lipophilicity) was found to be in the range 2.87–3.84 (≤5) which suggest that these compounds upon administration possess good drug likeness properties (). All the compounds except 14 and 21 exhibited high gastro-intestinal absorption and they are impermeable to brain. Furthermore, all the compounds showed nROTB in the range 5–7 (<10) suggesting good bioavailability. All the tested compounds except 14 revealed TPSA range 110.83–132.95 Å2 (<140 Å2), which indicates good intestinal absorption. From these parameters, these synthesised compounds exhibited good drug likeness properties.
Table 1. Pharmacokinetics/ADME predictions of the target compounds 7–21.
Biological evaluation
Cytotoxicity assay
The newly synthesised compounds 7–21 were tested for their cytotoxicity against human cancer cell line viz. breast and colorectal using MTT assay. The IC50 value of the synthesised compounds is presented in . From the results, it has been observed that the synthesised compounds showed significant to moderate growth inhibition compared to the reference drug, 5-fluorouracil (5FU). The cytotoxicity results revealed that compound 9, 10 and 15 showed promising activity. Compound 9 and 10 showed significant cytotoxicity with IC50 = 7.47 µM and IC50 = 7.87 µM, respectively against MCF-7 whereas reference drug 5-FU exhibited IC50 = 34.82 µM. Compound 9 (IC50 = 7.47 µM) and 10 (IC50 = 7.87 µM) displayed 4.5 and 4.4 folds activity of the 5-FU, respectively against MCF-7 cell line. Against HCT-116 cell line, four compounds 9, 10, 14 and 15 showed excellent cytotoxicity with IC50 value of 12.84, 16.14, 42.21 and 39.60 µM, respectively whereas reference drug 5-FU exhibited IC50 = 40.51 µM. It was noticed that the same compound 9 and 10 showed 3.1 and 2.5 folds cytotoxicity compared to5-FU against HCT-116 while compound 14 and 15 was found to be equipotent to the reference drug with IC50 = 42.2 µM and IC50 = 39.60 µM, respectively. The moderate cytotoxicity was observed by compounds 7, 8, 11, 12, 13, 16, 17, 19 and 21 with IC50 in the range 43.37–65.17 µM and 55.5–69.50 µM against MCF-7 and HCT-116, respectively while compound 18 and 20 were found to be inactive (IC50 > 100) against the tested cell lines. Among the synthesised compounds, compound 9 and 10 were the most potent against both MCF-7 and HCT-116 cell line. From the cytotoxicity results, it was noted that the 1,3,4-oxadiazole-thiazolidienedione hybrids derived from substituted aromatic acids hydrazides (7–15) were found to exhibit significant cytotoxicity (IC50 < 50 µM) than 1,3,4-oxadiazole-thiazolidienedione hybrids obtained from substituted phenoxy acetic acids hydrazides (16–21) against both the tested cell line. Among the synthesised hybrids (7–15), compound 9 and 10 bearing halogen at ortho and para position (o-Cl, p-Br) showed highest cytotoxicity.
Table 2. The IC50 (µM) of the synthesised compounds (7–21) against tested human cancer cell line (MCF-7 and HCT-116)Table Footnotea.
In vitro thymidylate synthase activity
Compounds (9, 10, 14 and 15) which showed better cytotoxicity in MTT assay were further tested for their in vitro TS activity to confirm its mechanism of action. The TS inhibition results are presented in . Interestingly, these compounds exhibited remarkable inhibition on the TS enzyme. Compound 9 and 10 revealed TS inhibition with IC50 = 1.67 and 2.21 µM, respectively compared to Pemetrexed (IC50 = 8.2 µM,). The results of TS inhibition were found to be in agreement with the cytotoxicity results.
Table 3. In vitro thymidylate synthase (TS) activity of the active compounds 9, 10, 14, 15 and PTX.
Molecular docking studies
Molecular docking is an intriguing technique to delineate the mechanism of drug-receptor binding in a time and cost-effective way. Among a number of software’s, AutoDock is known for fast speed, easy availability, and high accuracy (rmsd = 2 Å) results.Citation32–33 To support our human TS inhibition results and to get an idea about the binding modes, we carried out shape-based molecular docking studies of compounds 9 and 10 against TS protein (PDB = 6QXG) using Autodock tools. The results of the findings are depicted in and .
Table 4. Docking scores of active compounds 9 and 10 against human thymidylate synthase protein 6QXG.
It has been reported that TS-inhibitors act by interacting with the active sites of the receptor through multiple amino acid residues (viz. Arg50, Ser216, Asn 226, Asp218, His256, Tyr258, Arg50 and Arg215).Citation34–35 Interestingly, our docking results are completely corroborated the literature. We found that the compounds showed good binding affinity towards TS protein (−9.09 and −8.67 kcal/mol for 9 and 10, respectively) and interacted through multiple H-bonds. Oxygen atom of 4-methoxy group was involved in hydrogen bonding interaction with Cys 195, Tyr 135 and oxadiazole ring oxygen atom with Asn 226 in compound 9.Whereas in compound 10, oxygen atom of methoxy group showed hydrogen bonding interaction with Tyr 230, thiazolidinedione C=O group at 4-position showed hydrogen bonding interaction with Asn 226 and nitrogen atom of oxadiazole ring at 4-position with Leu 221. Contrary to this, reference drug 5-fluorouracil (5-FU) exhibited lower affinity (−4.22 kcal/mol) and interacted with Thy 306, Phe 80, Ile307 and Arg 78 via H-bonding interaction with C=O group at 2- and 4-position, N–H group at 1- and 3-position respectively. Compounds 9 and 10 have different substituent in the phenyl ring attached to the oxadiazole ring, bearing chlorine at ortho position and bromine at para position, respectively but due to flexibility of the molecule, compound 10 bend and interact with amino residue Tyr 230, Asn 226 and Leu 221. Whereas, 5 fluorouracil is a small ligand in which all the electronegative atoms interact with the amino acid residues except fluorine. These results were in agreement with the in vitro results of compound 9 (IC50 = 1.67) and 10 (IC50 = 2.21 µM).
Conclusions
A library of synthesised thiazolidinedione-1,3,4-oxadiazole hybrids have been screened for in vitro antiproliferative as well as TS activities. All the synthesised hybrids follow Lipinski and Veber rules indicating good drug likeness properties upon oral administration. Among the synthesised hybrids, compound 9 and 10 showed excellent cytotoxicity against MCF-7 and HCT-116 cells. From the in vitro TS activity, Compound 9 and 10 were found to be potent inhibitors against TS protein with IC50 = 1.67 and 2.21 µM, respectively. The docking studies of compounds 9 and 10 were found to be consistent with in vitro cytotoxicity and TS results. From these studies, it can be concluded that compounds 9 and 10 have the potential to be developed as TS inhibitors.
Supplemental Material
Download PDF (2.5 MB)Acknowledgement
The authors thanks Chemistry department, Albaha university for providing the necessary facilities to carry out this work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Goud NS, Ghouse SM, Arifuddin M, et al. Synthesis and biological evaluation of coumarin-1,3,4-oxadiazole hybrids as selective carbonic anhydrase IX and XII inhibitors. Bioorg Chem 2019;87:765–72.
- Patel K, Karthikeyan C, Solomon VR, et al. Synthesis of some coumarinyl chalcones and their antiproliferative activity against breast cancer cell lines. Lett Drug Design Disc 2011;8:308–11.
- Lambratu R, Donna V, Monzur R, et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell 2004;5:341–51.
- Young DW. Studies on thymidylate synthase and dihydrofolate reductase-two enzymes involved in the synthesis of thymidine. Chem Soc Rev 1994; 23:119–28.
- Kumar VP, Cisneros JA, Frey KM, et al. Structural studies provide clues for analog design of specific inhibitors of Cryptosporidium hominis thymidylate synthase-dihydrofolate reductase. Bioorg Med Chem Lett 2014;24:4158–61.
- Catalano A, Luciani R, Carocci A, et al. X-ray crystal structures of Enterococcus faecalis thymidylate synthase with folate binding site inhibitors. Eur J Med Chem 2016;123:649–64.
- Onen FE, Boum Y, Jacquement C, et al. Design, synthesis and evaluation of potent thymidylate synthase X inhibitors. Bioorg Med Chem Lett 2008;18:3628–31.
- Friedkin M, Kornberg A. The enzymatic conversion of deoxyuridylic acid to thymidylic acid and the participation of tetrahydrofolic acid. In: Mc Elroy WD and Glass B, eds. The Chemical Basis of Heredity. Baltimore: Johns Hopkins Press; 1957:609–614.
- Navalgund LG, Rossana C, Muench AJ, et al. Cell cycle regulation of thymidylate synthetase gene expression in cultured mouse fibroblasts. J Biol Chem 1980;255:7386–90.
- Nazreen S, Alam MS, Hamid H, et al. Design, synthesis, in silico molecular docking and biological evaluation of novel oxadiazole based thiazolidine-2,4-diones bis-heterocycles as PPAR-γ agonists. Eur J Med Chem 2014;87:175–85.
- Unlusoy MC, Verspohl EJ, Ertan R. Synthesis and antidiabetic activity of some new chromonyl-2,4-thiazolidinediones. J EzyInhib Med Chem 2010; 25:784–9.
- Chen R, Yan J, Liu P, et al. Effects of thiazolidinedione therapy on inflammatory markers of type 2 diabetes: a meta-analysis of randomized controlled trials. PLoS ONE 2015;10:e0123703.
- Hu CF, Zhang PL, Sui YF, et al. Ethylenic conjugated coumarin thiazolidinediones as new efficient antimicrobial modulators against clinical methicillin-resistant Staphylococcus aureus. Bioorg Chem 2020; 94:103434.
- Shankar S, Vuppu S. In vitro drug metabolism and pharmacokinetics of a novel thiazolidinedione derivative, a potential anticancer compound. J Pharma Biomed Anal 2020;179:113000.
- Tokala R, Thatikonda S, Sana S, et al. Synthesis and in vitro cytotoxicity evaluation of β-carboline-linked 2,4-thiazolidinedione hybrids: potential DNA intercalation and apoptosis-inducing studies. New J Chem 2018;42:16226–36.
- Trotsko N, Przekora A, Zalewska Z, et al. Synthesis and in vitro antiproliferative and antibacterial activity of new thiazolidine-2,4-dione derivatives. J EzyInhib Med Chem 2018;33:17–24.
- Shimazaki N, Togashi N, Hanai M, et al. Anti-tumour activity of CS-7017, a selective peroxisome proliferator-activated receptor gamma agonist of thiazolidinedione class, in human tumour xenografts and a syngeneic tumour implant model. Eur J Cancer 2008;44:1734–43.
- Smallridge RC, Copland JA, Brose MS, et al. Efatutazone, an oral PPAR-γ agonist, in combination with paclitaxel in anaplastic thyroid cancer: results of a multicenter phase 1 trial. J Clin Endocrin Metab 2013;98:2392–400.
- Belfiore A, Genua M, Malaguarnera R. PPAR-γ agonists and their effects on IGF-i receptor signaling: implications for cancer. PPAR Res 2009;2009:1–18.
- Lee WH, Kim SG. AMPK-dependent metabolic regulation by PPAR agonists. PPAR Res 2010;2010:1–10.
- Knight SD, Adams ND, Burgess JL, et al. Discovery of GSK2126458, a highly potent inhibitor of pi3k and the mammalian target of rapamycin. Med Chem Lett 2010;1:39–43.
- Liu K, Rao W, Parikh H, et al. 3,5-Disubstituted-thiazolidine-2,4-dione analogs as anticancer agents: design, synthesis and biological characterization. Eur J Med Chem 2012;47:125–36.
- James ND, Growcott JW. Zibotentan. Drugs Future 2009;34:624–33.
- Shen W, Xi H, Li C, et al. Endothelin‐A receptor in gastric cancer and enhanced antitumor activity of trastuzumab in combination with the endothelin—A receptor antagonist ZD4054. Ann N Y Acad Sci 2019;1448:30–41.
- Du QR, Li DD, Pi YZ, et al. Novel 1,3,4-oxadiazole thioether derivatives targeting thymidylate synthase as dual anticancer/antimicrobial agents. Bioorg Med Chem 2013;21:2286–97.
- Abbot V, Sharma P, Dhiman S, et al. Small hybrid heteroaromatics: resourceful biological tools in cancer research. RSC Adv 2017;7:28313–49.
- WahbaAJ FM. The enzymatic synthesis of thymidylate: i. early steps in the purification of thymidylate synthetase of Escherichia coli. JBiol Chem 1962;237:3794.
- Davisson VJ, Sirawaraporn W, Santi DV. Expression of human thymidylate synthase in Escherichia coli. J Biol Chem 1989;264:9145–8.
- Morris GM, Goodsell GS, Halliday RS, et al. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639–62.
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev 2001;46:3–26.
- Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23.
- Patrick AH, Jonathan BC, John OT. Molecular docking of intercalators and groove- binders to nucleic acids using autodock and surflex. J Chem Inf Model 2008;48:1602–15.
- Oleg T, Arthur JO. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 2010;31:455–61.
- Lamia HTA, Taghreed ZS, Abeer MN. Design, synthesis, anticancer evaluation and docking studies of new pyrimidine derivatives as potent thymidylate synthase inhibitors. Bioorg Chem 2019;91:103159.
- Ahmed FE, Qasem MAA, Emad S. Design, synthesis, molecular docking of new thiopyrimidine-5-carbonitrile derivatives and their cytotoxic activity against HepG2 cell line. J Appl Pharm Sci 2014;4:102–11.