
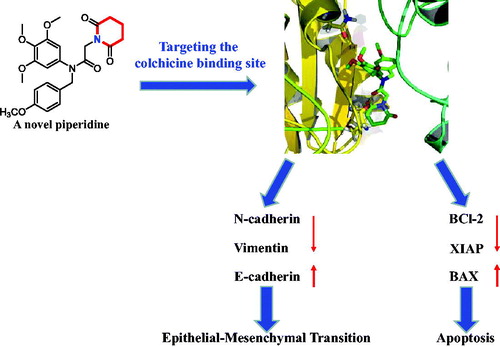
Abstract
Tubulin polymerisation inhibitors that target colchicine binding site were powerful anticancer agents. Although along the years many colchicine binding site inhibitors (CBSIs) have been reported, few piperidine derivatives were identified as CBSIs. In this regard, we focussed efforts on the piperidine as a promising chemotype to develop potent CBSIs. Herein, novel piperidine derivatives were synthesised and evaluated for their antiproliferative activities. Among them, compound 17a displayed powerful anticancer activity with the IC50 value of 0.81 µM against PC3 cells, which was significantly better than 5-fluorouracil. It could inhibit tubulin polymerisation binding at the colchicine site and inhibit the tumour growth in vitro and in vivo. Further biological studies depicted that 17a suppressed the colony formation, induced apoptosis, and inhibited epithelial-mesenchymal transition against PC3 cells. These results revealed that compound 17a is a promising colchicine binding site inhibitor for the treatment of cancer and it is worthy of further exploitation.
Graphical Abstract
1. Introduction
Microtubules are dynamic cytoskeletal polymers of tubulin, which involved in essential cellular functions, such as cell growth, mitosis, motility, intracellular transport, and divisionCitation1,Citation2. Because of the important role of microtubules, they have become an important target for the design of new anticancer agentsCitation3,Citation4. Microtubule-targeting agents interact with tubulin through at least four binding sites: the laulimalide site, paclitaxel site, vinca site, and colchicine siteCitation5. In contrast to the paclitaxel site and vinca site, the colchicine binding site is located between the α- and β-monomers of the α,β-tubulin heterodimerCitation6. Colchicine binding site inhibitors (CBSIs) exhibit the potently inhibitory effects of tumour cell proliferation and metastasisCitation7. Therefore, an increased attention has been focussed on the discovery of CBSIs.
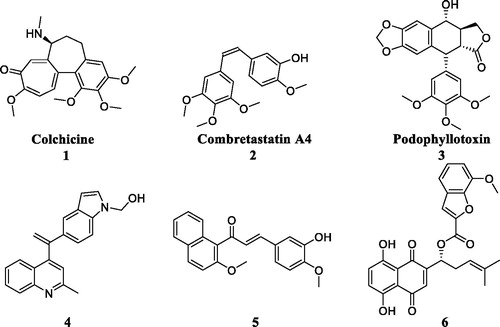
In recent years, some colchicine binding site inhibitors have been reported as anticancer agents ()Citation8. Colchicine (1), combretastatin A4 (2) and podophyllotoxin (3) as anticancer agents could induce apoptosis and cell death by targeting colchicine binding siteCitation9. Quinoline-indole derivative 4 disrupted cell microtubule networks and arrested the cell cycle at G2/M phase against K562 cellsCitation10. Chalcone 5 as a colchicine binding site inhibitor displayed the potent antiproliferative activity with an IC50 value of 1.42 µM against MCF7 cellsCitation11. Shikonin-benzo[b]furan 6 could regulate the expression of apoptosis related proteins in HT29 cells by targeting the colchicine binding siteCitation12. Although these CBSIs demonstrated potent antitumor activity against various cancer cell lines, there clinical trials were halted due to serious side effects, low solubility and low bioavailabilityCitation13. As to overcome the limitations, it is imperative to discover novel colchicine binding site inhibitors.
Figure 1. Reported colchicine binding site inhibitors (CBSIs).
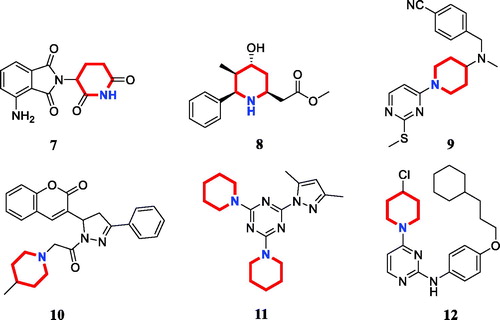
Piperidine is a privileged scaffold in the field of drug discovery which provides numerous opportunities in exploring this moiety as an anticancer agent by acting on various receptors of utmost importance ()Citation14. Pomalidomide (7) was a FDA approved drug to treat multiple myeloma by inhibiting TNFαCitation15. Piperidine 8 displayed the antiproliferative activity with IC50 values from 5.4 µM to 8.5 µM against A549, MCF7, DU145, and HeLa cell linesCitation16. Piperidine derivative 9 was a novel human heat shock protein 70 inhibitor for the treatment of drug-resistant tumoursCitation17. 5-Phenyl-N-piperidine ethanone containing 4,5-dihydropyrazole derivative 10 occupied high antiproliferative activities against SGC-7901, MGC-803 and Bcap-37 cellsCitation18. Piperidine 11 affected the cell viability and induced G2/M phase cell cycle arrest in K562 cellsCitation19. Piperidine 12 was a selective inhibitor against triple-negative breast cancer cell line MDA-MB-468Citation20.
Figure 2. Chemical structures of anticancer piperidine derivatives.
Our research group has long been involved in designing new colchicine binding site inhibitors with the aim to discover various anticancer agentsCitation21–24. In this work, we report the synthesis and anticancer evaluation of novel piperidine derivatives as colchicine binding site inhibitors. Importantly, their effects on apoptosis and epithelial-mesenchymal transition (EMT) were also revealed.
2. Experimental
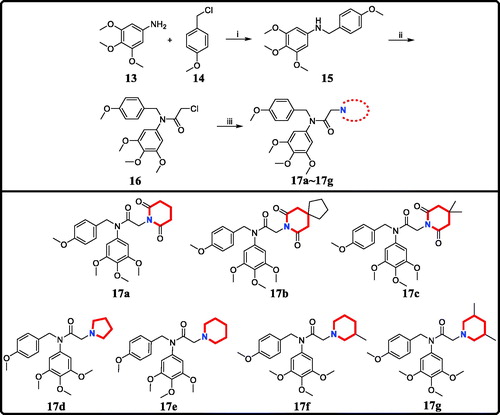
2.1. General procedure for the synthesis of compound 16
To a solution of 3,4,5-trimethoxyaniline (5 mmol, 1.0 eq) and 1-(chloromethyl)-4-methoxybenzene (5.5 mmol, 1.1 eq) in N,N-dimethylformamide (20 ml) was added triethylamine (7.5 mmol, 1.5 eq) at room temperature. The mixture was stirred at room temperature for eight hours. Upon completion, ethyl acetate and water were added. The aqueous layer was extracted with ethyl acetate for several times and the combined organic layers were evaporated to give the crude product 15. 2-Chloroacetyl chloride (3 mmol, 1.5 eq) and potassium carbonate (3 mmol, 1.5 eq) were added to the solution of crude product 15 (2 mmol, 1eq) in acetone. After stirring at room temperature for four hours, the reaction mixture was concentrated to remove then treated with a solution of ethyl acetate and H2O. The organic layer was washed with brine, dried over anhydrous sodium sulphate, and then concentrated to provide compound 16 without purification. The NMR analyses were described in Supplemental data.
2.2. General procedure for the synthesis of piperidine and pyrrolidine derivatives 17a∼17g
Cyclamine (2 mmol, 2 eq) was added to the mixture of compound 16 (1 mmol, 1 eq) and sodium hydroxide (1.5 mmol, 1.5 eq) in acetone (5 ml). The mixture was stirred at room temperature for 12 h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with brine and concentrated under reduced pressure. The residue was purified with column chromatography (hexane: EtOAc = 9:1) to obtain analogues 17a∼17g.
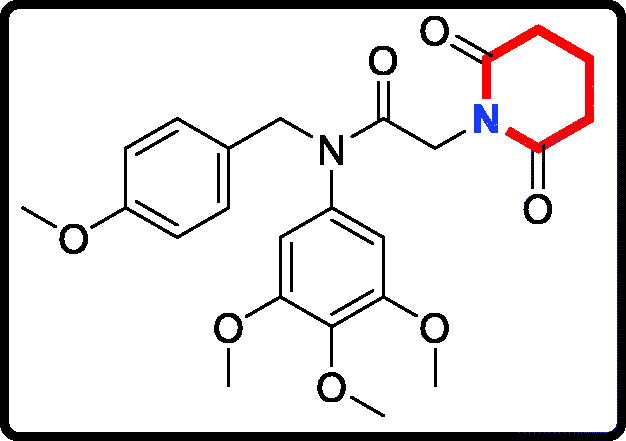
2–(2,6-Dioxopiperidin-1-yl)-N-(4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl) acetamide (17a)
Yield: 62%. White solid, m.p.:1 5 6 ∼ 158 °C. 1H NMR (400 MHz, CDCl3) δ 7.05 (d, J = 8.5 Hz, 2H), 6.74 (d, J = 8.6 Hz, 2H), 6.22 (s, 2H), 4.69 (s, 2H), 4.26 (s, 2H), 3.76 (s, 3H), 3.71 (s, 3H), 3.66 (s, 6H), 2.64 (t, J = 6.5 Hz, 4H), 1.99–1.90 (m, 2H). 13 C NMR (100 MHz, CDCl3) δ 171.35, 165.46, 158.02, 152.59, 136.88, 135.29, 129.35, 128.31, 112.72, 104.74, 59.92, 55.19, 54.26, 51.65, 40.23, 31.45, 16.02. HRMS (m/z) [M + H]+ calcd for C24H29N2O7, 457.1975; found, 457.1979.
2–(7,9-Dioxo-8-azaspiro[4.5]decan-8-yl)-N-(4-methoxybenzyl)-N- (3,4,5-trimethoxyphenyl)acetamide (17 b)
Yield: 52%. White solid, m.p.:9 5 ∼ 97 °C. 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 6.21 (s, 2H), 4.70 (s, 2H), 4.25 (s, 2H), 3.76 (s, 3H), 3.71 (s, 3H), 3.65 (s, 6H), 2.58 (s, 4H), 1.66 (t, J = 6.8 Hz, 4H), 1.54 (d, J = 6.4 Hz, 4H). 13 C NMR (100 MHz, CDCl3) δ 171.11, 165.45, 158.01, 152.57, 136.86, 135.27, 129.37, 128.32, 112.71, 104.77, 59.91, 55.18, 54.25, 51.52, 43.35, 40.22, 38.59, 36.65, 23.10. HRMS (m/z) [M + H]+ calcd for C28H35N2O7, 511.2444; found, 511.2448.
2–(4,4-Dimethyl-2,6-dioxopiperidin-1-yl)-N-(4-methoxybenzyl)-N- (3,4,5-trimethoxyphenyl)acetamide (17c)
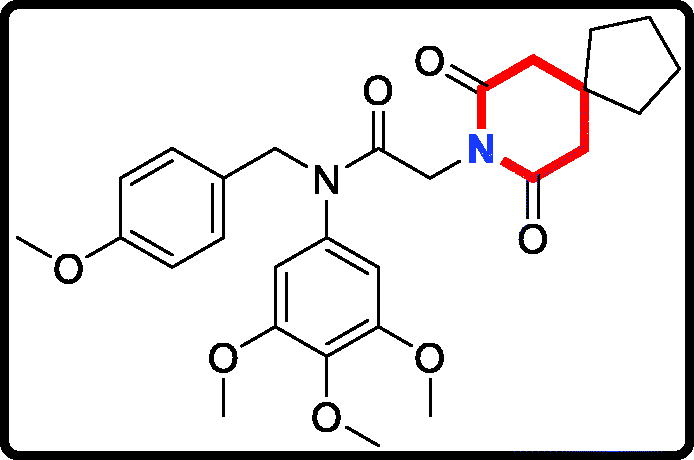
Yield: 46%. White solid, m.p.:1 4 1 ∼ 143 °C. 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 8.6 Hz, 2H), 6.21 (s, 2H), 4.70 (s, 2H), 4.26 (s, 2H), 3.76 (s, 3H), 3.71 (s, 3H), 3.65 (s, 6H), 2.49 (s, 4H), 1.11 (s, 6H). 13 C NMR (100 MHz, CDCl3) δ 170.88, 165.52, 158.01, 152.58, 136.87, 135.26, 129.37, 128.31, 112.71, 104.76, 59.91, 55.18, 54.25, 51.53, 45.03, 40.13, 28.32, 26.77. HRMS (m/z) [M + H]+ calcd for C26H33N2O7, 485.2288; found, 485.2294.
N-(4-methoxybenzyl)-2-(pyrrolidin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (17d)
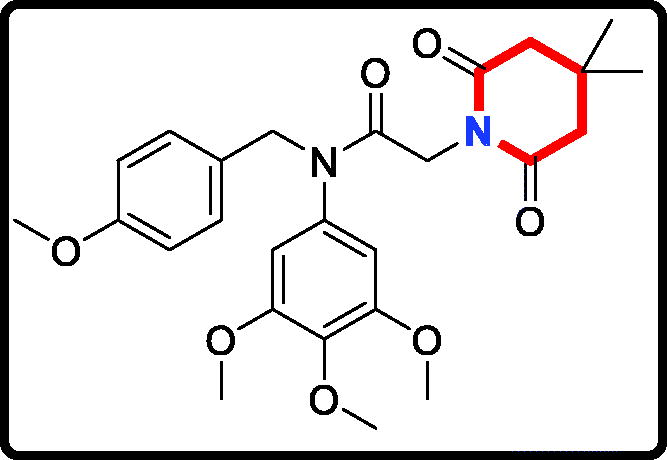
Yield: 75%. White solid, m.p.:9 4 ∼ 96 °C. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 8.5 Hz, 2H), 6.05 (s, 2H), 4.70 (s, 2H), 3.78 (s, 3H), 3.71 (s, 3H), 3.63 (s, 6H), 3.02 (s, 2H), 2.51 (s, 4H), 1.70 (s, 4H). 13 C NMR (100 MHz, CDCl3) δ 168.54, 157.97, 152.39, 136.63, 136.08, 129.59, 128.92, 112.60, 104.82, 59.94, 56.07, 55.12, 54.25, 53.23, 51.24, 22.66. HRMS (m/z) [M + H]+ calcd for C23H31N2O5, 415.2233; found, 415.2237.
N-(4-methoxybenzyl)-2-(piperidin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (17e)

Yield: 61%. White solid, m.p.:7 5 ∼ 77 °C. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 8.5 Hz, 2H), 6.04 (s, 2H), 4.69 (s, 2H), 3.78 (s, 3H), 3.71 (s, 3H), 3.63 (s, 6H), 2.85 (s, 2H), 2.35 (s, 4H), 1.57–1.42 (m, 4H), 1.32 (s, 2H). 13 C NMR (100 MHz, CDCl3) δ 168.38, 157.96, 152.34, 136.60, 136.20, 129.57, 128.93, 112.59, 104.88, 59.95, 59.30, 55.13, 54.25, 53.61, 51.28, 24.86, 22.98. HRMS (m/z) [M + H]+ calcd for C24H33N2O5, 429.2389; found, 429.2392.
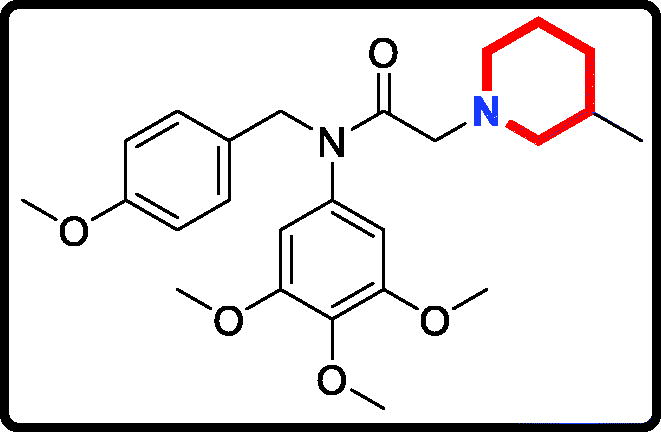
N-(4-methoxybenzyl)-2–(3-methylpiperidin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (17f)

Yield: 80%. White solid, m.p.:7 2 ∼ 74 °C. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 8.4 Hz, 2H), 6.04 (s, 2H), 4.69 (s, 2H), 3.78 (s, 3H), 3.71 (s, 3H), 3.63 (s, 6H), 2.86 (s, 2H), 2.74 (s, 2H), 1.85 (s, 1H), 1.59 (d, J = 10.3 Hz, 3H), 1.51 (s, 2H), 0.75 (s, 1H), 0.75 (d, J = 5.6 Hz, 3H). 13 C NMR (100 MHz, CDCl3) δ 168.44, 157.97, 152.34, 136.60, 136.20, 129.58, 128.93, 112.59, 104.89, 61.00, 59.95, 59.09, 55.13, 54.25, 53.04, 51.27, 31.65, 30.06, 24.50, 18.64. HRMS (m/z) [M + H]+ calcd for C25H35N2O5, 443.2546; found, 443.2548.
2–(3,5-Dimethylpiperidin-1-yl)-N-(4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (17 g)
Yield: 62%. White solid, m.p.:9 6 ∼ 98 °C. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.4 Hz, 2H), 6.72 (d, J = 8.4 Hz, 2H), 6.04 (s, 2H), 4.70 (s, 2H), 3.78 (s, 3H), 3.71 (s, 3H), 3.63 (s, 6H), 2.87 (s, 2H), 2.74 (d, J = 9.5 Hz, 2H), 1.61 (s, 2H), 1.58 (s, 1H), 1.48 (s, 2H), 0.74 (d, J = 6.3 Hz, 6H), 0.40 (q, J = 11.8 Hz, 1H). 13 C NMR (100 MHz, CDCl3) δ 168.43, 157.98, 152.35, 136.61, 136.15, 129.60, 128.92, 112.59, 104.88, 60.52, 59.95, 58.79, 55.12, 54.25, 51.27, 40.79, 30.02, 18.51. HRMS (m/z) [M + H]+ calcd for C26H37N2O5, 457.2702; found, 457.2706.
2.3. Mtt assay
MGC803 (gastric cancer cells), PC3 (prostate cancer cells) and MCF7 (breast cancer cells) were obtained from the Chinese Academy of Sciences (Shanghai, China). Dimethyl sulfoxide was used to mix the piperidine derivatives 17a∼17g to different concentrations (0.1 µM, 0.5 µM, 1 µM, 2 µM, 4 µM, 8 µM, 16 µM, 32 µM, and 64 µM). Tumour cells were seeded into 96-well plates in 100 µL of culture medium. Then, cells were treated in triplicate with a gradient concentration of 17a∼17g and incubated at 37 °C, 5% CO2 for 48 h. For all cell lines, MTT (thiazolyl blue tetrazolium bromide) assay was performed to measure the antiproliferative activityCitation25.
2.4. Colony formation assay
PC3 cells were seeded in a 6-well plate and incubated for 24 h, then treated with 17a at different concentrations (0, 0.125 µM, 0.25 µM and 0.5 µM). After seven days, the culture medium was removed and cells were washed with phosphate buffer saline. Then, the system was fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The cells image was captured with the camera (Jinghua Co. Ltd, Beijing, China).
2.5. Dapi staining
PC3 cells were seeded in a 6-well plate and incubated for 24 h, then treated with 17a at different concentrations (0, 0.5 µM and 1 µM). After 48 h incubation, remove the medium, wash the cells with phosphate buffer saline gently, and fix the cells with 4% paraformaldehyde for 20 min. Cells were then stained with 4′,6-diamidino-2-phenylindole (DAPI) in the dark for 20 min.
2.6. Cell apoptosis assay
PC3 cells were seeded in a 6-well plate and incubated for 24 h, then treated with 17a at different concentrations (0, 0.5 µM and 1 µM) for 48 h. Then, cells were harvested and the AnnexinV-FITC/PI kit (Jiangsu KeyGEN BioTECH Corp., Ltd, Zhilan Road, Jiangning District, Nanjing, China) was used according to the manufacturer’s instructions to perform the apoptosis assay.
2.7. Wound healing
PC3 cells were seeded in a 6-well plate and incubated for 24 h. The cell surface was scratched using a pipet tip. Then, the cells were cultured with fresh medium containing 1% foetal bovine serum and different concentrations of 17a (0, 0.5 µM and 1 µM) for 24 h.
2.8. Migration assay
PC3 cells were seeded in a Transwell plate (CORNING, Corning City, State of New York, USA). 1% heat-inactivated foetal bovine serum and 20% foetal bovine serum were added into upper and lower chamber separately. Different concentrations of 17a (0, 0.5 µM and 1 µM) were added in the chambers. After the incubation for 48 h, remove the medium and wash the chambers with phosphate buffer saline. The migrating cells were fixed with methanol for 20 min and stained with Hoechst-33258 (Innochem, Beijing, China). Each chamber was photographed using Thermo Fisher Cellomics High Content System (Heqi Co. Ltd, Shanghai, China).
2.9. In vitro tubulin polymerisation assay
5.6 mg/ml tubulin was resuspended in PEM buffer (containing 80 mmol/L PIPES, 1 mmol/L EGTA, 0.5 mmol/L MgCl2, 1 mmol/L ATP and 10.2% (v/v) glycerol). The system was incubated with 17a (0, 1 µM, 2 µM and 4 µM) and colchicine on ice. The reaction was monitored by a spectrophotometer (JB-750, Jiangsu, China) in absorbance at 420 nm at 37 °C every minute (excitation wavelength is 340 nm).
2.10. Ebi competition assay
N,N’-ethylenebis(iodoacetamide) (EBI) was purchased from Innochem company (Beijing, China). PC3 cells were seeded in a 6-well plate and incubated for 24 h. Cells were first incubated with compound 17a, colchicine (15 µM) or dimethyl sulfoxide for 2 h and afterward treated with EBI (100 µM) for 2 h. Then, the cells were harvested and lysed to perform western blotting analysis.
2.11. Western blot analysis
PC3 cells were seeded in a 6-well plate and incubated for 24 h, then treated with 17a at different concentrations (0, 0.5 µM, 1 µM and 1.5 µM). After the the incubation for 48 h, PC3 cells were harvested and lysed. Protein lysates were resolved by SDS-PAGE and transferred to nitrocellulose membrane. The membrane was incubated with appropriate antibodies at 4 °C for 12 h in 5% skimmed milk. After conjugated with secondary antibodies, the detection of proteins was carried out with an ECL Western Blotting Substrate Kit (Amyjet Scientific, Wuhan, China).
2.12. Docking studies
Piperidine derivative 17a was selected to do molecular docking study. For the receptor preparation, the PDB code (1SA0) was downloaded from the Protein Data Bank. The docking studies were performed by Autodock 4.2 (The Scripps Research Institute, San Diego, California, USA).
2.13. In vivo anti-tumour activity
Animals were treated according to the protocols established by the ethics committee of Zhengzhou University and the in vivo experiments were carried out in accordance with the approved guidelines and approved by the ethics committee of Zhengzhou University. BALB/c nude mice were purchased from Hunan Slack Scene of Laboratory Animal Co. Ltd. (Hunan, China). Prostate cancer PC3 xenograft model was established in BALB/c mice. The tumour-bearing mice were randomised into three groups (six mice in each group) and subcutaneous injection with normal saline or compound 17a (50 mg/kg) or colchicine (0.25 mg/kg) for 21 days. Compound 17a was firstly resolved with the mixture of ethyl alcohol and castor oil, and then diluted in normal saline. Tumour size was determined by calliper measurement. The tumour volume was calculated using the ellipsoid volume formula (Length × Width2/2).
2.14. Statistical evaluation
Data were presented as means ± SD. Statistical analyses were performed by SPSS 17.0 and GraphPad.
3. Results and discussion
3.1. Chemistry
The target derivatives 17a∼17g were synthesised from the starting material 3,4,5-trimethoxyaniline in a sequence of reactions and characterised by NMR and HRMS. Nucleophilic substitution reaction of 3,4,5-trimethoxyaniline with 1-(chloromethyl)-4-methoxybenzene afforded the secondary amine 15, which was reacted with 2-chloroacetyl chloride leading to the key intermediate 16 (Scheme 1). Compound 16 reacted with piperidines and pyrrolidine in the presence of sodium hydroxide to give derivatives 17a∼17g.
Scheme 1. Reagents and conditions: (i) N,N-dimethylformamide, triethylamine; (ii) 2-chloroacetyl chloride, K2CO3, acetone; (iii) Piperidines or pyrrolidine, NaOH, acetone.
3.2. Antiproliferative activity
To discover novel antitumor agents, we evaluated the antiproliferative activity of all piperidine and pyrrolidine derivatives 17a∼17g against three cancer cell lines PC3 (prostate cancer cell line), MGC803 (gastric cancer cell line) and MCF7 (breast cancer cell line) using the 3–(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Based on the previous reference Citation5,Citation26-fluorouracil and colchicine as well-known antitumor agents were selected as the reference drugs in this work.
The antiproliferative activity results of candidate derivatives 17a∼17g against all three cancer cell lines were shown in . Novel piperidine derivatives 17a∼17c exhibited more potent antiproliferative activity against all selected cancer cell lines with IC50 values from 0.81 µM to 9.37 µM than 5-fluorouracil. Among all target derivatives, compound 17a displayed the best antiproliferative activity with the IC50 value of 1.09 µM, 0.81 µM, and 1.30 µM against MGC803 cells, PC3 cells and MCF7 cells, respectively. These antiproliferative activity results indicate that piperidine-2,6-dione fragment might display potent anticancer activity in vitro and could be used as the leading unit to design more similar derivatives.
Table 1. Antiproliferative activity of candidate derivatives 17a∼17g.
In addition, candidate derivatives attaching different cyclamine units (pyrrolidine, piperidine, 3-methylpiperidine, and 3,5-dimethylpiperidine) were synthesised and evaluated for their antiproliferative activity. By this way, the molecular diversity of piperidine derivatives were explored. Changing the piperidine unit (17e) to a pyrrolidine unit (17d) led to a decrease of the inhibitory activity against all the tested cell lines, indicating that the piperidine fragment was unfavourable for the antiproliferative activity. When the 3-methylpiperidine group (17f) and the 3,5-dimethylpiperidine group (17 g) were replaced by a piperidine group (17e), the antiproliferative activity was improved against all cancer cell lines. All these modifications revealed that the piperidine scaffold was important for their inhibitory activity.
3.3. Compound 17a inhibited the colony formation and cell viability
Based on the screening activity results of all synthetic derivatives, compound 17a as the most potent compound was chosen to perform colony formation to investigate whether piperidine derivatives could inhibit the proliferation of tumour cells. As shown in , the cell viability of PC3 cells were decreased with the treatment of compound 17a in a time-dependent manner and a concentration-dependent manner.
Figure 3. (A) The cell viability of PC3 cells with the treatment of compound 17a at different concentrations for 48 h. (B) The cell viability of PC3 cells with the treatment of compound 17a at 1 μM for different hours. (C) Colony formation of PC3 cells with the treatment of 17a at various concentrations for 7 days. (D) Colony formation rate. *p < .05 verse control, **p < .01 verse control, ***p < .001 verse control and ****p < .0001 verse control.
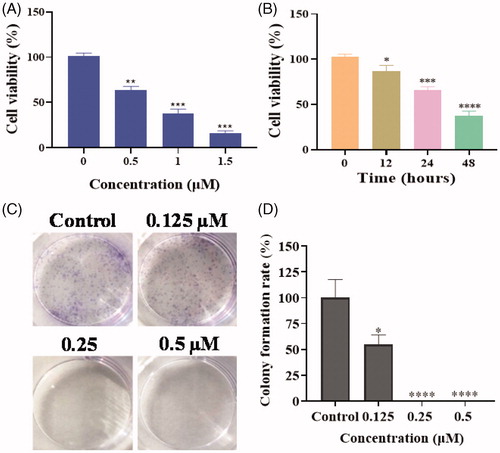
PC3 cells with the treatment of 17a exhibited fewer and smaller colonies compared to the control (), which indicated that piperidine derivative 17a could significantly inhibit the proliferation of PC3 cells in a concentration-dependent manner.
3.4. Compound 17a induced morphological changes and apoptosis
Inspired by the potent inhibition of the piperidine derivative 17a against PC3 cells, we then investigated whether 17a was able to induce morphological changes. After being incubated with the piperidine derivative 17a for 48 h at different concentrations (0 µM, 0.5 µM and 1 µM), the morphological changes of PC3 cells were recorded. As shown in , significant changes of cell morphology such as rounding up and cell debris were observed at the high concentration (yellow arrows). The effects of compound 17a on the apoptosis was also investigated using the propidium iodide (PI) and Annexin V-FITC. As illustrated in , compound 17a induced apoptosis of PC3 cells in a concentration-dependent manner. Specifically, the percentage of apoptotic cells was about 2.0% for the control group. When treated with the high concentration (1 µM) of compound 17a, around 30.4% of apoptosis rate was observed. To further investigate the mechanism of apoptosis, western blot analysis was performed to explore the expression levels of apoptosis-related proteins in PC3 cells. As shown in , the treatment of PC3 cells with 17a resulted in decreased expression levels of BCl-2 and XIAP in a concentration-dependent manner.
Figure 4. (A) Morphological changes analysis with DAPI staining after 48 h of compound 17a in PC3 cells. (B&C) Quantitative analysis of apoptotic cells using Annexin V-FITC/PI double staining. (D) Expression levels of 17a on apoptosis-related proteins.
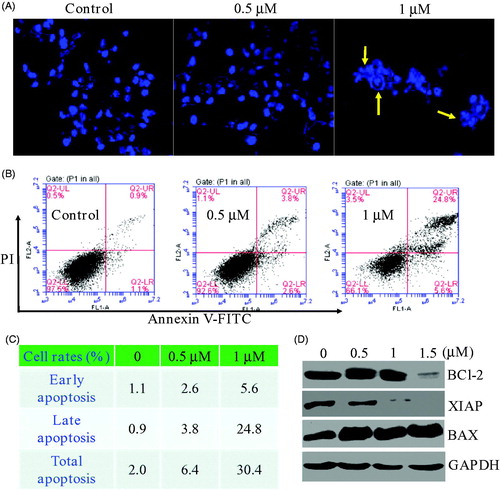
Meanwhile, the expression of BAX increased accordingly. Therefore, compound 17a could induce morphological changes and apoptosis in PC3 cells.
3.5. Compound 17a inhibited the epithelial-mesenchymal transition in PC3 cells
The epithelial-mesenchymal transition (EMT) is a key developmental programme that is often activated during cancer migration and metastasisCitation27. In this work, we evaluated the migration ability of PC3 cells by wound healing and migration assay. As shown in , the piperidine derivative 17a inhibited the wound healing obviously. The migration assay () also demonstrated that compound 17a hindered the PC3 cells migration through the biological membrane. We also examined the expression of the typical proteins of EMT process by western blot experiments. showed that compound 17a could upregulate the epithelial cells’ biomarker E-Cadherin while the mesenchymal cells’ biomarkers, N-Cadherin and Vimentin, were down-regulated correspondingly. All these results indicated that compound 17a inhibited the epithelial-mesenchymal transition in PC3 cells.
Figure 5. (A) Wound healing assay. (B&C) The migration ability of PC3 cells after the treatment of compound 17a. (D) Expression levels of N-cadherin, Vimentin, and E-Cadherin.
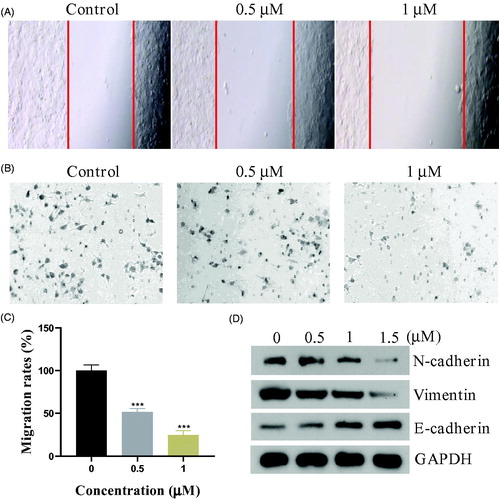
3.6. Compound 17a was a novel colchicine binding site inhibitor
The tubulin polymerisation inhibition activity in vitro of compound 17a was evaluated. When tubulin was incubated with the piperidine derivative 17a at different concentrations (1 µM, 2 µM and 4 µM), the increased tendency of the fluorescence intensity was obviously slowed down. The IC50 value of compound 17a was 2.03 µM against tubulin (). It revealed that the piperidine derivative 17a was a novel tubulin polymerisation inhibitor. In the N,N’-ethylenebis(iodoacetamide) (EBI) assay, compound 17a at 10 µM prevented the formation of EBI: β-tubulin adduct comparing with DMSO and EBI treatment. These results in indicated that the piperidine derivative 17a directly bind to the colchicine binding site.
Figure 6. (A) Tubulin polymerisation inhibitory activity of 17a. (B) EBI competition assay on PC3 cells.
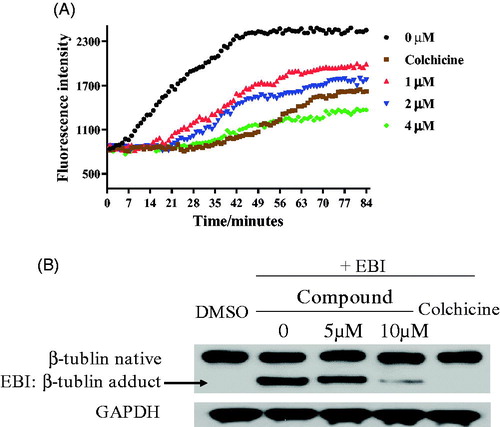
3.7. Molecular docking studies of compound 17a
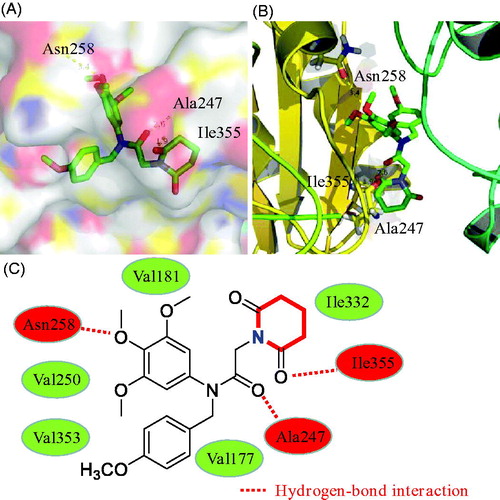
In this work, molecular docking methodologies were also used to explore any molecular interactions between the piperidine derivative 17a and tubulin. We used the autodock software as an automated tool to perform docking and selected PDB code 1SA0. As shown in , 3,4,5-trimethoxyphenyl ring, amide group, and piperidine-2,6-dione formed three hydrogen bonds with residues Ala247, Asn258 and Ile355, respectively. In addition, derivative 17a formed hydrophobic interactions with residues Val250, Val353, Val181, Val177 and Ile332. These docking results of compound 17a may provide a basis for further optimisation.
Figure 7. (A,B) 3D binding models of compound 17a in the active sites of tubulin. (C) 2D binding models and hydrogen-bond interactions.
3.8. In vivo antitumor study of compound 17a
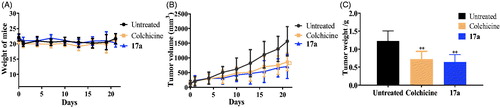
Due to the best inhibitory activity of compound 17a against PC3 cells, we also evaluated the in vivo antitumor effects of compound 17a on xenograft models bearing PC3 cells by the sub-cutaneous implantation. After the treatment of compound 17a, the weight of mice, the tumour weight and the tumour volume were measured and recorded. As shown in , the piperidine derivative 17a inhibited tumour growth, while the body weight was almost unchanged, indicating the antitumor efficacy and low toxicity.
Figure 8. In vivo antitumor effects after the treatment of normal saline (untreated group), colchicine (0.25 mg/kg), compound 17a (50 mg/kg). (A) Weight of mice. (B) Tumour volume. (C) Tumour weight of mice. **p < .01 verse control.
4. Conclusions
In the present study, we investigated the anticancer effects in vitro and in vivo of the piperidine derivative 17a. Our findings showed that compound 17a could induce apoptosis and inhibit the epithelial-mesenchymal transition progress in PC3 cells as a novel colchicine binding site inhibitor. Thus, the piperidine derivative 17a could be a lead compound for further anti-prostate cancer drug discovery.
Supplemental Material
Download PDF (869.1 KB)Disclosure statement
The authors declare no conflict of interests.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Barrett I, Carr M, O'Boyle N, et al. Lead identification of conformationally restricted benzoxepin type combretastatin analogs: synthesis, antiproliferative activity, and tubulin effects. J Enzyme Inhib Med Chem 2010;25:180–94.
- Tripathi A, Durrant D, Lee RM, et al. Hydropathic analysis and biological evaluation of stilbene derivatives as colchicine site microtubule inhibitors with anti-leukemic activity. J Enzyme Inhib Med Chem 2009;24:1237–44.
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004;4:253–65.
- Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 2015;16:711–26.
- Lu Y, Chen J, Xiao M, et al. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 2012;29:2943–71.
- Tian C, Chen X, Zhang Z, et al. Design and synthesis of (2-(phenylamino)thieno[3,2-d]pyrimidin-4-yl)(3,4,5-trimethoxyphenyl)methanone analogues as potent anti-tubulin polymerization agents. Eur J Med Chem 2019;183:111679.
- Banerjee S, Arnst KE, Wang Y, et al. Heterocyclic-fused pyrimidines as novel tubulin polymerization inhibitors targeting the colchicine binding site: structural basis and antitumor efficacy. J Med Chem 2018;61:1704–18.
- Li W, Sun H, Xu S, et al. Tubulin inhibitors targeting the colchicine binding site: a perspective of privileged structures. Future Med Chem 2017;9:1765–94.
- Bueno O, Tobajas G, Quesada E, et al. Conformational mimetics of the α-methyl chalcone TUB091 binding tubulin: design, synthesis and antiproliferative activity. Eur J Med Chem 2018;148:337–48.
- Li W, Shuai W, Sun H, et al. Design, synthesis and biological evaluation of quinoline-indole derivatives as anti-tubulin agents targeting the colchicine binding site. Eur J Med Chem 2019;163:428–42.
- Wang G, Liu W, Gong Z, et al. Synthesis, biological evaluation, and molecular modelling of new naphthalene-chalcone derivatives as potential anticancer agents on MCF-7 breast cancer cells by targeting tubulin colchicine binding site. J Enzyme Inhib Med Chem 2020;35:139–44.
- Shao Y-Y, Yin Y, Lian B-P, et al. Synthesis and biological evaluation of novel shikonin-benzo[b]furan derivatives as tubulin polymerization inhibitors targeting the colchicine binding site. Eur J Med Chem 2020;190:112105.
- Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 2010;9:790–803.
- Goel P, Alam O, Naim MJ, et al. Recent advancement of piperidine moiety in treatment of cancer - a review. Eur J Med Chem 2018;157:480–502.
- Zeidner JF, Knaus HA, Zeidan AM, et al. Immunomodulation with pomalidomide at early lymphocyte recovery after induction chemotherapy in newly diagnosed AML and high-risk MDS. Leukemia 2020;34:1563–76.
- Kumaraswamy G, Kumar RS, Sampath B, et al. A concise diastereoselective approach to enantioenriched substituted piperidines and their in vitro cytotoxicity evaluation. Bioorg Med Chem Lett 2014;24:4439–43.
- Zeng Y, Cao R, Zhang T, et al. Design and synthesis of piperidine derivatives as novel human heat shock protein 70 inhibitors for the treatment of drug-resistant tumors. Eur J Med Chem 2015;97:19–31.
- Liu X-H, Li J, Shi JB, et al. Design and synthesis of novel 5-phenyl-N-piperidine ethanone containing 4,5-dihydropyrazole derivatives as potential antitumor agents. Eur J Med Chem 2012;51:294–9.
- Farooq M, Sharma A, Almarhoon Z, et al. Design and synthesis of mono-and di-pyrazolyl-s-triazine derivatives, their anticancer profile in human cancer cell lines, and in vivo toxicity in zebrafish embryos. Bioorg Chem 2019;87:457–64.
- Jo J, Kim H, Oh JY, et al. SAR optimization studies on a novel series of 2-anilinopyrimidines as selective inhibitors against triple-negative breast cancer cell line MDA-MB-468. Bioorg Med Chem Lett 2019;29:126752.
- Fu DJ, Fu L, Liu YC, et al. Structure-activity relationship studies of β-lactam-azide analogues as orally active antitumor agents targeting the tubulin colchicine site. Sci Rep 2017;7:12788.
- Fu DJ, Liu JF, Zhao RH, et al. Design and antiproliferative evaluation of novel sulfanilamide derivatives as potential tubulin polymerization inhibitors. Molecules 2017;22:1470.
- Fu D-J, Yang J-J, Li P, et al. Bioactive heterocycles containing a 3,4,5-trimethoxyphenyl fragment exerting potent antiproliferative activity through microtubule destabilization. Eur J Med Chem 2018;157:50–61.
- Fu D-J, Li P, Wu B-W, et al. Molecular diversity of trimethoxyphenyl-1,2,3-triazole hybrids as novel colchicine site tubulin polymerization inhibitors. Eur J Med Chem 2019;165:309–22.
- Fu DJ, Zhang SY, Liu YC, et al. Design, synthesis and antiproliferative activity studies of novel dithiocarbamate-chalcone derivates. Bioorg Med Chem Lett 2016;26:3918–22.
- Fu DJ, Hou YH, Zhang SY, et al. Efficient click reaction towards novel sulfonamide hybrids by molecular hybridization strategy as antiproliferative agents. J Chem Sci 2018;130:6.
- Mani SA, Guo W, Liao M-J, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–15.